

Abstract
In utero exposure to valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, causes neural tube, heart, and limb defects. Valpromide (VPD), the amide derivative of VPA, does not inhibit HDAC activity and is a weak teratogen in vivo. The detailed mechanism of action of VPA as a teratogen is not known. The goal of this study was to test the hypothesis that VPA disrupts regulation of the expression of genes that are critical in chondrogenesis and osteogenesis during limb development. Murine gestation day-12 embryonic forelimbs were excised and exposed to VPA or VPD in a limb bud culture system. VPA caused a significant concentration- dependent increase in limb abnormalities, which was correlated with its HDAC inhibitory effect. The signaling of both Sox9 and Runx2, key regulators of chondrogenesis, was downregulated by VPA. In contrast, VPD had little effect on limb morphology and no significant effect on HDAC activity or the expression of marker genes. Thus, VPA exposure dysregulated the expression of target genes directly involved in chondrogenesis and osteogenesis in the developing limb. Disturbances in these signaling pathways are likely to be a consequence of HDAC inhibition because VPD did not affect their expressions.
Key Words: teratogen, histone deacetylase inhibitor, Sox9, Runx2
Valproic acid (VPA) is commonly used to treat epilepsy and bipolar disorders; moreover, it is currently being tested as an anticancer agent (Duenas-Gonzalez et al., 2008). VPA is also a teratogen; in utero exposures may result in the induction of neural tube defects, heart abnormalities, craniosynostosis, and skeletal malformations, such as ectrodactyly and syndactyly (Lajeunie et al., 2001; Ornoy, 2006; Sharony et al., 1993; Vajda and Eadie, 2005). Although a number of mechanisms have been proposed for the anticonvulsant and antidepressant actions of VPA (reviewed in Terbach and Williams, 2009), the molecular signaling pathways by which VPA exposures lead to malformations are not known. VPA is an inhibitor of class I and II histone deacetylases (HDACs) (Phiel et al., 2001). Furthermore, recent studies have suggested that the teratogenicity of several compounds is linked to their ability to inhibit HDACs; apicidin, MS-275, sodium butyrate, and sodium salicylate are all HDAC inhibitors that induce defects of the axial skeleton in mice (Di Renzo et al., 2007, 2008). Valpromide (VPD), a structural analog of VPA, has been reported to have little potency either as an HDAC inhibitor or as a teratogen (Okada et al., 2004; Phiel et al., 2001). The mechanisms by which HDAC inhibitors interfere with bone development remain unknown.
The long bones of the limb are formed by endochondral ossification (reviewed in Kawakami et al., 2006). In the first step, chondrogenesis leads to the formation of a cartilage template that is subsequently replaced in the second step by bone mineralization during the osteogenesis phase. Chondrogenesis is a very tightly regulated process during which chondrocytes undergo sequential proliferation, differentiation, and hypertrophy. Each of these steps is characterized by the expression of specific marker genes. Sex-determining region Y (SRY)-box 9 (Sox9) and its two downstream target genes, Sox5 and Sox6, are expressed in early proliferative chondrocytes and form a complex that regulates the transcription of type II collagen (Col2a1) and Aggrecan, two proteins involved in extracellular matrix formation. Sox9 heterozygote mice exhibit limb malformations; conditional Sox9 knockout in the limb mesenchyme results in a complete failure to form any cartilage, clearly illustrating the pivotal role this gene plays in chondrogenesis (Akiyama et al., 2002; Bi et al., 2001). Once the proliferative stage is terminated, the chondrocytes undergo hypertrophy and begin to express Runt-related factor 2 (Runx2), a key regulator of hypertrophic differentiation, that induces the transcription of collagen 10a1 and the mineralization of the surrounding matrix (Ding et al., 2012). In a recent review, Bradley et al. (2011) highlighted the importance of the different roles of HDACs in both endochondral and intramembranous ossification. However, little is known about the effects of VPA, or any other known teratogen, on the regulation of chondrogenesis in the developing limb.
