

Abstract
Mesotrypsin displays unusual resistance to inhibition by polypeptide trypsin inhibitors and cleaves some such inhibitors as substrates, despite a high degree of conservation with other mammalian trypsins. Substitution of Arg for the generally conserved Gly-193 has been implicated as a critical determinant of the unusual behavior of mesotrypsin toward protein protease inhibitors. Another relatively conserved residue near the trypsin active site, Tyr-39, is substituted by Ser-39 in mesotrypsin. Tyr-39, but not Ser-39, forms a hydrogen bond with the main chain amide nitrogen of the P4′ residue of a bound protease inhibitor. To investigate the role of the Tyr-39 H-bond in trypsin-inhibitor interactions, we reciprocally mutated position 39 in mesotrypsin and human cationic trypsin to Tyr-39 and Ser-39, respectively. We assessed inhibition constants and cleavage rates of canonical protease inhibitors bovine pancreatic trypsin inhibitor (BPTI) and the amyloid precursor protein Kunitz protease inhibitor domain by mesotrypsin and cationic trypsin variants, finding that the presence of Ser-39 relative to Tyr-39 results in a 4- to 13-fold poorer binding affinity and a 2- to 18-fold increase in cleavage rate. We also report the crystal structure of the mesotrypsin-S39Y•BPTI complex, in which we observe an H-bond between Tyr-39 OH and BPTI Ile-19 N. Our results indicate that the presence of Ser-39 in mesotrypsin, and corresponding absence of a single H-bond to the inhibitor backbone, makes a small but significant functional contribution to the resistance of mesotrypsin to inhibition and the ability of mesotrypsin to proteolyze inhibitors.
Keywords: peptidase, serine protease, proteolysis, protein protease inhibitor, Kunitz protease inhibitor domain, protein-protein binding interactions
Introduction
Mesotrypsin was first isolated and characterized by Rinderknecht et al. in the early 1980s.1, 2 It is one of three human trypsins encoded by different genes; the genes for cationic trypsinogen (PRSS1) and anionic trypsinogen (PRSS2) are located on chromosome 7q35, while the gene for mesotrypsinogen (PRSS3) is found on chromosome 9p13.3 The three pretrypsinogen isoforms are synthesized in pancreatic acinar cells, where the signal peptides are removed cotranslationally. The resulting trypsinogens are secreted into the pancreatic duct and finally discharged into the duodenal lumen where they are activated by enteropeptidase.4 Cationic trypsinogen is the most abundant isoform in pancreatic juice constituting ∼60% of total trypsin activity, followed by anionic trypsinogen constituting ∼30% of total trypsin activity, while mesotrypinogen is a minor isoform, representing ∼10% of the total trypsin activity.1, 2, 5
Trypsins are regulated in vivo by protein protease inhibitors. Canonical trypsin inhibitors feature binding loops of characteristic backbone conformation that bind to the trypsin active site extremely tightly, mimicking a substrate, but are cleaved extremely slowly.6–8 Although mesotrypsin shows high sequence homology with other trypsins, its interactions with protein protease inhibitors are very different. In sharp contrast to cationic and anionic trypsins, mesotrypsin shows almost total resistance to polypeptide trypsin inhibitors including pancreatic secretory trypsin inhibitor (SPINK1), soybean trypsin inhibitor (SBTI), amyloid precursor protein Kunitz protease inhibitor domain (APPI), bovine pancreatic trypsin inhibitor (BPTI), lima bean trypsin inhibitor (LBTI), and ovomucoid inhibitor, as well as the serpin α1-antitrypsin inhibitor.2, 9–11 Although mesotrypsin does bind to canonical trypsin inhibitors, it does so orders of magnitude more weakly than other trypsins.10, 11 Furthermore, it displays unusual catalytic activity in the accelerated cleavage of canonical inhibitors including SPINK1, SBTI, APPI, and BPTI.9–11 In previous work, we have found that mesotrypsin specifically targets polypeptide substrates constrained in a canonical conformation,11 which represents a distinctive element of specificity that differs fundamentally from that of other trypsins.
Mutagenesis studies have revealed that the resistance of mesotrypsin to polypeptide inhibitors is in large part attributable to the presence of active site Arg-193, which replaces the highly conserved Gly-193 of other trypsins.9, 12, 13 Structural studies have revealed that Arg-193 influences the shape and electrostatic potential of the “primed-side”14 subsites that interact with the C-terminal amino acids of the substrate on the leaving group side of the scissile bond.12 Our crystal structures of mesotrypsin bound to canonical inhibitors BPTI and APPI have revealed how steric conflict between Arg-193 and the P2′ residue of a bound inhibitor diminishes affinity by disrupting favorable primed-side interactions.10, 15, 16 Ser-39 is another atypical residue of mesotrypsin, located within loop A of the trypsin fold, a surface loop that borders the substrate binding cleft and contributes to primed-side substrate specificity.17 Residue 39 of mammalian trypsins is most frequently Tyr, but substitutions are observed in a small subset of trypsins [Fig. 1(A)]. The hydroxyl oxygen of Tyr-39, but not Ser-39, donates a hydrogen bond to the backbone of a bound inhibitor [Fig. 1(B)], and we hypothesized that this H-bond stabilizes the inhibitory interaction between classic trypsins and their canonical inhibitors, slowing inhibitor proteolysis. In the present study, we probe the functional significance of the loss of this H-bond in mesotrypsin as a potential contributing factor to the weakened binding and accelerated cleavage of canonical inhibitors by mesotrypsin.
Figure 1.
