

Abstract
Gaining more insight into the mechanisms governing the behavior of proteins at solid/liquid interfaces is particularly relevant in the interaction of high-value biologics with storage and delivery device surfaces, where adsorption-induced conformational changes may dramatically affect biocompatibility. The impact of structural stability on interfacial behavior has been previously investigated by engineering nonwild-type stability mutants. Potential shortcomings of such approaches include only modest changes in thermostability, and the introduction of changes in the topology of the proteins when disulfide bonds are incorporated. Here we employ two members of the aldo-keto reductase superfamily (alcohol dehydrogenase, AdhD and human aldose reductase, hAR) to gain a new perspective on the role of naturally occurring thermostability on adsorbed protein arrangement and its subsequent impact on desorption. Unexpectedly, we find that during initial adsorption events, both proteins have similar affinity to the substrate and undergo nearly identical levels of structural perturbation. Interesting differences between AdhD and hAR occur during desorption and both proteins exhibit some level of activity loss and irreversible conformational change upon desorption. Although such surface-induced denaturation is expected for the less stable hAR, it is remarkable that the extremely thermostable AdhD is similarly affected by adsorption-induced events. These results question the role of thermal stability as a predictor of protein adsorption/desorption behavior.
Keywords: protein adsorption, protein desorption, thermal stability, alcohol dehydrogenase, human aldose reductase, circular dichroism
Introduction
Over the past few decades, extensive focus has been applied to understanding the mechanisms governing the behavior of proteins at solid/liquid interfaces. Due to wide-spread implications of this phenomenon in numerous applications, a large number of systems have been studied. To gain more insight into how protein structure affects interfacial behavior, the importance of surface charge,1, 2 hydrophobicity,3 and structural stability4, 5 have been investigated.
Approaches to study the effects of these parameters can be categorized as follows: the study of well-defined model proteins,6–10 genetic variants,11, 12 mutants of single proteins,13–15 and most recently, synthetic polypeptides.16–18 Significant effort in the two former categories has pioneered our overall understanding of the mechanisms governing protein adsorption; however, it is the latter groups in which the effect of subtle molecular effects can be assessed. Thus, ideal studies could be envisioned in which a single parameter, such as stability or surface charge, are studied in isolation.
Many of these studies have explored the structural consequences of adsorption. Elbaum et al.19 used stability variants of hemoglobin to hypothesize a strong structural basis for observed differences in adsorption kinetics and protein unfolding at the air/water interface. Kato and Yutani20 strengthened this correlation between conformational stability and interfacial affinity and level of surface-induced perturbation using tryptophan synthase mutants. More recently, bacteriophage T4 lysozyme has been the protein of choice for similar studies because it is extremely well characterized and synthesis of stability mutants is well documented. McGuire et al.14 created T4 variants with enhanced stability by introducing cysteine residues to form additional intramolecular disulfide linkages. Following the kinetics of adsorption and elutability, they postulated a correlation between protein stability and time scale of attachment and binding strength. CD spectroscopy revealed that both the rate and extent of unfolding (characterized by α-helical loss) upon adsorption to silica nanoparticles was most pronounced for the least stable mutants.4
The general hypothesis emerging from these studies is a strong correlation between thermostability and affinity of surface attachment, structural perturbation, and desorbability. However, there are two potential shortcomings with the application of stability mutants to evaluate surface activity. First, only incremental changes in stability can be assessed. The range of stabilities is limited by the number of residue substitutions which alter stability without impacting secondary structure. Second, stability is artificially optimized in these systems. For example, common approaches involve strategically placed disulfide bonds or the insertion of residues with side chains that cause steric disruptions in critical locations. However, these approaches change the topology of the peptide chain and it is difficult to decouple the effect of the stability-altering mutations from the effect on protein-surface interactions.
The goal of our investigation is to explore the relationship between stability and adsorption behavior using naturally evolved homologs with very similar three-dimensional structures yet vastly different intrinsic stabilities. We have chosen two members of the aldo–keto reductase (AKR) superfamily21 which share high structural homology with low primary sequence homology (Fig. 1). The members of the AKR superfamily are monomeric, do not contain disulfide bonds, and fold into well-known eight stranded, TIM-like, α/β barrels. The first enzyme, alcohol dehydrogenase D (AdhD) from the hyperthermophilic archaea Pyrococcus furiosus, is highly thermostable22, 23 while the second enzyme, human aldose reductase (hAR), is mesostable.24 We believe this is the first time that homologous stability variants have been used to explore the effects of protein stability on interfacial behavior. Adsorption and desorption were investigated using a hydrophilic silica surface which was previously used in adsorption studies of model proteins.25–27
Figure 1.
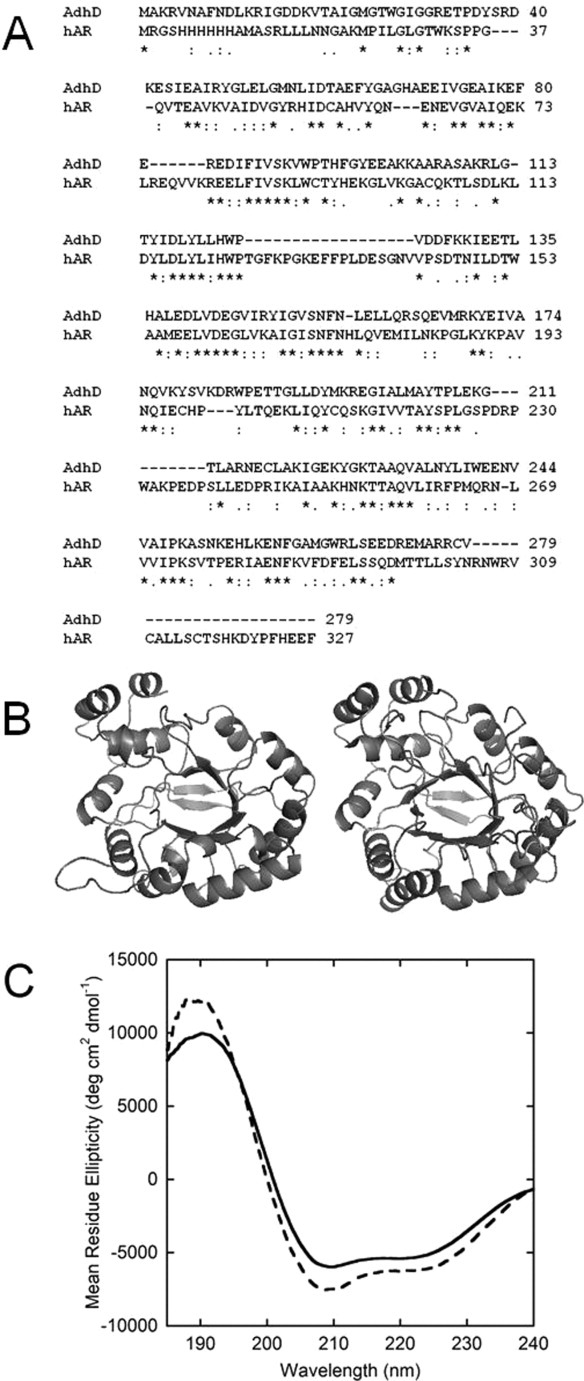
(A) Amino acid sequence alignment of AdhD and hAR showing <30% sequence homology between the selected proteins; (B) three-dimensional homology model of AdhD (left) and crystal structure of hAR (PDB: 2ACQ) (right) showing similarities in secondary and tertiary structure; (C) Far-UV CD spectra of native AdhD (—) and native hAR (- - -) quantitatively demonstrating the similarities in secondary structure between the two.