In this study, we tested the hypothesis that the inhibition of HDAC activity by VPA disturbs the expression of key genes that regulate chondrogenesis and osteogenesis, leading to limb malformations. To elucidate how VPA affects these processes, we exposed mouse embryonic limb buds to increasing concentrations of VPA and its analog, VPD, in an in vitro limb bud culture system. We then analyzed the effects of these compounds on histone acetylation, the morphology and differentiation of limb cartilage, and the expression of genes involved in limb bone formation. Our results show that VPA induces histone hyperacetylation; this epigenetic modification is directly correlated with inhibition of the expression of markers of both chondrogenesis and osteogenesis at early time points after exposure and with the subsequent limb development defects that are observed.
MATERIALS AND METHODS
Limb bud cultures and drug treatments.
Time-pregnant CD1 mice (20–25g) purchased from Charles River Canada Inc., (St-Constant, QC, Canada) were euthanized by cervical dislocation on gestation day-12 and their embryos were explanted. All animal studies complied with the guidelines established by the Canadian Council on Animal Care under protocol 1825. The embryonic forelimbs were cultured as previously described (Huang and Hales, 2002). Briefly, limbs were excised in Hank’s balanced salt solution (HBSS), pooled and cultured in vitro in 6 ml culture medium consisting of 75% BGJb medium (GIBCO BRL Products, Burlington, ON, Canada), 25% salt solution supplemented with ascorbic acid (160 μg/ml), and gentamicin (1 μl/ml, GIBCO BRL Products). Each culture was gassed with 50% O2, 5% CO2, and 45% N2. Different concentrations of sodium valproate (VPA, Sigma, St Louis, MO, no. P4543), dissolved in distilled water, or VPD (Katwijk Chemie, The Netherlands), dissolved in dimethyl sulfoxide (DMSO), were added to designated cultures. No differences were observed between limbs cultured with DMSO or water (data not shown).
Limb morphology.
Forelimbs were cultured for 6 days at 37°C with a change of medium and reoxygenation on day 3; VPA was not added to the culture medium at this time. Limbs were then fixed overnight in Bouin’s fixative, stained with 0.1% toluidine blue (Fisher Scientific, Nepean, ON, Canada) in 70% ethanol for 24h, dehydrated using a gradient of ethanol, and stored in cedarwood oil (Fisher Scientific). Limbs were observed using a dissection microscope, and the morphology and differentiation of each limb was assessed using a limb morphogenetic differentiation scoring system (Neubert and Barrach, 1977). Briefly, this system attributes a score to the radius, the ulna, the carpalia, and each one of the five digits according to their differentiation status. Five separate replicates (n = 5 bottles for each treatment and time, with 7–10 limbs per bottle) were done.
Real-time qRT-PCR.
Total RNA from homogenized limbs (4–5 limbs per group) was extracted using an RNeasy Microkit (Qiagen, Mississauga, ON, Canada). The RNA concentration and purity of each sample were assessed by spectrophotometry using a NanoDrop1000 spectrophotometer (Fisher Scientific). The samples were diluted to a working concentration of 10ng/μl, and transcripts were quantified using Quantitect One-Step SYBR Green RT-PCR (Qiagen) and the Roche LightCycler. The primer sets were designed using Primer 3 software (Table 1) and produced by alpha DNA (Montreal, QC, Canada), with the exception of the Runx2 primers that were purchased from Qiagen (no. QT00102193). The reaction was done in a final volume of 20 μl that was composed of 10 μl SYBR Green Master Mix, 2 μl of each forward and reverse primer in a 10μM solution, 0.2 μl Quantitect Reverse Transcriptase mix, 4.8 μl RNase-DNase-free water, and 1 μl sample. PCR was done under the following conditions: 20min at 50°C followed by 50 cycles of 95°C for 15min, 94°C for 15 s, 55°C for 30 s, and 72°C for 20 s. Serial dilutions of nontreated hindlimb RNA samples were used to create a standard curve. Each reaction was done in duplicate, averaged, and normalized to the amount of 18S rRNA transcripts. Each experiment was replicated 6–12 times per group.
Table 1.