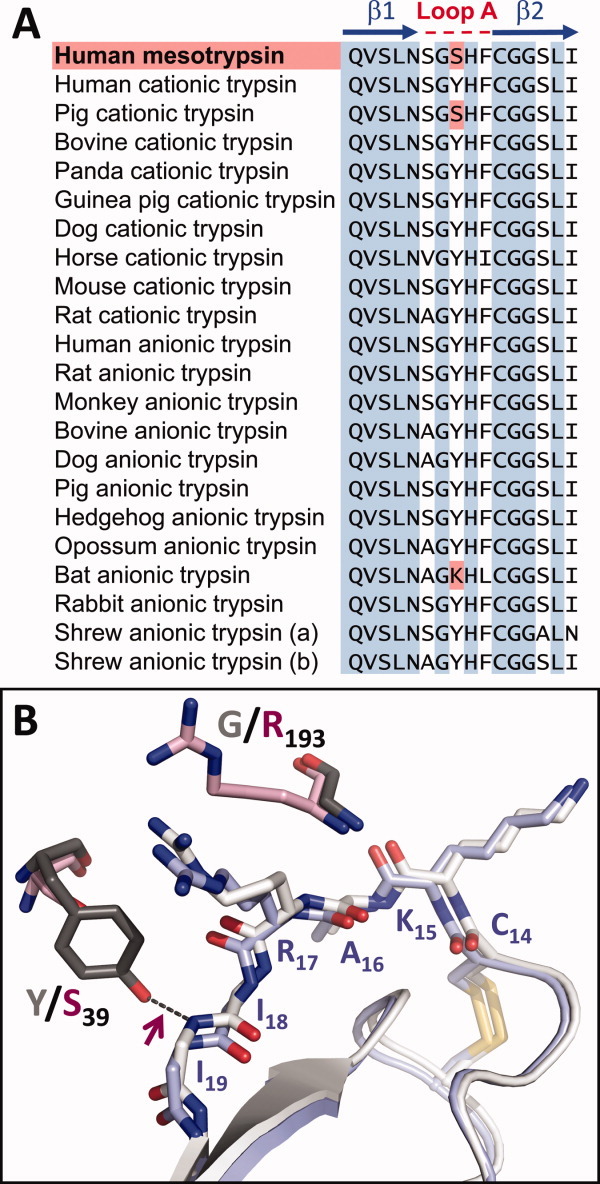
Sequence and structural variation among trypsins at residue 39. (A) A sequence alignment of mesotrypsin with mammalian cationic and anionic trypsins, spanning β-strands 1 and 2 and the intervening A-loop, reveals Ser at mesotrypsin residue 39 where a majority of trypsins possess Tyr. Sequences were obtained from MEROPS: the peptidase database for mesotrypsin (S01.119), cationic trypsins (S01.127, S01.151 and orthologs) and anionic trypsins (S01.119, S01.120, S01.258, and orthologs). (B) Cationic trypsin and mesotrypsin differ at residues 193 (Gly vs. Arg, respectively) and 39 (Tyr vs. Ser, respectively). Positions of these residues relative to a bound BPTI molecule are shown for human cationic trypsin·BPTI (gray/white; PDBID 2RA3) and mesotrypsin·BPTI (pink/light blue; PDBID 2RP9) structures. Tyr-39 in cationic trypsin forms an H-bond (maroon arrow) to the amide nitrogen of BPTI Ile-19, whereas Ser-39 in mesotrypsin does not. Only the main chain of BPTI residues 18 and 19 is shown so as not to obscure this interaction.
Results
The impact of trypsin residue 39 on inhibition by canonical inhibitors
Because residue 39 forms an H-bond with the primed side of a bound canonical inhibitor in trypsins possessing Tyr-39, but not in mesotrypsin that possesses Ser-39, we anticipated that this position may play a significant role in the weakened binding of inhibitors to mesotrypsin and/or in the accelerated cleavage of inhibitors by mesotrypsin. To dissect the contribution of this residue, we reciprocally mutated residue 39 in mesotrypsin and cationic trypsin to generate mesotrypsin-S39Y and cationic trypsin-Y39S, and measured the impact of these mutations on trypsin interactions with the canonical inhibitors BPTI and APPI.
To assess binding of mesotrypsin-S39Y to BPTI and APPI, we obtained equilibrium inhibition constants (Ki) using classic competitive inhibition experiments as we have described for WT mesotrypsin.10, 11, 16 We monitored cleavage of different concentrations of the chromogenic substrate carboxybenzyl-Gly-Pro-Arg-p-nitroanilide (zGPR-pNA) in the presence of varying concentrations of inhibitor and fit our data to the competitive inhibition model; an example is shown for the inhibition of mesotrypsin-S39Y by APPI [Fig. 2(A,B)]. Equilibrium inhibition constants of cationic trypsin-Y39S toward BPTI and APPI, which fit a slow, tight binding model of inhibition, were measured as described previously for WT cationic trypsin.10, 11 We monitored cleavage of zGPR-pNA in the presence of different concentrations of inhibitor for an extended reaction period to allow equilibrium rates to be achieved, and then analyzed data as described in the Materials and Methods section; an example is shown for inhibition of cationic trypsin-Y39S by BPTI [Fig. 2(C,D)].
Figure 2.
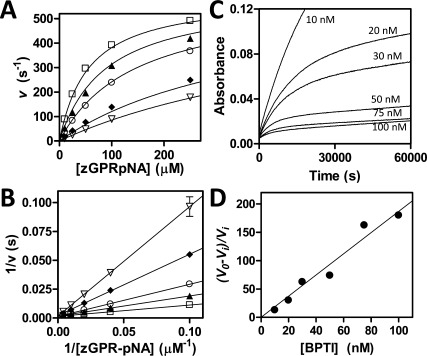
Inhibition of trypsin variants by BPTI and APPI. (A) Competitive inhibition of mesotrypsin-S39Y by APPI. Substrate concentration ranged from 10 to 250 μM and enzyme concentration was 0.25 nM; APPI concentrations were 0 (open squares), 7.5 nM (filled triangles), 20 nM (open circles), 75 nm (diamonds), and 120 nM (open triangles). Data were fit globally to the competitive inhibition equation as described in the Materials and Methods. (B) The Lineweaver-Burk double reciprocal transform displays convergence on the y-axis as is characteristic of the competitive model. (C) Slow, tight binding inhibition of cationic trypsin-Y39S by BPTI. A 16-h time course shows attainment of binding equilibrium by a series of parallel reactions with varying [BPTI] as indicated on the figure; substrate concentration was 150 μM and enzyme concentration was 0.1 nM. (D) Ki was determined from the slope of the replot of (v0 − vi)/vi versus [BPTI], where v0 is the uninhibited initial rate and vi corresponds to steady-state rates after attainment of enzyme-inhibitor binding equilibrium, as described in the Materials and Methods.
Values of Ki for inhibition of both enzymes and their mutants by BPTI and APPI are summarized in Table I. The presence of Tyr-39 strengthened the inhibition of both mesotrypsin and cationic trypsin by BPTI by ∼5-fold (relative to the analogous Ser-39 enzymes). Similar trends were observed with APPI, where mesotrypsin-S39Y was inhibited 12.5-fold more potently than WT mesotrypsin and WT cationic trypsin was inhibited 4-fold more potently than cationic trypsin-Y39S. Thus, we find that the identity of residue 39 (Tyr vs. Ser) has a significant and roughly similar impact on the inhibition of both enzymes.
Table I.