Based on previous results, we hypothesized that AdhD and hAR would exhibit different adsorption behavior due to their drastically different thermostabilities. Unexpectedly, our results indicate that both proteins have similar affinities to the substrate and undergo nearly identical levels of structural perturbation. Interesting differences between AdhD and hAR are observed during desorption, with respect to elutability, refolding pathways, and distribution of desorbed structural states. Both proteins exhibit some level of activity loss and irreversible conformational change upon desorption. Although surface-induced denaturation and activity loss is expected for the less stable hAR, it is remarkable that AdhD, which retains native-like structure and activity at even at 100°C, is significantly affected by adsorption-induced events. Therefore, our results suggest that intrinsic structural stability may not be the most accurate predictor of adsorption behavior, and that other predictors, such as electrostatics, may have a greater impact on the extent of surface affinity and unfolding. These results may call into question the commonly held belief that increasing thermostability should reduce surface activity.
Results
Structural characterization of model system
Two homologous AKR superfamily members, AdhD and hAR, were chosen due to their similar secondary structures and different intrinsic structural stabilities. The sequence alignment [ClustalW2, EBI; Fig. 1(A)] and ribbon structures [Fig. 1(B)] show that while there is low sequence homology (<30%) between the proteins, the tertiary structures are very comparable. Overlaying the CD spectra for both proteins more clearly demonstrates these similarities [Fig. 1(C)]. The high degree of spectral overlap indicates similar proportions of α-helical and β-sheet content. The fraction of secondary structure assigned to various domains from primary sequence predictions, the homology model (AdhD) or crystal structure (hAR; PDB: 2ACQ), and deconvolution of CD spectra are tabulated in Table I. For both proteins, the fractions of secondary structure assigned to the helix and sheet domains are similar between the sequence prediction and homology model, but there is a discrepancy compared with experimental deconvolution values. This discrepancy is explained further below. However, the similarity in each secondary structure domain of the two proteins, with all three enumeration methods, is obvious from Table I. The CD-based method was used as the principle experimental tool to assess relative changes in secondary structure. While the CD data demonstrate the similar structures of the proteins, the significant difference in structural stability is demonstrated by the melting curve shown in Supporting Information Figure S2, where the CD signal at 222 nm (indicative of helical content) is measured over a thermal excursion from 25 to 95°C. The curve shows that hAR loses a considerable amount of helical structure, while AdhD is minimally affected and no noticeable unfolding occurs below 95°C.
Table I.
Secondary Structure Domain Allocation
| AdhD | hAR | |||||
|---|---|---|---|---|---|---|
| Sequence predictiona | Homology modelb | CDc | Sequence predictiona | Crystal structured | CDc | |
| Helix | 0.41 (115)e | 0.43 (118) | 0.18 | 0.35 (112) | 0.36 (119) | 0.21 |
| Sheet | 0.14 (39) | 0.14 (39) | 0.28 | 0.13 (48) | 0.12 (40) | 0.27 |
| Disordered | 0.45 (125) | 0.42 (115) | 0.54 | 0.52 (154) | 0.52 (169) | 0.52 |
Predictions based on six different algorithms averaged together (JPred, Porter, PsiPred, Prof, SCRATCH, 3DJigsaw).
Homology model obtained as previously described.22
Deconvolutions of CD Spectra performed using CDPro Software.
Crystal structure obtained from PDB file 2ACQ.
Where applicable, the number of amino acids assigned to each domain is indicated in parentheses.
Adsorption behavior
The surface coverage of protein molecules adsorbed to the nanoparticles following a 16-h isothermal incubation period is plotted against the equilibrium (supernatant) concentration (Fig. 2). For both proteins, there are three well-defined regions: a rising linear portion, a transition region, and a maximum coverage plateau. At least one point from each of these regions is represented in subsequent data sets (letters in Fig. 2 indicate these coverages). The adsorption data are described using the Langmuir adsorption isotherm defined as:
Figure 2.
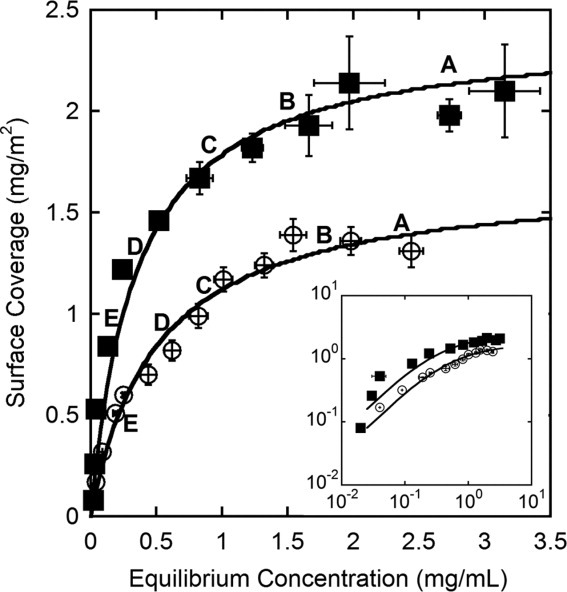
Adsorption isotherms for AdhD, ○ (K = 0.50 mg mL−1, Γmax = 1.68 mg m−2) and hAR, ▪ (K = 0.35 mg mL−1, Γmax = 2.40 mg m−2) with least-squared fits to a Langmuir isotherm. Inset contains adsorption isotherms on logarithmic axes. (A)–(E) Points selected for subsequent data sets. Error bars represent the standard deviation of three independent replicates.
| (1) |
where Γ is surface coverage, Γmax is plateau surface coverage, Ceq is equilibrium protein concentration, and K is the apparent dissociation constant. Although assumptions underlying the Langmuirian model are not all necessarily appropriate for the adsorption of complex biomacromolecules (absence of conformational change and lateral interaction on the surface), the model describes this high-affinity protein-surface interaction well. The maximum surface coverage and affinity obtained from this fit are 1.68 mg m−2 and 0.50 mg mL−1 for AdhD, and 2.40 mg m−2 and 0.35 mg mL−1 for hAR, respectively. The lower affinity coefficient and higher maximum surface coverage of hAR, as compared with AdhD, indicate an increased affinity and adsorbed amount. Based on the molecular volume of each protein, theoretical surface coverages of 2.20 and 2.26 mg m−2 for AdhD and hAR, respectively, were obtained. In order to account for the packing of molecules on the surface, the random sequential adsorption (RSA)28 packing density (θ = 0.547) was multiplied by the total surface area of the particles to obtain the available surface area.29 Since these calculations only account for size and not intermolecular interactions, the variance from experimental values is not concerning.