Primer Sequences Used for qRT-PCR
| Gene | Sequence | |
|---|---|---|
| Sox9 | Forward | GATGCAGTGAGGAGCACTGA |
| Reverse | TATCCACGGCACACACACTT | |
| Sox5 | Forward | aagcagcaacaggagcaga |
| Reverse | aaaccaggaggaaggaggaa | |
| Sox6 | Forward | aacgaccacaccatcacctc |
| Reverse | aggcttccatttcatcctctc | |
| Col2a1 | Forward | GAAGGTGCTCAAGGTTCTCG |
| Reverse | CTTTGGCTCCAGGAATACCA | |
| Col10a1 | Forward | ACCCCAAGGACCTAAAGGAA |
| Reverse | CCCCAGGATACCCTGTTTTT | |
| 18S rRNA | Forward | AAACGGCTACCACATCCAAG |
| Reverse | CCTCCAATGGATCCTCGTTA |
Western blotting.
Cultures were ended at specified times. Limbs were homogenized by sonication in lysis buffer containing protease inhibitors: 150mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50mM Tris pH 7.5, 40 μg/ml bestatin, 0.2M phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 6 μg/ml aprotinin. Total protein was extracted and quantified using spectrophotometric Bio-Rad protein assays (Bio-Rad Laboratories, Mississauga, ON, Canada). Proteins (15–20 μg per sample) were separated by SDS-PAGE and transferred on polyvinylidene difluoride membranes (BioSciences Inc., Baie d’Urfé, QC, Canada). Precision standards (Bio-Rad Laboratories) were used as molecular weight markers. Membranes were blocked in 5% nonfat milk in Tris-buffered saline with Tween 20 (TBS-T) (137mM NaC1, 20mM Tris pH 7.4, 0.05% Tween 20) for 1h at room temperature, probed overnight at 4°C with primary antibodies, washed, and incubated with the secondary antibody for 2h at room temperature. Immunoblotting was done using polyclonal antibodies against histone 4 lysine 12 acetylation (H4K12ac, EMD Millipore, Billerica, MA, 1:5000), SOX9 (Abcam, Cambridge, MA, 1:500) and beta-actin (Santa Cruz Biotechnology Inc., Santa Cruz, CA, 1:10,000). The secondary antibodies, conjugated to horseradish peroxidase, were donkey anti-rabbit antibodies (1:5000, GE Healthcare Limited, Baie d’Urfé, QC, Canada) for H4K12ac and SOX9 and anti-goat antibodies (Santa Cruz Biotechnology Inc., 1:10,000) for beta-actin. Western blots were visualized with the Enhanced Chemiluminescence Plus Kit (GE Healthcare Limited) and quantified by densitometry using a Chemi-Imager 400 imaging system (Alpha Innotech, San Leandro, CA).
Primary cell cultures.
Embryonic forelimbs were collected on gestation day-12 and washed with low calcium HBSS (Hank’s Balanced Salt Solution Ca/Mg free [Gibco, Life Technologies, Burlington, ON], 1M HEPES pH 7.4, 0.15M CaCl2). Limbs were cut and incubated in a collagenase solution (1.5mg/ml collagenase type II, 300mg/ml bovine serum albumin in low calcium HBSS) for 2h at 36°C. Cells were sedimented from the suspension by centrifugation at 1000 × g for 5min and incubated with 2.5 g/ml pancreatin for 15min at room temperature. Cells were then washed with complete medium 199 (Medium 199, GIBCO, Life Technologies, 26.2mM NaHCO3, 25mM HEPES pH 7.4, 0.06 mg/ml gentamicin sulfate, 1% 100X antibiotic-antimycotic, 10% fetal bovine serum) and passed through a 40-μm filter cloth. Cells were counted with a hemocytometer, and approximately 1 × 106 cells were plated in a 12-well culture plate for 24h. Cells were then treated with vehicle or VPA (3.6mM) with or without 5 μg/ml actinomycin D (Sigma). Cultures were stopped at designated times, and total RNA was extracted and analyzed by qRT-PCR. The experiment was repeated 9 times.
Alcian blue staining.
Limbs were cultured for 24h and fixed overnight in 95% ethanol. Limbs were then transferred to alcian blue solution (15 mg alcian blue, 80 ml 95% ethanol, 20 ml glacial acetic acid) for 24h at room temperature and again to 95% ethanol for 24h. Limbs were transferred to 1 part 95% ethanol: 1 part glycerol solution and visualized by light microscopy; staining was qualitatively assessed. The experiment was repeated 5 times with 7–10 limbs/concentration and time in each experiment.
Statistical analyses.