Impact of Trypsin Residue 39 on Inhibition by BPTI and APPI
| Enzymes compared | Inhibitor | Ki (M) (S39) | Ki (M) (Y39) | Fold diff. | ΔΔGa° (kcal/mol) |
|---|---|---|---|---|---|
| Cationic-Y39S/Cationic-wt | BPTI | 1.1 ± 0.3 × 10−10 | 2.0 ± 0.3 × 10−11a | 5.5 | −1.04 |
| Mesotrypsin-wt/Mesotrypsin-S39Y | BPTI | 1.4 ± 0.3 × 10−5 a | 2.7 ± 0.4 × 10−6 | 5.1 | −1.00 |
| Cationic-Y39S/Cationic-wt | APPI | 7.5 ± 1.3 × 10−10 | 1.7 ± 0.3 × 10−10b | 4.4 | −0.91 |
| Mesotrypsin-wt/Mesotrypsin-S39Y | APPI | 1.4 ± 0.2 × 10−7b | 1.1 ± 0.3 × 10−8 | 12.5 | −1.55 |
Because Ki approximates the reciprocal of the equilibrium association constant Ka, we can use our data to calculate the changes in the free-energy of association (ΔΔGa°) to an inhibitor between pairs of trypsin variants, revealing the thermodynamic stabilization or destabilization conferred on the enzyme-inhibitor complex by mutation of residue 39. From these calculations we estimate that the presence of Tyr-39 (relative to Ser-39) stabilizes trypsin complexes with APPI or BPTI by 0.9–1.6 kcal/mol (Table I).
The impact of trypsin residue 39 on cleavage of canonical inhibitors
We next used HPLC assays to monitor inhibitor cleavage in time course incubations with trypsins and to calculate rates of catalysis (kcat), as we have described previously.10, 15 For samples collected at different time points, intact BPTI was chromatographically resolved from cleavage products as shown in Figure 3(A); plots of residual intact BPTI (calculated from peak area) versus time allowed calculation of hydrolysis rates as shown in Figure 3(B). Similarly, chromatographic resolution of intact APPI from cleaved APPI* [Fig. 3(C)] enabled determination of APPI hydrolysis rates [Fig. 3(D)]. Because time courses were carried out at BPTI or APPI concentrations sufficient to saturate the enzyme, these hydrolysis rates can be considered approximations of kcat.10, 15 Hydrolysis rates are compiled in Table II.
Figure 3.
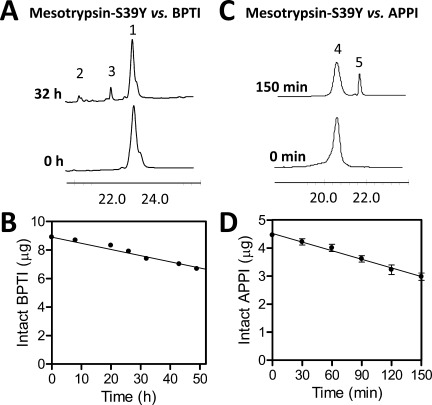
Hydrolysis of BPTI and APPI by mesotrypsin-S39Y. (A) HPLC chromatograms for samples taken at time zero and 32 h show resolution of intact BPTI (peak 1) from reduced cleavage products (peaks 2 and 3); the x-axis shows retention time in minutes and the y dimension represents absorbance at 210 nm. Reaction contained 60 μM BPTI and 2 μM mesotrypsin-S39Y. (B) Depletion of intact BPTI over time was quantified by peak integration, plotted versus reaction time, and fitted by linear regression to calculate hydrolysis rates. (C) HPLC traces from time zero and 4 h show resolution of intact APPI (peak 4) from nonreduced cleaved APPI* (peak 5). Reaction contained 25 μM APPI and 0.5 μM mesotrypsin-S39Y. (D) Depletion of intact APPI over time was quantified by peak integration, plotted vs. time, and fitted by linear regression to calculate hydrolysis rates.
Table II.
Impact of Trypsin Residue 39 on Cleavage of Inhibitors BPTI and APPI
| Enzymes compared | Inhibitor | kcat (s−1) (S39) | kcat (s−1) (Y39) | Fold diff. |
|---|---|---|---|---|
| Cationic-Y39S/Cationic-wt | BPTI | 6.3 ± 0.6 × 10−7 | 3.7 ± 0.4 × 10−7a | 1.7 |
| Mesotrypsin-wt/Mesotrypsin-S39Y | BPTI | 1.9 ± 0.2 × 10−4a | 3.1 ± 0.3 × 10−5 | 5.9 |
| Cationic-Y39S/Cationic-wt | APPI | 3.3 ± 1.1 × 10−5 | 1.8 ± 0.3 × 10−5b | 1.9 |
| Mesotrypsin-wt/Mesotrypsin-S39Y | APPI | 4.2 ± 0.3 × 10−2b | 2.4 ± 0.2 × 10−3 | 17.6 |
Differences between WT and cationic trypsin-Y39S in cleavage of BPTI and APPI are small (∼ 1.8-fold), suggesting that mutating residue 39 to Ser is insufficient to confer upon this enzyme significantly enhanced function for cleavage of canonical inhibitors (Table II). By contrast, mesotrypsin-S39Y revealed much more significant reductions in cleavage rates of both BPTI and APPI relative to WT mesotrypsin (6- and 18-fold, respectively). Thus, while cationic trypsin possesses minimal activity for inhibitor cleavage irrespective of the identity of residue 39, this residue does make a significant contribution to the ability of mesotrypsin to hydrolyze canonical inhibitors as substrates.
The impact of trypsin residue 39 on inhibition by modified canonical inhibitors
Canonical inhibitors inhibit their cognate proteases via a common “standard mechanism” or “Laskowski mechanism,” in which inhibitors bind to proteases extremely tightly in a substrate-like manner but are cleaved extremely slowly.6, 8, 18 An additional characteristic of the Laskowski mechanism is that inhibitor hydrolysis is reversible; a ‘modified’ inhibitor, singly-cleaved at the reactive site, can bind to the cognate protease and can undergo religation to produce a thermodynamic equilibrium mixture of intact and cleaved inhibitor.6, 8, 18 Although the intact and cleaved inhibitors, being reversibly interconvertible, are equivalent thermodynamic inhibitors, a cleaved inhibitor often binds much more slowly,19, 20 and shows a poorer apparent Ki on the typical experimental time scale.
Having found that the presence of Tyr-39 versus Ser-39 has a substantial impact on the affinity of trypsin toward APPI and BPTI, we next sought to determine whether this residue is similarly important for affinity toward the modified canonical inhibitors BPTI* and APPI*. We purified mesotrypsin-cleaved BPTI* and APPI* by HPLC and compared the ability of these modified inhibitors to inhibit human cationic trypsin, mesotrypsin, and their variants. All kinetic data are summarized in Table III. Similar to our results with the intact inhibitors, we found that cationic trypsin was inhibited ∼5-fold more strongly by BPTI* and APPI* than was cationic trypsin-Y39S. This result suggests that cationic trypsin complexes with BPTI* and APPI* feature the H-bond between Tyr-39 and the inhibitor backbone, that this H-bond is eliminated upon mutation of Tyr-39 to Ser, and that this H-bond confers a similar degree of stabilization to these complexes as to the analogs complexes with intact inhibitors. By contrast, the S39Y mutation had little impact on the inhibition of mesotrypsin by BPTI* or APPI*, suggesting that either the H-bond does not form between mesotrypsin-S39Y and the modified inhibitors, or that if it does form it does so only weakly or transiently, and does not confer significant stabilization to the complex.