Adsorbed protein structure
Supporting Information Figure S3 shows the CD spectra of the different populations of proteins following surface interaction. A combination of these spectra was used to calculate adsorbed protein spectra. Figure 3 shows the adsorbed spectra for five different coverages for AdhD [Fig. 3(A)] and hAR [Fig. 3(B)]. Two important conclusions emerge from these plots: first, for both proteins, the adsorption induces a significant level of unfolding compared with native; and second, the adsorbed spectra do not vary significantly as a function of surface coverage. To further compare the level of unfolding that occurs upon adsorption, all spectra were deconvoluted and the results plotted in Figure 3(C). The tabulation of results can be found in Supporting Information Table SI. The results show that both surface-bound proteins converge on similar α-helical (∼0.06) and β-sheet (∼0.35) content.
Figure 3.
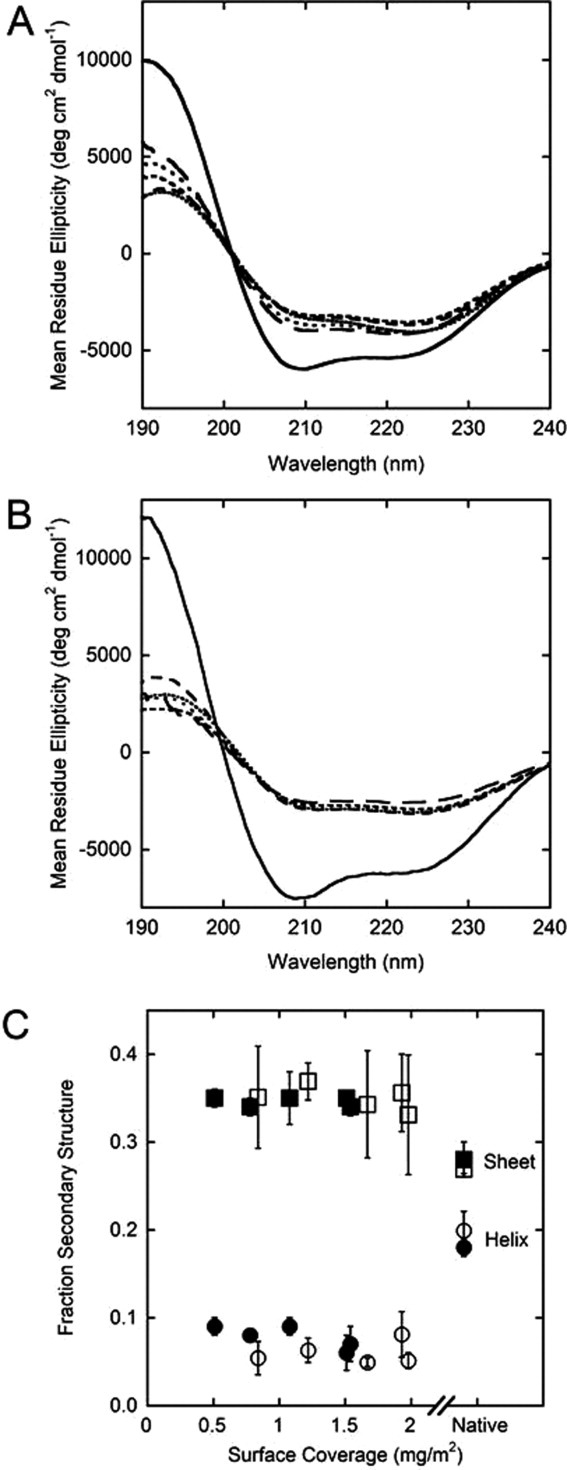
Adsorbed protein spectra for AdhD (A) and hAR (B) as compared with the native spectra (—) of each protein. Starting with the highest surface coverage, the line types indicate (surface coverage and Ceq follow each line type): …….. (AdhD: 1.54 mg m−2, 2.44 mg mL−1; hAR: 1.98 mg m−2, 2.73 mg mL−1), …….. (AdhD: 1.51 mg m−2, 1.62 mg mL−1; hAR: 1.93 mg m−2, 1.66 mg mL−1), ---- (AdhD: 1.08 mg m−2, 0.85 mg mL−1; hAR: 1.67 mg m−2, 0.83 mg mL−1), ––– (AdhD: 0.78 mg m−2, 0.50 mg mL−1; hAR: 1.22 mg m−2, 0.24 mg mL−1), and —— (AdhD: 0.51 mg m−2, 0.24 mg mL−1; hAR: 0.84 mg m−2, 0.13 mg mL−1); (C) fraction of secondary structure that exists as α-helical (circle) and β-sheet (square) domains for AdhD (• and ▪) and hAR (○ and □) for native protein and adsorbed protein.
Desorption behavior
The pellets obtained from the protein-particle mixtures were resuspended in fresh buffer to induce desorption. Previously, eluents such as morpholine25 or dodeclytrimethylammonium bromide1 were used to desorb bound proteins. In this study, we relied on the protein concentration gradient between the surface and bulk as the driving force for desorption. Introducing fresh buffer creates a transient difference in chemical potential (Δμs) at the interface, which is then eliminated by spontaneous desorption. Following resuspension and incubation of the particles, the supernatant protein concentration was measured, which represents the desorbed amount. This sequence of resuspension and supernatant collection was repeated for 10 cycles for each protein [Fig. 4(A,B)]. Data with varying protein: particle ratios were collected to assess the effect of coverage on desorption. The most striking difference in behavior between the two proteins is that the desorbed amount is affected by surface coverage much more significantly for hAR [Fig. 4(B)] than the AdhD [Fig. 4(A)]. For AdhD, there is only a slight decrease in desorbed amount as coverage decreases, while for hAR, desorbed amount varies by over an order of magnitude for the lowest coverage shown, 0.08 mg m−2, than for coverages above 0.52 mg m−2. This indicates the propensity to desorb is decreased when fewer proteins coat the surface. A more subtle difference between the two desorption curves is that while desorbed amount levels off after the third cycle for hAR, this decreases continuously from cycle to cycle for AdhD. This indicates that the arrangement of particles for hAR reaches a steady state value earlier in the experiment, while for AdhD a possible redistribution on the surface manifests as a variability of the desorbed amount from cycle to cycle. Also, because elutability is expected to increase with molecular weight,9 the higher concentration values of hAR in the supernatant after cycle 5 are consistent with this hypothesis (MWhAR = 37.2 kDa, MWAdhD = 31.9 kDa).
Figure 4.