All morphology and gene expression data sets were analyzed statistically using Systat 10.2 (Systat Software, Point Richmond, CA). The Mann-Whitney U-test with Bonferroni’s multiple-comparison correction was used to compare all concentration groups within a given time point. The minimum level of significance was p < 0.05.
RESULTS
Effects of VPA and VPD on Limb Morphology
The effects of VPA on the morphology and differentiation of embryonic forelimbs cultured in vitro are shown in Figure 1A. The gross appearance of limbs may vary slightly due to distortions in the rotating culture system. However, differentiation of the control limbs was normal; the cartilage template was properly formed, and long-elongated digits with phalanges were observed. Although the low-dose VPA treatment group showed minimal effects on morphology, the limbs exposed to 1.8 or 3.6mM VPA exhibited a marked decrease in growth and differentiation. The long bones were reduced in size; the staining of the carpalia was decreased; and the metacarpals were short and thick. Phalanges were often missing, and in the highest concentration group, several limbs with oligodactyly were observed. The morphology and state of differentiation of the limbs was assessed quantitatively using a limb morphogenetic differentiation scoring system (Fig. 1B). A significant concentration-dependent decrease in the limb score was observed in all VPA treatment groups. These results showed that VPA inhibited the development of the limbs in vitro and caused a pronounced decrease in cartilage formation. In contrast, limbs exposed to 0.6 or 1.8mM VPD were not different from control limbs (Fig. 1A). Limbs exposed to 3.6mM VPD exhibited a mild phenotype of under development of the phalanges; this resulted in a small but significant reduction in limb score (Fig. 1C).
Fig. 1.

Embryonic day-12 forelimbs were cultured in the presence of increasing concentrations of VPA or VPD (0, 0.6, 1.8, or 3.6mM) for 6 days. They were stained with 0.1% toluidine blue (A) to visualize cartilage formation and were scored according to their morphology (B and C). *p < 0.05.
Effects of VPA and VPD on Histone Acetylation
To determine whether the phenotype observed following VPA exposure was associated with HDAC inhibition, we examined the level of histone-4 acetylation as an indicator of global HDAC activity. VPA caused a rapid and concentration-dependent hyperacetylation of histone 4, as early as 1h following exposure; this was maintained up to 6h (Fig. 2A). VPD did not cause detectable changes in histone-4 acetylation (Fig. 2B).
Fig. 2.

VPA induction of histone 4 lysine 12 acetylation. H4K12 acetylation was used as a marker of HDAC inhibition and normalized to β-actin. Western blot of whole-cell protein extracts from VPA-treated (A) and VPD-treated (B) limbs at 3h following exposure (left). Limbs were cultured for 1, 3, or 6h, and proteins were quantified by densitometry (right). *p < 0.05.
Effects of VPA and VPD on Markers of Chondrogenesis
VPA exposure resulted in the downregulation of Sox9 mRNA expression by 1h in all VPA-exposed limbs (Fig. 3A). This inhibition peaked at 3h; transcripts returned to control levels in the limbs exposed to higher concentrations by 12h although histone-4 acetylation remained upregulated up to 24h (Supplementary fig. S1), suggesting that a feedback mechanism helps to maintain homeostasis. VPD-treated samples did not show any changes in Sox9 expression (Fig. 3B).
Fig. 3.

Sox9 gene expression following VPA (A) or VPD (B) treatment. Limbs were cultured with VPA for 1, 3, 6, 12, or 24h and with VPD for 1, 3, or 6h. Sox9 mRNA was quantified by qRT-PCR and normalized to 18S rRNA. Results were subsequently normalized to the control. *p < 0.05.
SOX9 downstream targets, Sox5 and Sox6, were downregulated after 3 and 6h, respectively (Figs. 4A and 4B). Col2a1 expression was significantly diminished in all VPA-treated limbs by 6h; this inhibition persisted up to 24h following exposure (Fig. 4C). This sequential inhibition of SOX9 downstream targets is consistent with the disruption of the Sox9-Sox5-Sox6 complex regulating collagen 2 expression previously reported, suggesting that Sox signaling is inhibited by VPA treatment. Interestingly, although its three downstream targets are downregulated at 6h, SOX9 protein expression was unchanged at this time point (Supplementary fig. S2). SOX9 also regulates Vegf gene expression in the limb, as the limb mesenchyme-specific Sox9 knockout mice showed a lack of Vegf expression and limb vasculature defects (Eshkar-Oren et al., 2009). However, Vegf expression remained constant in our model (Supplementary fig. S3), suggesting that VPA affects markers of chondrogenesis but not vascularization at this time during limb development.