Table III.
Kinetic Constants of Cationic Trypsin and Mesotrypsin with BPTI* and APPI*
| Enzymes compared | Inhibitor | Ki (M) (S39) | Ki (M) (Y39) | Fold diff. | ΔΔGa° (kcal/mol) |
|---|---|---|---|---|---|
| Cationic-Y39S/Cationic-wt | BPTI* | 1.2 ± 0.3 × 10−8 | 1.8 ± 0.4 × 10−9 | 6.5 | −1.15 |
| Mesotrypsin-wt/Mesotrypsin-S39Y | BPTI* | 9.8 ± 1.2 × 10−4 | 7.0 ± 0.6 × 10−4 | 1.4 | −0.21 |
| Cationic-Y39S/Cationic-wt | APPI* | 1.1 ± 0.1 × 10−7 | 2.2 ± 0.3 × 10−8a | 5.0 | −1.00 |
| Mesotrypsin-wt/Mesotrypsin-S39Y | APPI* | 1.4 ± 0.1 × 10−5 | 1.3 ± 0.1 × 10−5 | 1.1 | −0.03 |
Value previously reported in Ref.12.
Insights from crystal structure of mesotrypsin-S39Y•BPTI complex
To gain insights into the interaction of Tyr-39, introduced by mutagenesis into mesotrypsin, with a bound canonical inhibitor, we solved the crystal structure of the mesotrypsin-S39Y·BPTI complex. To avoid heterogeneity associated with proteolysis of BPTI and/or trypsin autoproteolysis, we used an inactive double mutant of mesotrypsin featuring a Ser-195 to Ala mutation in the active site in addition to the S39Y mutation. The structure was solved by molecular replacement and refined against data extending to 1.4 Å resolution; Table IV summarizes the crystal, data collection, and refinement statistics. As discussed under Materials and Methods, the mesotrypsin-S39Y/S195A·BPTI structure was obtained from a pseudo-merohedrally twinned crystal, requiring the use of twin-specific refinement procedures. The model contains four highly similar complexes in the asymmetric unit, each featuring a molecule of BPTI bound in the canonical fashion at the mesotrypsin active site. All four of the complexes are well-defined and near identical, with only small differences in the positioning of a number of flexible side chains on the periphery of the complex, distant from the enzyme-inhibitor interface. Aside from the presence of the mutated Tyr-39 side chain, the main structural change in the mesotrypsin-S39Y/S195A·BPTI complex compared to the mesotrypsin-S195A·BPTI structure is a series of backbone adjustments in mesotrypsin residues 37–39, including a flipped peptide bond between Ser-37 and Gly-38.
Table IV.
Data Collection and Refinement Statistics for Mesotrypsin-S39Y·BPTI Complex
| PDB ID | 4DG4 |
|---|---|
| Complexes per ASU | 4 |
| Space group | P 1 21 1 |
| Unit cell (Å) | 73.5, 109.9, 81.1 |
| 90°, 116.8°, 90° | |
| Resolution range (Å) | 30.2–1.4 |
| Unique reflections | 180,220 |
| Completeness (%) | 80.0 (29.9a) |
| Multiplicity | 4.5 (2.7) |
| I/S.D. | 14.4 (3.7) |
| Rsym | 0.115 (0.483) |
| Rcryst/Rfree | 16.63/21.14 |
| RMSD bonds (Å) | 0.003 |
| RMSD angles (°) | 0.557 |
Completeness at 1.76 Å is 95.5%.
At the enzyme-inhibitor interface in the vicinity of Tyr-39, we observe spacing and geometry that satisfy the standard criteria for H-bond formation between Tyr-39 OH and the backbone nitrogen of BPTI Ile-19 [Fig. 4(A)]. The distance between hydrogen donor and acceptor atoms ranges from 2.99 to 3.11 Å in the four copies of the mesotrypsin-S39Y/S195A·BPTI complex, slightly longer than the analogs 2.88 Å H-bond formed in the cationic trypsin·BPTI structure (PDBID 2RA3),10 the 2.89 Å H-bond formed in the bovine trypsin·BPTI structure (PDBID 2FTL),21 or the 2.96 Å H-bond formed in the rat anionic trypsin·BPTI structure (PDBID 3FP6),22 all of which were refined at similarly high resolution. The mesotrypsin-S39Y/S195A·BPTI structure supports the attribution of observed changes in free-energy of complex association to the energetic contribution of this single hydrogen bond, since we note no other compensatory changes in structure or intermolecular interactions that would complicate interpretation [Fig. 4(B)].
Figure 4.
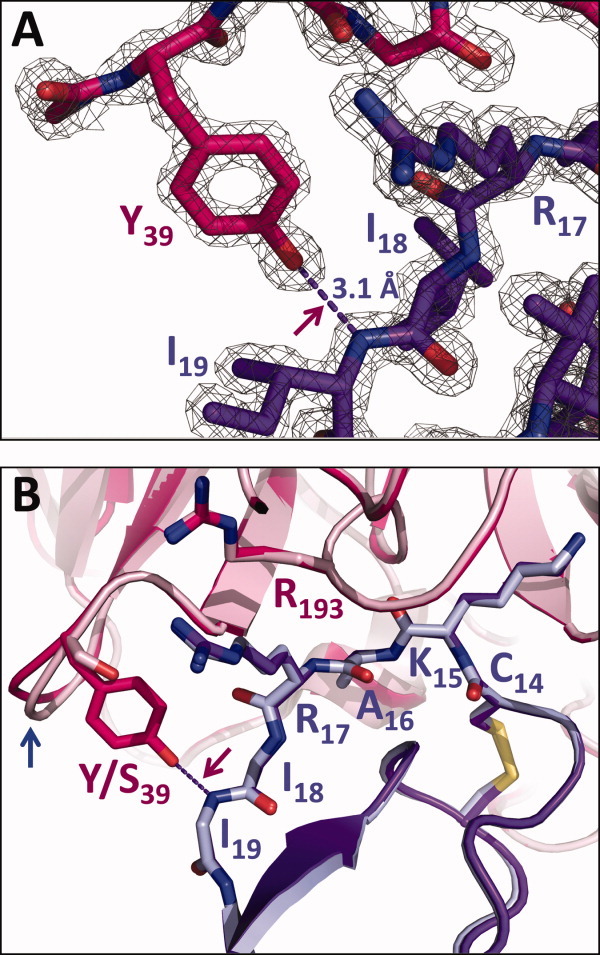
Tyr-39 H-bond formation in the mesotrypsin-Y39S·BPTI complex. (A) A close view of the Tyr-39 side chain with 2Fo-Fc map contoured at 2 sigma shows very clearly defined electron density supporting H-bond formation between Tyr-39 OH and Ile-19 N, with an interatomic spacing of 3.1 Å. (B) The mesotrypsin-S39Y·BPTI complex (hot pink/purple) superposed with the mesotrypsin·BPTI complex (light pink/light blue; PDBID 2RP9) show no observable structural change other than a small adjustment in residues 37–39 (blue arrow). Only the main chain of BPTI residues 18 and 19 is shown for clarity.