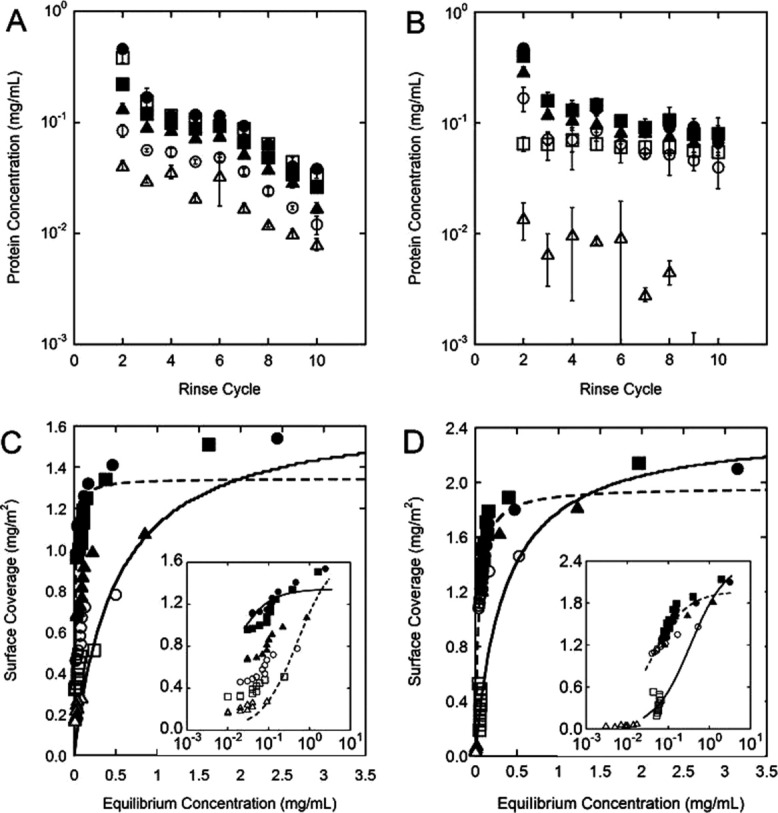
Desorbed protein concentration as a function of rinse cycle for (A) AdhD and (B) hAR. Data shown for rinse cycles 2 to 10. Protein: particle ratios corresponding to five different regions along the adsorption isotherm (as labeled A–E in Fig. 2) were used (surface coverage and Ceq follow each sample). AdhD: • Sample A: 1.4 mg m−2, 1.9 mg mL−1; ▪ Sample B: 1.2 mg m−2, 1.3 mg mL−1; ▴ Sample C: 0.82 mg m−2, 0.62 mg mL−1; □ Sample D: 0.60 mg m−2, 0.25 mg mL−1; ○ Sample E: 0.32 mg m−2, 0.09 mg mL−1; hAR: • Sample A: 2.1 mg m−2, 3.2 mg mL−1; ▪ Sample B: 2.1 mg m−2, 2.0 mg mL−1; ▴ Sample C: 1.8 mg m−2, 1.23 mg mL−1; □ Sample D: 1.46 mg m−2, 0.52 mg mL−1; ○ Sample E: 0.53 mg m−2, 0.04 mg mL−1. Each point is the average of three distinct replicates and error bars represent their standard deviation. Not shown on the graphs is the limit of detection of each assay, as all concentration values shown are above this limit. Desorption isotherms are shown in (C) AdhD and (D) hAR. Data for all 10 cycles are depicted, and symbols are consistent with those used in (A) and (B). The solid line represents the Langmuir fit for the adsorption isotherm data and the dashed line represents a Langmuir fit for the desorption data. Insets show the same data sets on a logarithmic x-axis.
To assess adsorption/desorption reversibility, surface coverage following each rinse cycle is calculated and desorption isotherms are created, as shown in Figure 4(C) (AdhD) and (D) (hAR). In these figures, the lines represent the Langmuir fits of the adsorbed data (solid line) and desorbed data (dashed line). For AdhD, all six desorbed data sets are used for the Langmuir fit, as each group follows similar trends. This desorption curve in Figure 4(C) varies distinctly from the adsorption isotherm. For reversible adsorption, the ascending and descending branches of the isotherm must overlap at all values of ceq. Therefore, our results indicate irreversible adsorption, and also coverage-dependent desorption (namely a path-dependent hysteresis of desorption). Conversely, for hAR, two desorbed populations are found: at higher coverages (corresponding to transition and plateau values), a single Langmuirian desorption curve can be fit to all the data sets. As before, this desorption curve in Figure 4(D) deviates significantly from the adsorption isotherm, indicating nonreversible adsorption. At low coverages (below 0.53 mg m−2), two data sets reside in a different desorption regime as seen from their position relative to the Langmuir desorption curve. Interestingly, these data sets align closely with the Langmuir adsorption curve.
Desorbed protein characterization
To characterize secondary structure of the desorbed protein, far-UV CD spectra were taken from the third resuspension cycle. Native and desorbed protein spectra are shown in Figure 5. Three different coverages (corresponding to the plateau, transition, and rising portions of the adsorption curve) are shown for the desorbed protein. The results are consistent with the previously described desorption curves: for AdhD [Fig. 5(A)], little variation exists for desorbed protein structure with coverage, while for hAR [Fig. 5(B)], the structure at lower coverages is severely perturbed compared with the higher coverages. The results also show that all desorbed samples are slightly less structured than native for AdhD, while there is practically no difference between the high coverage desorbed spectra and native hAR.
Figure 5.
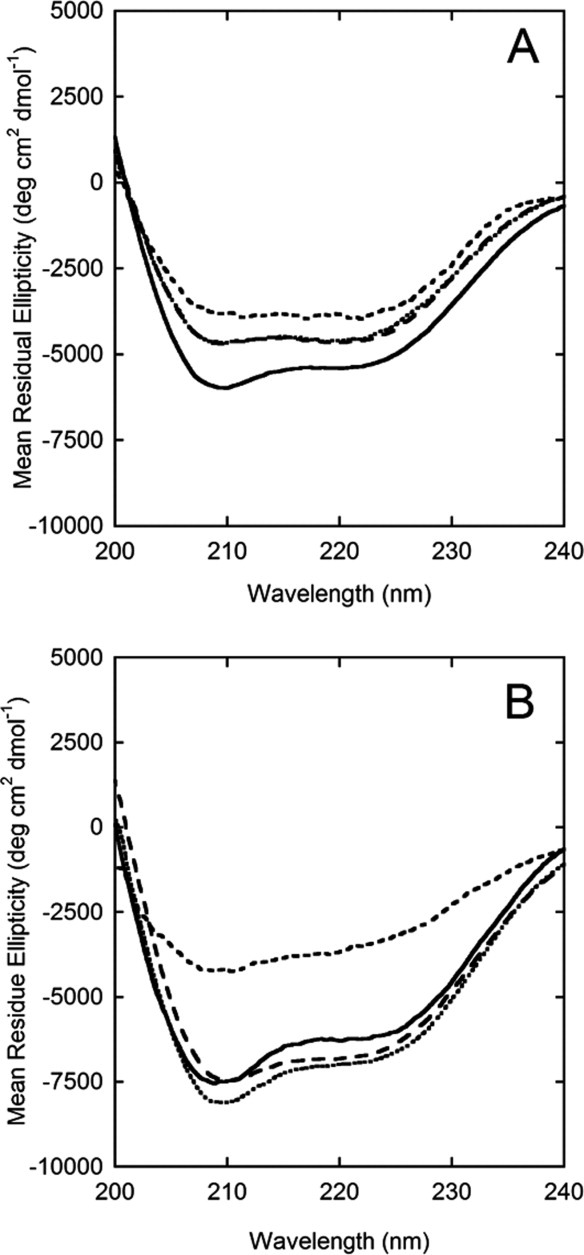
CD spectra of native protein (solid line) versus desorbed protein (obtained from cycle 3 of the desorption experiments captured at surface coverages along the plateau value ………., transition region ——, and rising portion of the adsorption isotherm ---) for (A) AdhD and (B)) hAR.