Fig. 4.

Effects of VPA on Sox9 signaling and the expression of its downstream target genes. Sox5 (A), Sox6 (B), and Col2a1 (C) transcripts were quantified by qRT-PCR at 1, 3, 6, 12, or 24h following exposure to VPA. Transcripts were normalized to the 18S rRNA transcript, and results were normalized to the control. *p < 0.05.
In order to determine whether the effects on Sox9 mRNA levels were due to an increase in the degradation of the transcripts or a decrease in transcription, we used embryonic forelimbs to obtain a primary cell culture that was subsequently cotreated with VPA and actinomycin D, a transcriptional inhibitor (Fig. 5). Steady-state concentrations of Sox9 mRNA were significantly decreased following VPA treatment in primary cultures. As expected, a decrease in transcripts was observed over time in samples treated with actinomycin D alone. No additional effect was observed when cultures were cotreated with VPA and actinomycin D; therefore, the effects of VPA on Sox9 mRNA expression are transcription dependent.
Fig. 5.

Effects of actinomycin D on VPA-induced downregulation in primary limb bud cell cultures. Cells were exposed to vehicle or 3.6mM VPA with or without actinomycin D for 1 or 3h. Total RNA was extracted, and qRT-PCR for Sox9 and 18S rRNA was done. *p < 0.05 compared with control. #p < 0.05 compared with 1-h homologous groups.
To further examine the functional implications of the rapid effects on the expression of marker genes, limbs were exposed to VPA for 24h and cartilage condensation was assessed using Alcian blue staining. Control limbs showed deep staining in the digital space of 3–4 digits and in the ulna and radius, indicating the initiation of chondrogenic condensations (Fig. 6). Several limbs exhibited condensations of the prospective phalanges. VPA-exposed limb exhibited a drastic decrease or an absence of staining in multiple digits, suggesting a delay in this process as early as 24h following exposure.
Fig. 6.

Effects of VPA exposure on early chondrocyte mesenchymal condensations. Limbs were cultured for 24h in the presence of increasing concentrations of VPA, and the cartilage proteoglycans were stained with alcian blue. Limbs were then mounted on slides and observed with light microscopy.
Effects of VPA and VPD on Markers of Osteogenesis
To determine the effects of VPA and VPD on the different iation of osteocytes, the mRNA transcripts of Runx2 and its downstream gene, Collagen10a1 (Col10a1), were analyzed by qRT-PCR. VPA treatment resulted in a downregulation of the expression of both genes at 3 and 24h following exposure in a concentration-dependent manner (Figs. 7A and 8). VPD did not significantly alter Runx2 expression (Fig. 7B) although a trend was observed which correlated with the subtle decrease in limb score in the highest VPD concentration group. In order to determine whether these changes in gene expression had an impact on bone mineralization, limbs were cultured for 6 days, as for the morphological analysis, and stained with alizarin red. However, no staining was observed, suggesting that this process is not detected in our in vitro system (data not shown).
Fig. 7.