Discussion
Since its discovery, mesotrypsin has been regarded as a biochemical curiosity because of its resistance to protein inhibitors,2, 23 but beyond representing a point of novelty, this characteristic may be central to the function of mesotrypsin. The catalytic resiliency of mesotrypsin may enable robust signaling activities impervious to the presence of endogenous trypsin inhibitors.24–28 Additionally, the facility with which mesotrypsin can digest protein inhibitors in the diet, including soy bean trypsin inhibitor,9 and endogenous protein inhibitors involved in physiological regulatory mechanisms, including SPINK1 and protease nexin 2 (a soluble form of APP possessing the APPI domain),9, 11 suggests that this function may be a purposeful evolutionary adaptation. The Arg-193 residue in the substrate-binding cleft is essential for inhibitor resistance;9, 12, 13 we hypothesize that mesotrypsin may possess additional sequence and structural features that play supporting roles in its optimization for inhibitor resistance and inhibitor cleavage. Here, we evaluated the significance of Ser-39, finding that incorporation of this residue, and consequent loss of a single intermolecular H-bond relative to other trypsin complexes, modestly but significantly weakens inhibitor binding and accelerates inhibitor cleavage.
In our mutagenesis studies with mesotrypsin and cationic trypsin, the presence of Ser-39 rather than Tyr-39 reduced enzyme-inhibitor affinity by a factor of 4–13, corresponding to an increase of 0.9–1.6 kcal/mol in the free-energies of association for the complexes (Table I). Because we solved the structure of the mesotrypsin-S39Y·BPTI complex and found no other intermolecular contacts significantly altered by the mutation, we suggest that this free-energy difference can be attributed to the loss of the H-bond between Tyr-39 OH and the P4′ residue backbone nitrogen of the bound protease inhibitor (Fig. 4). This value is highly consistent with previous reports that place the energetic value of an uncharged hydrogen bond in macromolecular binding in the range of 0.5–1.8 kcal/mol.29, 30
Trypsin inhibition by canonical inhibitors requires not only strong protein–protein association, but also that the bound inhibitor resist degradation. Trypsin catalysis follows a two-step mechanism in which the catalytic Ser first attacks the scissile peptide bond to generate an acyl-enzyme intermediate, and then subsequent attack of water hydrolyzes the acyl-enzyme. High resolution structures of trypsin catalytic intermediates demonstrate that the trajectory of this hydrolytic water requires first that the primed-side leaving group residues of the substrate depart from the active site.31 We have presented evidence previously that the rate-determining step in proteolysis of canonical inhibitors involves dissociation of the primed-side leaving group residues and hydrolytic attack on the acyl-enzyme.32 Multiple stabilizing contacts between the primed-side inhibitor residues, the primed-side subsites of the enzyme, and the inhibitor scaffold, including multiple H-bonds essentially lacing the leaving group peptide into place, counteract leaving group dissociation (Fig. 5).
Figure 5.
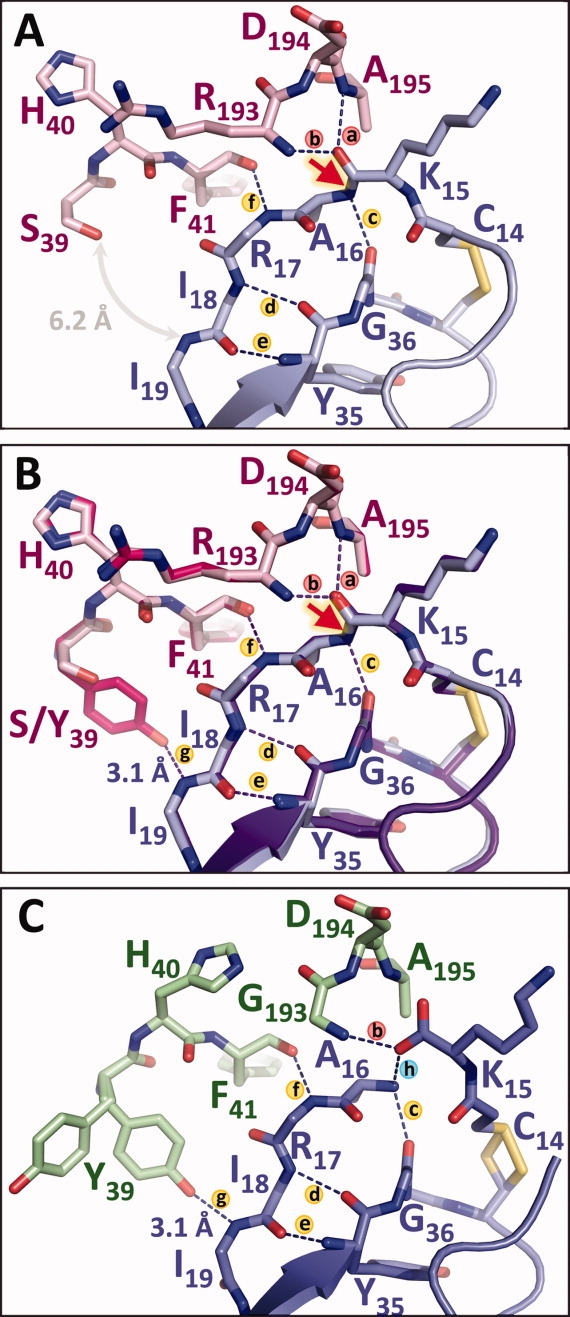
Primed-side H-bonds stabilize trypsin-inhibitor interactions and slow inhibitor hydrolysis. (A) In the mesotrypsin·BPTI structure (PDBID 2RP9), H-bonds (a and b) represent the “oxyanion hole” that will stabilize negative charge on the carbonyl oxygen during the cleavage of the scissile bond (red arrow). H-bonds (c–e) to the inhibitor scaffold and (f) to the enzyme counteract leaving group dissociation, but the absence of Tyr-39 leaves a large gap that may facilitate leaving group dissociation. (B) Mutation of mesotrypsin Ser-39 to Tyr restores H-bond g that is present in other trypsins, stabilizing the P3′ and P4′ residues between the enzyme and the inhibitor scaffold. (C) The structure of rat anionic trypsin bound to cleaved BPTI* (green/periwinkle; PDBID 3FP7) illustrates how H-bonds (c–g) may function cooperatively in retention of the primed-side leaving group after scissile bond cleavage. The opening of Tyr-39 seen in the alternate conformational rotamer in this structure may reflect the movement required of this residue prior to leaving group dissociation.