Activity assays were also performed on the same desorbed protein populations. The 2,3-butanediol oxidation activity of AdhD using NAD+ cofactor and the d,l-glyceraldehyde reduction activity of hAR using NADPH as cofactor were measured. The results of the kinetic assays are shown as percent of native activity in Figure 6. The specific activities are also reported in Supporting Information Table SII. These activity results correlate well with the secondary structure characterization. For AdhD, all desorbed protein, independent of surface coverage, lose ∼35% native activity. In contrast, for hAR, the four high coverage samples which were grouped together earlier based on lack of secondary structural modification upon desorption, have no apparent activity loss upon desorption. However, the lower coverage samples, which were found to be severely perturbed by CD, show ∼80% activity loss.
Figure 6.
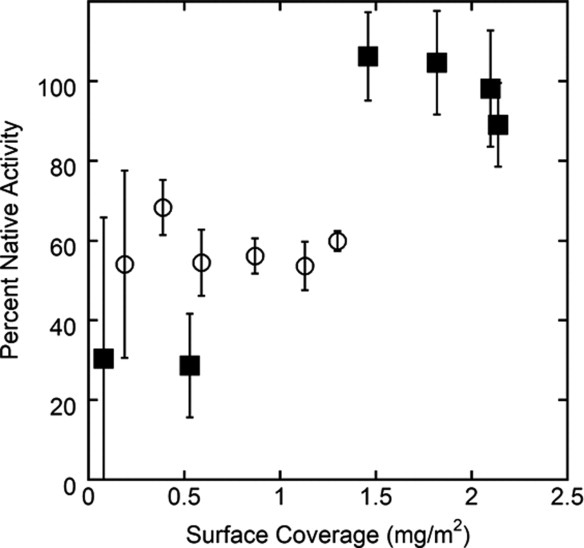
Kinetic data showing percent native activity for desorbed AdhD (○) and hAR (▪) at various levels of surface coverage. Desorbed protein has been taken from the third rinse cycle of the desorption experiments. Error bars represent standard deviations of three independent samples. Coverages targeted are the same five data sets as have been used in Figures 4 and 5, for direct comparison.
Discussion
Because proteins are naturally surface-active, adsorption can act as a catalyst to induce structural alteration, activity loss, or aggregation. Preventing such undesired surface-induced effects requires a fundamental understanding of the role of intrinsic protein parameters in adsorption behavior. Such knowledge is especially relevant in the pharmaceutical industry, where biologically active therapeutic agents contact numerous surfaces during manufacturing, storage, and delivery. During drug development, extensive effort is dedicated to improve the stability of such proteins, as various types of degradation pathways compromise their safety and efficacy. Although surface adsorption is not traditionally considered among the most common causes for physical instability, this phenomenon has been documented as a catalyst for denaturation.30 Designing greater stability into proteins is thought to be a key strategy to mitigate such surface effects.31, 32
It has long been recognized that a protein's structural stability influences its interfacial behavior, as the presence of a surface can disrupt intermolecular forces and render the protein susceptible to adsorption-induced conformational changes. In previous studies, this role of thermostability has been studied using engineered stability variants. Although using point mutations allows very specific changes to artificially stabilize (or destabilize) the protein without significantly changing its tertiary structure, we believe applying these systems in adsorption studies have inherent limitations. Only a limited range of thermostabilities can be studied and the approach used to alter stability often changes protein topology. Along these lines, artificially stabilized mutants may have other attributes that would not be favored in naturally evolved systems. To overcome these limitations, we assess the role of thermostability using naturally evolved homologs. Additionally, we significantly expand the range of surface coverages previously explored.
As shown in Figure 1, the use of AdhD and hAR is a fitting model system for the study of the effect of naturally occurring stability. These two proteins have similar tertiary structures but vastly different thermostabilities: hAR loses approximately half of its helical content at 57°C, while AdhD is practically unaffected even at 95°C. Regarding structural similarity of the proteins, it must be noted that Table I shows some discrepancies between secondary structure fractions determined theoretically from sequence prediction algorithms and direct CD measurement followed by spectral deconvolution. Although the two approaches result in similar trends in distribution of secondary structure, the CD-derived results are lower than the theoretical values. This is likely due to the inherent limitations of deconvolution algorithms used to quantify secondary structure.33 Although such issues have been documented, CD remains a powerful tool for evaluating comparative structural changes34, 35 and was used here to examine changes in protein structure upon adsorption/desorption.
The difference in thermal stabilities of the two reductases could be due to many factors. One possible explanation is that since AdhD has a more compact structure, there is more potential for close-range interactions that will stabilize its conformation. Another contributing factor could be the fraction of amino acids assigned to different secondary structure motifs. For example, AdhD and hAR have 118 and 112 amino acids involved in alpha helices, respectively. However, because AdhD has an overall lower residue count than hAR this corresponds to 42% amino acids assigned to helices (vs. 34% for hAR). This means that a larger fraction of AdhD is made of a motif that contains a high number of hydrogen bonds per amino acid, which could translate into increased intrinsic thermal stability of AdhD.
In this study, we chose hydrophilic, unmodified silicon dioxide nanoparticles as the surface for several reasons. On hydrophilic surfaces, in the absence of strong electrostatic forces, protein stability is a key parameter that determines surface activity, as structural rearrangement is the main entropic driving force favoring adsorption.25, 36 Colloidal silica also has favorable size, refractive index, and light scattering properties, which render the particles CD-compatible, allowing in situ structural analysis during adsorption. A final benefit of using colloidal system is our ability to target a wide range of surface coverages. This is a key parameter for our system of interest: proteins in delivery devices, where therapeutic concentration can vary by orders of magnitude. Thus, relevant coverages extend to the plateau region of the adsorption isotherm.37 To minimize and normalize the effect of electrostatics, experiments were performed at a pH slightly greater than the isoelectric point of each protein, (AdhD pI 5.5, hAR pI 6.9; system pH 5.7 for AdhD and 7.2 for hAR) thus imparting a slight negative charge on both proteins. The sodium cacodylate buffer (pKa 6.3) was selected as its effective buffering range was compatible with the desired conditions. Zeta-potential experiments shown in Supporting Information Figure S4 confirm this slight negative charge on each protein, and the data demonstrate that there is no significant charge difference between proteins (ζ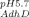 = −4.8 ± 1.7 mV; ζ
= −4.8 ± 1.7 mV; ζ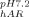 = −3.2 ± 0.6 mV). The same silica particles, with a point of zero charge between 2.0 and 3.0, were used in both systems. Therefore, the particles are negatively charged at both pHs and are in both cases more negatively charged than the proteins. However, as Supporting Information Figure S4 demonstrates, there is a difference in zeta potentials for the two suspensions: (ζ
= −3.2 ± 0.6 mV). The same silica particles, with a point of zero charge between 2.0 and 3.0, were used in both systems. Therefore, the particles are negatively charged at both pHs and are in both cases more negatively charged than the proteins. However, as Supporting Information Figure S4 demonstrates, there is a difference in zeta potentials for the two suspensions: (ζ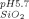 = −12.0 ± 5.0 mV; ζ
= −12.0 ± 5.0 mV; ζ = −26.6 ± 3.4 mV). The difference between the protein solution and the respective silica suspension is greater in the case of hAR (23.4 mV difference) than for AdhD (7.2 mV difference). The potential implications of this charge difference will be discussed in detail further below.