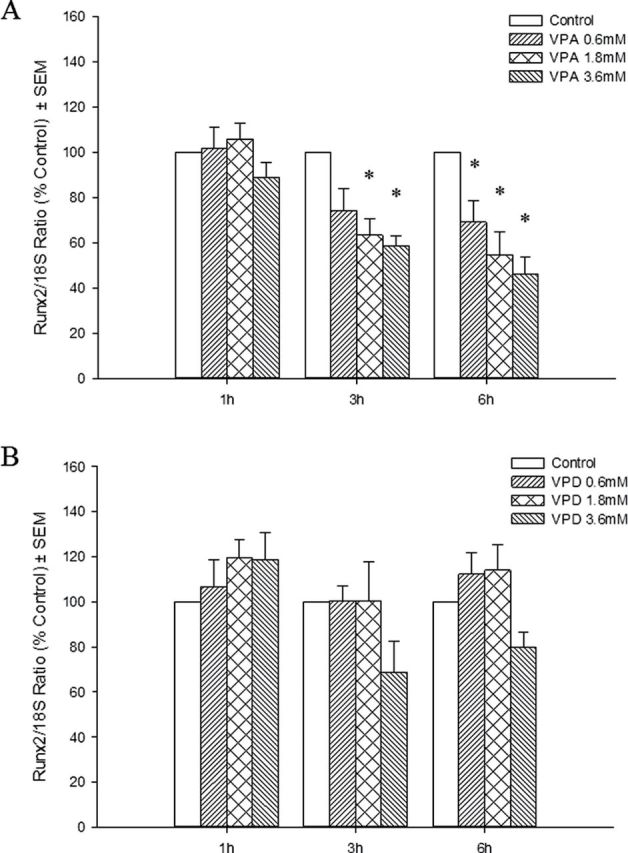
Effects of VPA and VPD on Runx2 gene expression. Total RNA was extracted from VPA-treated (A) and VPD-treated (B) limbs. Runx2 transcripts were quantified and normalized to 18S rRNA. Results were further normalized to the control. *p < 0.05.
Fig. 8.

Effects of VPA on Runx2 downstream signaling. At 6, 12, 24, and 48h, limb cultures were ended and total RNA was isolated. Col10a1 mRNA was quantified, and 18S rRNA was used for normalization. Results are expressed as % control. *p < 0.05.
DISCUSSION
Although VPA is both a commonly used anticonvulsant and an established human teratogen, the mechanisms underlying these actions are complex and have not been resolved. This study provides the first evidence that the Sox9 and Runx2 signaling pathways are targeted in VPA-induced limb developmental defects. As Sox9 is expressed in a number of tissues, VPA-induced downregulation of Sox9 may affect the formation of many organs during embryogenesis. In humans, a heterozygous deletion of the Sox9 gene results in a condition known as campomelic dysplasia that is often lethal due to thoracic cage defects and respiratory distress; survivors often exhibit sex reversal and severe skeletal and craniofacial malformations, such as cleft palate, scoliosis, and micrognathia. Some cases had spina bifida, global developmental delay and learning impairment, or developed seizures later in life, suggesting that neural development is impaired by Sox9 deletion (Mansour et al., 2002). Hence, the deregulation of this pathway may be involved in multiple defects.
The mechanism by which VPA exposure leads to the transcriptional downregulation of Sox9 expression is not known. The rapidity of the response suggests that it is independent of de novo protein synthesis. Our results showed that this effect is dependent on transcription and point toward a direct modulation of transcriptional regulation through posttranslational modifications. The VPA-induced induction of histone-4 hyperacetylation and inhibition of Sox9 expression are directly correlated, both with respect to timing and concentration dependence. Furthermore, VPD, a close analog of VPA, failed to affect either histone acetylation or Sox9 expression. HDAC inhibition may affect several pathways that are important in chondrogenesis. Two other HDAC inhibitors, trichostatin A and PDX101, were reported to repress collagen 2 expression in rabbit articular chondrocytes in a Wnt5a-dependent manner (Huh et al., 2007). These studies are consistent with our results as Col2a1 expression was repressed in VPA-exposed limbs.
In addition to Wnt signaling, several pathways regulating Sox9 expression have been identified; these include the bone-morphogenetic protein (BMP), nuclear factor kappa B (NFκB), and p38 MAPK pathways (Murakami et al., 2000; Pan et al., 2009). Downregulation of Bmp2 and upregulation of the inhibitory Smad7 were observed following trichostatin A–induced HDAC inhibition, ultimately leading to a downregulation of BMP signaling (Shakèd et al., 2008). Moreover, although histone tails and chromatin compaction have been considered the primary targets of HDACs, several nonhistone protein targets have now been identified. Among these are many Sox9 transcriptional regulators that are modulated by acetylation. For example, SMAD7 was shown to directly interact with HDAC1; trichostatin A treatment caused SMAD7 hyperacetylation and stabilization. Both the p65 and p52 subunits of the NFκB complex can be acetylated, and this acetylation modifies NFκB transcriptional activity (Hu and Colburn, 2005; Kim et al., 2012; Simonsson et al., 2005).