Mesotrypsin accelerates leaving group dissociation via steric repulsion between Arg-193 and the P2′ residue of the leaving group peptide, and via propagation of increased mobility to the primed-side residues of an inhibitor, destabilizing primed-side enzyme-inhibitor interactions.10, 15, 16 We found that mesotrypsin-S39Y cleaved canonical inhibitors about an order of magnitude more slowly than WT mesotrypsin (Table II), and thus our results indicate that the acquisition of Ser-39 contributes to the functional adaptation of mesotrypsin for digestion of protease inhibitors. These data suggest that an additional means by which mesotrypsin favors reaction progress is through eliminating the key Tyr-39 H-bond that helps to lace the leaving group in place [Fig. 5(A)], while the mesotrypsin-S39Y mutant restores this H-bond and thus diminishes mesotrypsin capacity for inhibitor hydrolysis [Fig. 5(B)]. We note that the reciprocal Y39S mutation of cationic trypsin does not similarly enhance cleavage of BPTI or APPI (Table II), suggesting Ser-39 (or rather the absence of Tyr-39) plays a supporting role to Arg-193 in destablizing the primed-side interface, and that in the context of Gly-193, the identity of trypsin residue 39 is far less important.
Another difference between cationic trypsin and mesotrypsin is that while for cationic trypsin mutation of residue 39 had a substantial impact on affinity toward cleaved inhibitors BPTI* and APPI*, the impact for mesotrypsin was minimal (Table III). We interpret this as an indication that the Tyr-39 H-bond forms readily and offers similar energetic stabilization to the cationic trypsin-cleaved inhibitor complexes as to the complexes with intact inhibitors whereas for mesotrypsin complexes with cleaved inhibitors this bond does not form readily or offers little stabilization. A structure of rat anionic trypsin bound to cleaved BPTI* has been reported,22 in which Tyr-39 is present in two rotameric conformations, one in which the H-bond to the backbone nitrogen of BPTI Ile-19 is formed with a distance of 3.07 Å, and the other in which the Tyr side chain has swung outward eliminating contact with the inhibitor [Fig. 5(C)]. We speculate that the primed-side contacts in an acyl-enzyme are similar to those observed in the anionic trypsin·cleaved BPTI* complex, and that the “opening” of Tyr-39 evidenced in this structure is an obligate first step in dissociation of the primed-side leaving group of an inhibitor. In mesotrypsin, the presence of Ser at residue 39 creates a more open channel enabling leaving group disengagement [Fig. 5(A)], while the presence of Arg-193 provides the major impetus promoting leaving group dissociation and reaction progression. In tandem, these adaptations optimize mesotrypsin for resistance to inhibition and for directed cleavage of trypsin inhibitors.
Materials and Methods
Production of recombinant proteins
Expression plasmids for human cationic trypsinogen and mesotrypsinogen have been described previously.9, 33 Mutations were introduced using the QuikChange kit (Stratagene) and verified by sequencing. Recombinant mesotrypsinogen, mesotrypsinogen-S39Y, human cationic trypsinogen, trypsinogen-Y39S, and a catalytically inactive mesotrypsinogen-S39Y/S195A double mutant were expressed in E. coli, isolated from inclusion bodies, refolded, purified, and activated with bovine enteropeptidase as previously described.10 Recombinant APPI was expressed in Pichia pastoris as a soluble secreted protein and purified essentially as described previously.15, 34 Modified BPTI* and APPI* singly cleaved at the reactive site bond were produced by preparative enzymatic cleavage using mesotrypsin and purified by HPLC essentially as described previously for APPI*.11
Inhibition studies
All active trypsin concentrations were quantified by active-site titration using 4-nitrophenyl 4-guanidinobenzoate (Sigma). BPTI (Aprotinin, Sigma) and APPI concentrations were determined by titration with a titrated bovine trypsin (Sigma) as described previously.10, 35 Concentrations of the chromogenic substrate zGPR-pNA (Sigma) were determined by end point assay. For determination of mesotrypsin and mesotrypsin-S39Y inhibition constants, enzyme assays performed at 37°C in the presence of varying concentrations of substrate and inhibitor were followed spectroscopically for 3–5 min, and initial rates were determined from the absorbance increase caused by the release of p-nitroaniline (ε410 = 8480 M−1 cm−1), as previously described.10, 16 Data were globally fitted by multiple regression to the classic competitive inhibition equation [Eq. (1)], using Prism (GraphPad Software, San Diego, CA). Reported inhibition constants are average values obtained from three or more independent experiments.
| (1) |
For measurement of inhibition constants with cationic trypsin and trypsin-Y39S, the observation of slow, tight-binding behavior required an alternative kinetic treatment, using methods that we have described previously.10, 36 Reactions were run at 25°C, and were followed spectroscopically for 16 h so that reliable steady-state rates could be obtained. Inhibition constants were calculated using Eq. (2) as previously described,10, 36 where vi and v0 are the steady-state rates in the presence and absence of inhibitor, KM is the Michaelis constant for substrate cleavage, and [S0] and [I0] are the initial concentrations of substrate and inhibitor. Equation (2) predicts that a plot of (v0 − vi)/vi versus [I0] will yield a straight line passing through the origin, with a slope of 1/Ki (1 + [S0]/Km), allowing calculation of Ki.10, 36 Calculations were performed using Km values of 36.5 μM for cationic trypsin and 43.3 μM for cationic trypsin-Y39S, determined from Michaelis–Menten kinetic studies performed in triplicate (not shown).
| (2) |
For inhibition of cationic trypsin and trypsin-Y39S by cleaved inhibitors BPTI* and APPI*, Ki values were found to be in the low nanomolar range where we have transitioned between the classic competitive and slow, tight binding kinetic models. These values were measured via both methods, which gave generally consistent results; the values reported here are fit from the classic competitive inhibition model, which gave smaller error margins between replicate experiments.