= −26.6 ± 3.4 mV). The difference between the protein solution and the respective silica suspension is greater in the case of hAR (23.4 mV difference) than for AdhD (7.2 mV difference). The potential implications of this charge difference will be discussed in detail further below.
Our results shed new light on the relative role of structural stability in the different kinetic processes depicted in Figure 7. During surface attachment (Step I, Fig. 7), the Langmuir curves in Figure 2 compare the surface affinity and adsorbed amounts of hAR and AdhD. Although we anticipated hAR to have a significantly greater affinity for the surface due to its lower thermostability, we find its K value is comparable with that of AdhD. Similarly, the differences in maximum surface coverage can most likely be attributed to the size difference between the proteins. The aforementioned differences in the electrostatic potentials could also contribute to the differences in the Langmuirian parameters between the two systems. However, these results indicate that structural stability does not play a significant role in protein-surface attachment. Figure 3 shows hAR and AdhD exhibit similar structural transitions on the surface (Step II, Fig. 7). While both proteins undergo significant structural perturbation, the extent of α-helical loss and concomitant β-sheet formation is nearly identical. Although such disruption of intermolecular forces upon adsorption was expected for hAR, it is remarkable that the extremely thermostable AdhD undergoes such drastic conformational changes on a hydrophilic surface. Another interesting finding is the lack of correlation between surface coverage and structural denaturation for either protein [Fig. 3(C)]. Coverage-dependence of adsorption-induced unfolding has been reported previously.25, 38
Figure 7.
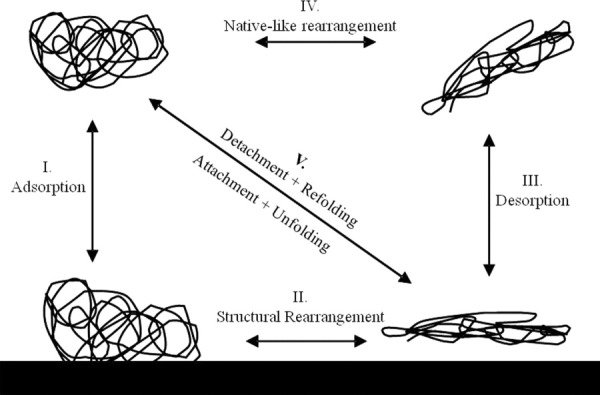
Classical four-state kinetic model, representing the native state in bulk before adsorption, the adsorbed state before unfolding, followed by structural rearrangement while on the surface, desorption into bulk, and refolding into native-like state.
The results of this study reveal differences between hAR and AdhD during detachment and refolding (Steps III and IV, Fig. 7). The desorption isotherms [Fig. 4(C,D)], and structural (Fig. 5) and kinetic (Fig. 6) characterization of the desorbed states indicate that while adsorption is irreversible in both cases, desorption follows different pathways for hAR and AdhD. For AdhD, some of the structural loss incurred upon adsorption is regained upon detachment; however, native-like refolding is not complete and ∼35% enzymatic activity is lost. These findings re-emphasize the surprising effect adsorption-induced instabilities have on a thermally stable protein. AdhD also exhibits hysteresis in its refolding pathways as shown in Figure 4(C). The pathways appear to be a function of surface coverage (possibly due to microscopic difference in surface arrangement). Although such examples of hysteresis are not commonly reported, Norde et al. found similar behavior for albumin.39 To schematically capture this incomplete refolding and pathway-dependent hysteresis behavior of AdhD, we believe that the single diagonal arrow in Figure 7 (Step V) can be replaced by multiple arrows representing different pathways. In contrast to AdhD, we find two distinct desorbed populations for hAR: at high coverages, the desorption isotherms follow a single pathway to native-like structural refolding and the desorbed protein has no activity loss. These results are unexpected due to the low thermal stability of hAR. At low coverages, desorbed hAR maintains high levels of structural loss and ∼80% activity loss, which more closely represents our hypothesis. Unlike AdhD, no hysteresis is found. This coverage-dependent desorption behavior can be represented in Figure 7 by a combination of arrows both at Step III (desorption to a perturbed state, the low coverage case) and at Step V (desorption to a native-like state, the high coverage case).
Two driving forces are likely to govern the adsorption and structural unfolding of the proteins on the silica surface. One driving force is enthalpic-based, due to charge differences between the protein and silica. Although we have set the pH of the system to minimize overall point charge of the proteins, the surface charge profiles of the two proteins vary, and localized charged patches can attribute to attractive/repulsive forces with the substrate. Three-dimensional molecular models are shown in Supporting Information Figure S5 to illustrate the distribution of surface charge on both hAR and AdhD. Based on these diagrams, qualitative assessment of the differences between the two proteins can be made. While both proteins have a few localized patches (both positive and negative) this charge distribution is generally homogeneous across the surface of the protein for hAR. By contrast, AdhD has more localized patches of both charges, with very few neutral areas. Specifically, positive patches seem to be larger for AdhD. The implication of these differences in surface charge distribution is discussed below. The other, entropic-based, driving force can be due to unfolding of secondary structure which increases conformational entropy of the protein molecule. This has been cited in previous studies.25 Both proteins demonstrate a helix-to-sheet transition, which indicates that once the protein comes into proximity of an interface, less favorable intermolecular interactions, which constrain the molecular dynamics of the protein, are lost in favor of a more relaxed conformation and more possible surface-interactions.
One possible explanation for the adsorption-induced denaturation of the thermophilic AdhD is the presence of the aforementioned positive patches on the surface of the protein, as seen in Supporting Information Figure S4. Because the surface is negatively charged, strong Coulombic attraction would lead to high levels of surface affinity, with possibly concomitant structural unfolding as favorable attractive forces lead to new protein-surface bond formation.
Unlike previous results that indicate only two states exist: a native-like desorbed and a highly perturbed adsorbed state,37 we see evidence of a non-native like desorbed state. There is also indirect evidence that differentially unfolded states exist on the surface. This hypothesis is supported as follows: first, hAR desorbs in a coverage-dependent manner. This may be a consequence of two (or more) adsorbed protein populations: at low coverages, proteins arriving early have longer residence times, allowing the occupation of more optimal attachment sites, formation of stronger surface bonds, and lateral intermolecular interactions. The detachment of clusters would require greater free energy consumption, resulting in decreased desorbability.40 Conversely, at high coverages, a greater packing density may inhibit optimal protein-surface interactions.41 Second, previous results show differences in elutability, bound fraction, and interfacial area in adsorbed proteins, indicating that the existence of a single adsorbed population is unlikely.14, 42 Finally, evidence for molten globule-like adsorbed states is postulated by studies involving carbonic anhydrase,43 lysozyme,44 and human growth hormone.45
Our results indicate that overall adsorption behavior of these homologous proteins on silica does not correlate strongly with thermostability. These results are surprising, as the structural stability of mutants has been previously hypothesized to strongly influence their surface affinity, degree of structural perturbation, and desorbability. The main difference in our study is that we assess a greater difference in thermostability, by using proteins with naturally evolved structures that have been optimized for their environment. As such, these homologs have similar topology and tertiary structure, but vastly different primary sequences. In contrast, stability mutants have similar primary and tertiary structure, but often different topologies. Both approaches introduce parameters into the system whose effect on adsorption behavior is unknown: for stability mutants, we hypothesize that altered topology may inhibit optimal protein/surface interaction, thus artificially reducing (or promoting) extent of surface-induced perturbation. However, with naturally occurring homologs, we cannot be certain that observed adsorption behavior is not due to differences in specific amino acid and surface interactions (due to the differences in primary structure). We argue that although both systems have some limitations, the use of physiologically relevant systems which allows a wider range of thermostabilities to be studied provides valuable insight which may have been previously overlooked.