VPA treatment repressed the expression of Runx2, downstream of the Sox9 pathway in chondrocyte differentiation and limb development. Although VPD had no effect on Sox9 expression, limbs exposed to the highest concentration of VPD showed a nonsignificant decrease in Runx2 expression at both 3 and 6h. It is notable that there was a small decrease in limb score among this treatment group in the absence of any detectable increase in histone acetylation. It is possible that any effect of VPD on Runx2 expression does not involve HDAC inhibition. Although we have observed a repression in Runx2 expression in our limb buds in culture, other groups have reported that VPA and other HDAC inhibitors induce Runx2 in mesenchymal stem cells and calvarial-derived primary osteoblasts cells, thus promoting osteogenic differentiation (Cho et al., 2005; Schroeder and Westendorf, 2005). This discrepancy suggests that Runx2 regulation differs in whole limb buds and in cell culture, and it may be tissue and developmental-stage specific. Altogether, this study provides valuable insight into the mechanism of action of VPA during limb development. Identifying molecular pathways affected by this teratogen and other birth defect–inducing drugs is pivotal for the development of assays to test the potential of drugs as teratogens before they are prescribed to women who are pregnant or of child-bearing age.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
Canadian Institutes of Health Research (CIHR) (MOP-86511); F.P. is the recipient of training awards from CIHR and the Fonds de recherche du Québec - Santé (FRQS); B.F.H. is the recipient of a James McGill Professorship.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Katwijk Chemie for providing Valpromide.
References
- Akiyama H., Chaboissier M. C., Martin J. F., Schedl A., de Crombrugghe B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W., Huang W., Whitworth D. J., Deng J. M., Zhang Z., Behringer R. R., de Crombrugghe B. (2001). Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. U.S.A. 98, 6698–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley E. W., McGee-Lawrence M. E., Westendorf J. J. (2011). Hdac-mediated control of endochondral and intramembranous ossification. Crit. Rev. Eukaryot. Gene Expr. 21, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. H., Park H. T., Kim Y. J., Bae Y. C., Suh K. T., Jung J. S. (2005). Induction of osteogenic differentiation of human mesenchymal stem cells by histone deacetylase inhibitors. J. Cell. Biochem. 96, 533–542. [DOI] [PubMed] [Google Scholar]
- Di Renzo F., Broccia M. L., Giavini E., Menegola E. (2007). Relationship between embryonic histonic hyperacetylation and axial skeletal defects in mouse exposed to the three HDAC inhibitors apicidin, MS-275, and sodium butyrate. Toxicol. Sci. 98, 582–588. [DOI] [PubMed] [Google Scholar]
- Di Renzo F., Cappelletti G., Broccia M. L., Giavini E., Menegola E. (2008). The inhibition of embryonic histone deacetylases as the possible mechanism accounting for axial skeletal malformations induced by sodium salicylate. Toxicol. Sci. 104, 397–404. [DOI] [PubMed] [Google Scholar]
- Ding M., Lu Y., Abbassi S., Li F., Li X., Song Y., Geoffroy V., Im H. J., Zheng Q. (2012). Targeting Runx2 expression in hypertrophic chondrocytes impairs endochondral ossification during early skeletal development. J. Cell. Physiol. 227, 3446–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duenas-Gonzalez A., Candelaria M., Perez-Plascencia C., Perez-Cardenas E., de la Cruz-Hernandez E., Herrera L. A. (2008). Valproic acid as epigenetic cancer drug: Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat. Rev. 34, 206–222. [DOI] [PubMed] [Google Scholar]
- Eshkar-Oren I., Viukov S. V., Salameh S., Krief S., Oh C. D., Akiyama H., Gerber H. P., Ferrara N., Zelzer E. (2009). The forming limb skeleton serves as a signaling center for limb vasculature patterning via regulation of Vegf. Development. 136, 1263–1272. [DOI] [PubMed] [Google Scholar]