Changes in the ground state free-energy of association (ΔΔGa°) to each inhibitor upon alteration of residue 39 of mesotrypsin or cationic trypsin were calculated according to Eq. (3), where R is the gas constant, T is the absolute temperature, and the equilibrium association constant Ka is the reciprocal of our measured equilibrium inhibition constant (1/Ki).
| (3) |
Inhibitor hydrolysis studies
To quantitatively determine the catalysis rates (kcat) for cleavage of inhibitors BPTI or APPI by trypsin variants, we measured the depletion of intact APPI or BPTI in time course incubations with mesotrypsin-S39Y or cationic trypsin-Y39S as previously described.10, 11, 15 Incubations of mesotrypsin-S39Y (2 μM) or cationic trypsin-Y39S (6 μM) with BPTI (60 μM) were carried out in 0.1M Tris-HCl pH 8.0 and 1 mM CaCl2 at 37°C, and aliquots were withdrawn at periodic intervals and prepared for HPLC analysis as previously reported.10 Incubations with APPI (25 μM) were carried out similarly except that mesotrypsin-S39Y and cationic trypsin-Y39S concentrations were 0.5 and 2 μM, respectively, and the time point samples were prepared for HPLC analysis as previously described.11, 15 Enzyme, inhibitors, and hydrolysis products were resolved by HPLC and the disappearance of intact inhibitors over time was quantified by peak integration as described previously;10, 11, 15 initial rates were obtained by linear regression using a minimum of five data points within the initial linear phase of the reaction, and not exceeding 50% conversion of intact inhibitor to hydrolysis products. Hydrolysis rates reported for each inhibitor represent the average of three independent experiments.
Crystallization of mesotrypsin S39Y-BPTI complex
Mesotrypsin-S39Y/S195A dissolved in 1 mM HCl and BPTI dissolved in 10 mM NaOAc, pH 6.5, were mixed in a 1:1 stoichiometric molar ratio. The heterodimeric complex was further purified by HiLoad Superdex 75 gel filtration chromatography (GE Healthcare), exchanged into 10 mM NaOAc, pH 6.5, and concentrated to ∼ 5 mg/mL. The complex was crystallized at room temperature in hanging drops, over a reservoir of 1.6M (NH4)2SO4; drops (4 μL) were prepared by mixing equal volumes of protein and reservoir solutions. Crystals (0.2 × 0.3 × 0.2 mm3) appeared within 3 days and grew over the course of 6 days. Crystals were harvested, soaked in a 1.6M (NH4)2SO4 and 20% glycerol and cryo-cooled in liquid N2.
X-ray data collection, structure solution, and model refinement
Synchrotron X-ray data were collected from crystals at 100 K using an ADSC CCD detector at beam line X12-B at the National Synchrotron Light Source, Brookhaven National Laboratory. The software package HKL200037 was used for integrating, scaling, and merging the reflection data. The structure was solved by molecular replacement using the program Phaser38 operated by PHENIX,39 using the mesotrypsin·BPTI complex structure (PDBID 2R9P)10 as the search model. Crystals were pseudo-merohedrally twinned as estimated by the program Xtriage;40 a crystal with minimal twinning of twin fraction 0.08 was selected for structure solution. The successful solution contained four copies of the heterodimeric complex in the asymmetric unit. COOT41 and PHENIX42 were used for manual rebuilding and automated refinement, respectively. Using PHENIX, the structure was refined using a twin-specific target function based on the twin law “-h, -k, and h+l”. TLS refinement was employed, with each protein chain assigned to a separate TLS group. Waters, ions, and alternative conformations of protein residues were added using COOT. A test set comprised of 10% of the total reflections was excluded from refinement to allow calculation of the free R factor. All superpositions and structure figures were created using the graphics software Pymol.
Acknowledgments
The authors wish to thank Blake Fechtel, Kasia Ellsworth, and Chuhan Zong for technical assistance. Diffraction data were measured at beamline X12-B of the National Synchrotron Light Source, which is supported by the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and the National Center for Research Resources of the National Institutes of Health.
Glossary
Abbreviations
- APPI
amyloid precursor protein Kunitz protease inhibitor domain
- APPI*
APPI that has been cleaved at the Arg-15–Ala-16 (P1–P1′) reactive site bond. Inhibitor residues are designated by the nomenclature of Schechter and Berger14: starting from the reactive site bond, residues are numbered P1, P2, P3, etc. in the direction of the N terminus (collectively the “non-primed side” residues) and P1′, P2′P3′, etc. in the direction of the C terminus (collectively the “primed side” residues)
- BPTI
bovine pancreatic trypsin inhibitor
- BPTI*
BPTI that has been cleaved at the Lys-15 – Ala-16 (P1–P1′) reactive site bond
- H-bond
hydrogen bond
- LBTI
lima bean trypsin inhibitor
- SBTI
soybean trypsin inhibitor
- SPINK1
pancreatic secretory trypsin inhibitor
- zGPR-pNA
carboxybenzyl-Gly-Pro-Arg-p-nitroanilide
References
- 1.Rinderknecht H, Renner IG, Carmack C. Trypsinogen variants in pancreatic juice of healthy volunteers, chronic alcoholics, and patients with pancreatitis and cancer of the pancreas. Gut. 1979;20:886–891. doi: 10.1136/gut.20.10.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rinderknecht H, Renner IG, Abramson SB, Carmack C. Mesotrypsin: a new inhibitor-resistant protease from a zymogen in human pancreatic tissue and fluid. Gastroenterology. 1984;86:681–692. [PubMed] [Google Scholar]
- 3.Chen JM, Ferec C. Genes, cloned cDNAs, and proteins of human trypsinogens and pancreatitis-associated cationic trypsinogen mutations. Pancreas. 2000;21:57–62. doi: 10.1097/00006676-200007000-00052. [DOI] [PubMed] [Google Scholar]
- 4.Rinderknecht H. Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig Dis Sci. 1986;31:314–321. doi: 10.1007/BF01318124. [DOI] [PubMed] [Google Scholar]
- 5.Scheele G, Bartelt D, Bieger W. Characterization of human exocrine pancreatic proteins by two-dimensional isoelectric focusing/sodium dodecyl sulfate gel electrophoresis. Gastroenterology. 1981;80:461–473. [PubMed] [Google Scholar]
- 6.Laskowski M, Jr, Kato I. Protein inhibitors of proteinases. Annu Rev Biochem. 1980;49:593–626. doi: 10.1146/annurev.bi.49.070180.003113. [DOI] [PubMed] [Google Scholar]
- 7.Bode W, Huber R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur J Biochem. 1992;204:433–451. doi: 10.1111/j.1432-1033.1992.tb16654.x. [DOI] [PubMed] [Google Scholar]
- 8.Krowarsch D, Cierpicki T, Jelen F, Otlewski J. Canonical protein inhibitors of serine proteases. Cell Mol Life Sci. 2003;60:2427–2444. doi: 10.1007/s00018-003-3120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szmola R, Kukor Z, Sahin-Toth M. Human mesotrypsin is a unique digestive protease specialized for the degradation of trypsin inhibitors. J Biol Chem. 2003;278:48580–48589. doi: 10.1074/jbc.M310301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salameh MA, Soares AS, Hockla A, Radisky ES. Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J Biol Chem. 2008;283:4115–4123. doi: 10.1074/jbc.M708268200. [DOI] [PubMed] [Google Scholar]