We also find interesting differences in the desorption behavior of hAR and AdhD. One explanation is that desorption is sensitive to structural stability because the same intermolecular forces which drive protein folding in solution will determine whether desorption is energetically more favorable. However, we cannot be sure that the observed differences are not protein dependent. In their natural environments, we know that these proteins are cystolic and may fold with the help of chaperones. Furthermore, thermal stress causes irreversibly unfolding in hAR while having no effect on AdhD. Therefore other intrinsic parameters, such as primary sequence may be the dominant factor in determining desorption behavior.
For more insight into the role of surface adsorption as a catalyst for protein destabilization, we compare the denaturing effects of the surface to other well-known stresses. Supporting Information Figure S6 shows the far-UV CD spectra of hAR following thermal, chemical, and surface-induced stress. Thermal effects results in almost complete loss in secondary structure, while both surface- and chemically induced stress cause only partial structural loss. Such differences are likely due to the probability of breaking the hydrogen bonds which maintain secondary structure in the presence of various disrupting forces.46 Interestingly, thermal versus surface-induced stress have a markedly different effect on AdhD, while extreme temperature excursions cause essentially no structural alteration, the surface is capable of irreversibly perturbing its structure.
To the best of our knowledge, this study marks the first time that naturally occurring stability variants have been evaluated for interfacial behavior, by studying their differences and interdependencies along each step of the adsorption lifecycle. Our results show that there is little correlation between a protein's thermostability, surface affinity, and susceptibility to surface-induced unfolding. Additionally, we show interesting desorption behavior between the proteins and the importance of surface coverage in determining refolding pathway. Our results reveal that role of thermostability in interfacial behavior may be less dominant than previously thought while protein surface properties may be a more important determinant of this behavior. These conclusions shed some doubt on the notion that improving thermostability is the main way to reduce the effects of adsorption-induced changes of proteins.
Experimental
Materials
DNA oligonucleotides were synthesized by Integrated DNA Technology (IDT; Coralville, IA). Isopropyl β-d-1-thiogalactopyranoside (IPTG) was obtained from Promega (Madison, WI). Fumed colloidal silica particles (Cab-o-sil M-5, <99.8% purity) were purchased from Cabot Corp. (Boston, MA) and used without further treatment. Bicinchoninic acid (BCA) and QuantiPro BCA Assay kits were purchased from Thermo Fisher Scientific (Rockford, IL). Restriction enzymes NcoI and HindIII, T4 DNA ligase, and Phusion DNA polymerase were purchased from New England Biolabs (Ipswich, MA). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Cloning
The hAR gene was amplified from human placental cDNA (Clontech, Mountain View, CA) using overlap extension PCR to eliminate an internal NcoI restriction site present in the hAR gene. Detailed information about the primers can be found in the Supporting Information Table SI. The purified PCR fragment was doubly digested and ligated into a similarly digested pET-24d (Novagen, Gibbstown, NJ) vector containing a poly-His tag. Ligated plasmids were electroporated into BLR Escherichia coli cells (Novagen) and selected on LB agar plates supplemented with 50 ng μL−1 kanamycin. Colonies were grown in terrific broth (TB) with 50 ng μL−1 kanamycin and stored as glycerol stocks at −80°C. The correct insertion of the hAR gene was verified by DNA sequencing.
Protein purification
AdhD from the hyperthermophilic archaea P. furiosus was expressed and purified as previously described.22 hAR was expressed and purified as follows. Following transformation and expression as described above, the cells were induced with 0.2 mM IPTG at OD600 of 0.6. After incubating 18 h at 37°C with agitation, the cells were harvested by centrifugation. The pelleted cells were resuspended in one-tenth of the expression volume in 20 mM Tris-HCl, 150 mM NaCl, and 40 mM imidazole (pH 7.5) and supplemented with 1× HALT protease inhibitor cocktail (Thermo-Fisher). The cells were lysed by sonication with an 8-min run time with pulses of 5 s with a 5-s rest between each pulse. The cell debris was pelleted by centrifugation at 15,000g for 30 min. The hAR was purified from the clarified lysate using a HisTrap column (GE Healthcare) on a GE ÄKTA FPLC. The fractions containing hAR, (verified by SDS-PAGE) were pooled and concentrated using Amicon (EMD Millipore) centrifugal filters with a 30 kDa MWCO. Protein stocks were stored at 4°C.
Protein concentration
Concentrations were determined by UV absorbance at 280 nm and the BCA total protein assay (macro assay: 1–0.025 mg mL−1 and micro assay: 0.05–0.0005 mg mL−1). Absorbance measurements were conducted on a SpectraMax M2 spectrophotometer (Molecular Devices, Sunnyvale, CA). Extinction coefficients for AdhD (ε280 = 1.97 mL mg−1 cm−1) and hAR (ε280 = 1.1 mL mg−1 cm−1) were measured experimentally. Preparation of standards for the BCA assays and protein samples for extinction coefficient measurements are described in the Supporting Information Table SI.
Activity assay
The 2,3-butanediol oxidation activity of AdhD and d,l-glyceraldehyde reduction activity of hAR were measured using spectrophotometric assays under saturation conditions. NAD+ and NADPH were used as cofactors for AdhD and hAR, respectively. For AdhD, the absorbance at 340 nm (tracking NAD+ reduction) was measured at 45°C with final concentrations of reaction buffer (glycine, pH 8.8), substrate, and cofactor of 50 mM, 100 mM, and 1 mM, respectively. For hAR, the absorbance at 340 nm (tracking NADPH oxidation) was followed for reactions at 25°C with concentrations of reaction buffer (sodium phosphate, pH 7.0), substrate and cofactor of 100 mM, 100 mM, and 0.5 mM, respectively. Raw absorbance change was converted to specific activity using enzyme concentration and cofactor extinction coefficient (ε340nm = 6.22 × 103 cm−1M−1).