- Hu J., Colburn N. H. (2005). Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-kappaB/p65 DNA binding. Mol. Cancer Res. 3, 100–109. [DOI] [PubMed] [Google Scholar]
- Huang C., Hales B. F. (2002). Role of caspases in murine limb bud cell death induced by 4-hydroperoxycyclophosphamide, an activated analog of cyclophosphamide. Teratology. 66, 288–299. [DOI] [PubMed] [Google Scholar]
- Huh Y. H., Ryu J. H., Chun J. S. (2007). Regulation of type II collagen expression by histone deacetylase in articular chondrocytes. J. Biol. Chem. 282, 17123–17131. [DOI] [PubMed] [Google Scholar]
- Kawakami Y., Rodriguez-León J., Izpisúa Belmonte J. C. (2006). The role of TGF betas and Sox9 during limb chondrogenesis. Curr. Opin. Cell Biol. 18, 723–729. [DOI] [PubMed] [Google Scholar]
- Kim J. W., Jang S. M., Kim C. H., An J. H., Kang E. J., Choi K. H. (2012). New molecular bridge between RelA/p65 and NF-κB target genes via histone acetyltransferase TIP60 cofactor. J. Biol. Chem. 287, 7780–7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajeunie E., Barcik U., Thorne J. A., El Ghouzzi V., Bourgeois M., Renier D. (2001). Craniosynostosis and fetal exposure to sodium valproate. J. Neurosurg. 95, 778–782. [DOI] [PubMed] [Google Scholar]
- Mansour S., Offiah A. C., McDowall S., Sim P., Tolmie J., Hall C. (2002). The phenotype of survivors of campomelic dysplasia. J. Med. Genet. 39, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S., Kan M., McKeehan W. L., de Crombrugghe B. (2000). Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. U.S.A. 97, 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert D., Barrach H.-J. (1977). Techniques applicable to study morphogenetic differentiation of limb buds in organ culture. In Methods in Pre-Natal Toxicology. (Neubert D., Merker H.-J., Kwasigroch T, Eds.), pp. 241–251. Georg Thieme Publishers, Stuttgart, Germany. [Google Scholar]
- Okada A., Aoki Y., Kushima K., Kurihara H., Bialer M., Fujiwara M. (2004). Polycomb homologs are involved in teratogenicity of valproic acid in mice. Birth Defects Res. Part A Clin. Mol. Teratol. 70, 870–879. [DOI] [PubMed] [Google Scholar]
- Ornoy A. (2006). Neuroteratogens in man: An overview with special emphasis on the teratogenicity of antiepileptic drugs in pregnancy. Reprod. Toxicol. 22, 214–226. [DOI] [PubMed] [Google Scholar]
- Pan Q., Wu Y., Lin T., Yao H., Yang Z., Gao G., Song E., Shen H. (2009). Bone morphogenetic protein-2 induces chromatin remodeling and modification at the proximal promoter of Sox9 gene. Biochem. Biophys. Res. Commun. 379, 356–361. [DOI] [PubMed] [Google Scholar]
- Phiel C. J., Zhang F., Huang E. Y., Guenther M. G., Lazar M. A., Klein P. S. (2001). Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276, 36734–36741. [DOI] [PubMed] [Google Scholar]
- Schroeder T. M., Westendorf J. J. (2005). Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 20, 2254–2263. [DOI] [PubMed] [Google Scholar]
- Shakèd M., Weissmüller K., Svoboda H., Hortschansky P., Nishino N., Wölfl S., Tucker K. L. (2008). Histone deacetylases control neurogenesis in embryonic brain by inhibition of BMP2/4 signaling. PLoS ONE. 3, e2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharony R., Garber A., Viskochil D., Schreck R., Platt L. D., Ward R., Buehler B. A., Graham J. M., Jr (1993). Preaxial ray reduction defects as part of valproic acid embryofetopathy. Prenat. Diagn. 13, 909–918. [DOI] [PubMed] [Google Scholar]
- Simonsson M., Heldin C. H., Ericsson J., Grönroos E. (2005). The balance between acetylation and deacetylation controls Smad7 stability. J. Biol. Chem. 280, 21797–21803. [DOI] [PubMed] [Google Scholar]
- Terbach N., Williams R. S. (2009). Structure-function studies for the panacea, valproic acid. Biochem. Soc. Trans. 37,(Pt 5)1126–1132. [DOI] [PubMed] [Google Scholar]
- Vajda F. J., Eadie M. J. (2005). Maternal valproate dosage and foetal malformations. Acta Neurol. Scand. 112, 137–143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.