- 11.Salameh MA, Robinson JL, Navaneetham D, Sinha D, Madden BJ, Walsh PN, Radisky ES. The amyloid precursor protein/protease nexin 2 Kunitz inhibitor domain is a highly specific substrate of mesotrypsin. J Biol Chem. 2010;285:1939–1949. doi: 10.1074/jbc.M109.057216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katona G, Berglund GI, Hajdu J, Graf L, Szilagyi L. Crystal structure reveals basis for the inhibitor resistance of human brain trypsin. J Mol Biol. 2002;315:1209–1218. doi: 10.1006/jmbi.2001.5305. [DOI] [PubMed] [Google Scholar]
- 13.Szepessy E, Sahin-Toth M. Human mesotrypsin exhibits restricted S1′ subsite specificity with a strong preference for small polar side chains. FEBS J. 2006;273:2942–2954. doi: 10.1111/j.1742-4658.2006.05305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 15.Salameh MA, Soares AS, Navaneetham D, Sinha D, Walsh PN, Radisky ES. Determinants of affinity and proteolytic stability in interactions of Kunitz family protease inhibitors with mesotrypsin. J Biol Chem. 2010;285:36884–36896. doi: 10.1074/jbc.M110.171348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salameh MA, Soares AS, Hockla A, Radisky DC, Radisky ES. The P(2)' residue is a key determinant of mesotrypsin specificity: engineering a high-affinity inhibitor with anticancer activity. Biochem J. 2011;440:95–105. doi: 10.1042/BJ20110788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laskowski M, Qasim MA. What can the structures of enzyme-inhibitor complexes tell us about the structures of enzyme substrate complexes? Biochim Biophys Acta. 2000;1477:324–337. doi: 10.1016/s0167-4838(99)00284-8. [DOI] [PubMed] [Google Scholar]
- 19.Quast U, Engel J, Steffen E, Tschesche H, Kupfer S. Kinetics of the interaction of alpha-chymotrypsin with trypsin kallikrein inhibitor (Kunitz) in which the reactive-site peptide bond Lys-15-Ala-16 is split. Eur J Biochem. 1978;86:353–360. doi: 10.1111/j.1432-1033.1978.tb12317.x. [DOI] [PubMed] [Google Scholar]
- 20.Quast U, Engel J, Steffen E, Tschesche H, Kupfer S. Stopped-flow kinetics of the resynthesis of the reactive site peptile bond in kallikrein inhibitor (Kunitz) by beta-trypsin. Biochemistry. 1978;17:1675–1682. doi: 10.1021/bi00602a015. [DOI] [PubMed] [Google Scholar]
- 21.Hanson WM, Domek GJ, Horvath MP, Goldenberg DP. Rigidification of a flexible protease inhibitor variant upon binding to trypsin. J Mol Biol. 2007;366:230–243. doi: 10.1016/j.jmb.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zakharova E, Horvath MP, Goldenberg DP. Structure of a serine protease poised to resynthesize a peptide bond. Proc Natl Acad Sci U S A. 2009;106:11034–11039. doi: 10.1073/pnas.0902463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nyaruhucha CN, Kito M, Fukuoka SI. Identification and expression of the cDNA-encoding human mesotrypsin(ogen), an isoform of trypsin with inhibitor resistance. J Biol Chem. 1997;272:10573–10578. doi: 10.1074/jbc.272.16.10573. [DOI] [PubMed] [Google Scholar]
- 24.Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. 2004;279:13532–13539. doi: 10.1074/jbc.M312090200. [DOI] [PubMed] [Google Scholar]
- 25.Grishina Z, Ostrowska E, Halangk W, Sahin-Toth M, Reiser G. Activity of recombinant trypsin isoforms on human proteinase-activated receptors (PAR): mesotrypsin cannot activate epithelial PAR-1, -2, but weakly activates brain PAR-1. Br J Pharmacol. 2005;146:990–999. doi: 10.1038/sj.bjp.0706410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Luo W, Wartmann T, Halangk W, Sahin-Toth M, Reiser G. Mesotrypsin, a brain trypsin, activates selectively proteinase-activated receptor-1, but not proteinase-activated receptor-2, in rat astrocytes. J Neurochem. 2006;99:759–769. doi: 10.1111/j.1471-4159.2006.04105.x. [DOI] [PubMed] [Google Scholar]
- 27.Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K, Peterson A, Chapman K, Hollenberg MD, Vergnolle N, Bunnett NW. Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem. 2007;282:26089–26100. doi: 10.1074/jbc.M703840200. [DOI] [PubMed] [Google Scholar]
- 28.Ceppa EP, Lyo V, Grady EF, Knecht W, Grahn S, Peterson A, Bunnett NW, Kirkwood KS, Cattaruzza F. Serine proteases mediate inflammatory pain in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1033–G1042. doi: 10.1152/ajpgi.00305.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MM, Winter G. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:2357–238. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 30.Fersht AR. The hydrogen bond in molecular recognition. Trends in Biochemical Sciences. 1987;12:301–304. [Google Scholar]
- 31.Radisky ES, Lee JM, Lu CJ, Koshland DE., Jr Insights into the serine protease mechanism from atomic resolution structures of trypsin reaction intermediates. Proc Natl Acad Sci U S A. 2006;103:6835–6840. doi: 10.1073/pnas.0601910103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radisky ES, Koshland DE., Jr A clogged gutter mechanism for protease inhibitors. Proc Natl Acad Sci U S A. 2002;99:10316–10321. doi: 10.1073/pnas.112332899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahin-Toth M. Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J Biol Chem. 2000;275:22750–22755. doi: 10.1074/jbc.M002943200. [DOI] [PubMed] [Google Scholar]
- 34.Navaneetham D, Jin L, Pandey P, Strickler JE, Babine RE, Abdel-Meguid SS, Walsh PN. Structural and mutational analyses of the molecular interactions between the catalytic domain of factor XIa and the Kunitz protease inhibitor domain of protease nexin 2. J Biol Chem. 2005;280:36165–36175. doi: 10.1074/jbc.M504990200. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski M, Jr, Kato I, Leary TR, Schrode J, Sealock RW. In: Proteinase inhibitors: proceedings of the 2nd international research conference. Fritz H, Tschesche H, Greene LJ, Truscheit E, editors. Berlin, New York: Springer-Verlag; 1974. p. 16. 751. [Google Scholar]
- 36.Radisky ES, King DS, Kwan G, Koshland DE., Jr The role of the protein core in the inhibitory power of the classic serine protease inhibitor, chymotrypsin inhibitor 2. Biochemistry. 2003;42:6484–6492. doi: 10.1021/bi034275d. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography, Part A. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 39.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 40.Zwart PH, Grosse-Kunstleve RW, Adams PD. 2005. Xtriage and Fest: automatic assessment of X-ray data and substructure structure factor estimation. CCP4 Newsletter 43.
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Afonine PV, Grosse-Kunstleve RW, Adams PD. 2005. The Phenix refinement framework. CCP4 Newsletter 42.