Adsorption isotherms
Silica particles were suspended in 20 mM sodium cacodylate buffer pH 5.0 (AdhD) or pH 6.9 (hAR) to achieve a concentration of 12 mg mL−1. Equal parts of protein solution and silica suspension were mixed. Protein concentrations of 0.1 to 6 mg mL−1were achieved by diluting with 20 mM Tris pH 7.5. Final pH resulted in 5.7 for AdhD and 7.2 for hAR. The samples were rotated on a rotisserie shaker for 16 h at room temperature. Supernatants were collected and surface coverage was calculated as described previously.37
Desorption
After removal of the supernatant, the particle pellet was resuspended in 0.8 mL of 10 mM sodium cacodylate buffer pH 5.0 (AdhD) or pH 6.9 (hAR). Samples were weighed to determine the particle loss over time. A solution density of 1.02 g cm−3 was used to determine the resuspension volume. After resuspension, the samples were incubated on a rotisserie shaker for 30 min. Then, supernatant was collected in the same as in the adsorption experiments. Resuspension was repeated for a total of 10 supernatants, from which a desorption curve was obtained. It was assumed that by the third resuspension cycle all the protein had interacted with the particles, and these samples were used for CD and kinetic activity evaluation.
Circular dichroism
A Jasco J-815 CD spectrometer (Jasco, Inc., Easton, MA) equipped with a Peltier junction temperature control was calibrated with 0.06% d-10-camphorsulfonate solution and used for far-UV CD measurements. Quartz cuvettes with path lengths of 0.01, 0.02, 0.05, or 0.1 cm were used. Protein solutions and protein-particle mixtures were measured using buffer or silica blanks, respectively, for subtraction of the baseline signal. Measurements were taken from 185 to 240 nm with a 0.1 nm interval, 1 nm bandwidth, 8 s response time, and scanning speed of 50 nm min−1. For desorbed protein structure, the 0.1 cm cuvette was used, and the scan was cut off at 200 nm due to buffer interference. For each sample, three accumulations were measured and averaged. Raw CD signal, θ, was converted to mean residue ellipticity (MRE) using the equation [θ]MRE = θ/(10 × Cr × l) where Cr is the protein concentration (M × residue number) and l is the cuvette path length (cm).
Measurement of adsorbed protein structure
The pellets from the adsorption isotherm were resuspended with gentle pipetting using a 1:1 mixture of 20 mM Tris HCl pH 7.5 and 20 mM sodium cacodylate pH 5.0 (AdhD) or pH 6.9 (hAR). An aliquot was removed for CD assessment, and the rest of the solution was pelleted through centrifugation (as described above) and supernatant concentration measured. The CD spectra of the supernatant were also recorded. Using the known amount of adsorbed protein, concentration in the resuspended pellet was determined. Using the MRE signal of the resuspended pellet, [θ]pell, and of the supernatant [θ]SN, and the fraction of total protein concentration in the supernatant, χSN, the adsorbed protein structure was calculated using Eq. (2):
| (2) |
Deconvolutions
The CD spectra were deconvoluted using the CD Pro software package.47 With each deconvolution algorithm (Continll, Selcon3, and Cdstr), three different reference sets were applied: SP37, SPD42, and SMP50. The average across all three algorithms and three bases are used to estimate the helical, sheet, and disordered content of each sample. The α-helix and β-sheet content is reported as the sum of the distorted and regular classes.
Electrophoretic mobility
A Malvern Zetasizer Nano-ZS was used to measure particle size distribution and zeta potential of the silica particle suspensions in the pH = 5.7 and pH = 7.2 tris/cacodylate buffers, or protein–silica mixtures. A 50 mW laser operating at a wavelength of 532 nm was used. Scattering intensities were recorded at a 90° angle. A clear, disposable zeta cell was used for zeta potential measurements. All measurements were performed at 25°C.
Acknowledgments
The authors are grateful for the laboratory-related assistance of Ms. Sara Chuang, the guidance of Dr. Elliot Campbell, and critical review of the manuscript by Mr. Kevin Dooley.
Glossary
Abbreviations
- AdhD
alcohol dehydrogenase D
- AKR
aldo-keto reductase
- BCA
bicinchoninic acid
- DTAB
dodeclytrimethylammonium bromide
- hAR
human aldose reductase
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- MRE
mean residue ellipticity
- RSA
random sequential adsorption
- TB
terrific broth
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.McGuire J, Wahlgren MC, Arnebrant T. The influence of net charge and charge location on the adsorption and dodecyltrimethylammonium bromide-mediated elutability of bacteriophage-T4 lysozyme at silica surfaces. J Colloid Interface Sci. 1995;170:193–202. [Google Scholar]
- 2.Kato K, Sano S, Ikada Y. Protein adsorption onto ionic surfaces. Colloids Surf B. 1995;4:221–230. [Google Scholar]
- 3.Malmsten M, Burns N, Veide A. Electrostatic and hydrophobic effects of oligopeptide insertions on protein adsorption. J Colloid Interface Sci. 1998;204:104–111. doi: 10.1006/jcis.1998.5568. [DOI] [PubMed] [Google Scholar]
- 4.Billsten P, Wahlgren M, Arnebrant T, McGuire J, Elwing H. Structural changes of T4 lysozyme upon adsorption to silica nanoparticles measured by circular dichroism. J Colloid Interface Sci. 1995;175:77–82. [Google Scholar]
- 5.Froberg JC, Arnebrant T, McGuire J, Claesson PM. Effect of structural stability on the characteristics of adsorbed layers of T4 lysozyme. Langmuir. 1998;14:456–462. [Google Scholar]
- 6.Haynes C, Norde W. Globular proteins at sold/liquid interfaces. Colloids Surfaces B. 1994;2:517–566. [Google Scholar]
- 7.Arai T, Norde W. The behavior of some model proteins at solid liquid interfaces. 1. Adsorption from single protein solutions. Colloids Surf. 1990;51:1–15. [Google Scholar]
- 8.Andrade JD, Hlady V. Plasma-protein adsorption—the Big 12. Ann NY Acad Sci. 1987;516:158–172. doi: 10.1111/j.1749-6632.1987.tb33038.x. [DOI] [PubMed] [Google Scholar]
- 9.Wahlgren MC, Paulsson MA, Arnebrant T. Adsorption of globular model proteins to silica and methylated silica surfaces and their elutability by dodecyltrimethylammonium bromide. Colloids Surf A. 1993;70:139–149. [Google Scholar]
- 10.Norde W. Adsorption of proteins from solution at the solid-liquid interface. Adv Colloid Interface Sci. 1986;25:267–340. doi: 10.1016/0001-8686(86)80012-4. [DOI] [PubMed] [Google Scholar]
- 11.Karlsson M, Ekeroth J, Elwing H, Carlsson U. Reduction of irreversible protein adsorption on solid surfaces by protein engineering for increased stability. J Biol Chem. 2005;280:25558–25558. doi: 10.1074/jbc.M503665200. [DOI] [PubMed] [Google Scholar]
- 12.Xu SQ, Damodaran S. Comparative adsorption of native and denatured egg-white, human, and T(4)-phage lysozymes at the air-water interface. J Colloid Interface Sci. 1993;159:124–133. [Google Scholar]
- 13.Tian MH, Lee WK, Bothwell MK, McGuire J. Structural stability effects on adsorption of bacteriophage T4 lysozyme to colloidal silica. J Colloid Interface Sci. 1998;200:146–154. [Google Scholar]
- 14.McGuire J, Wahlgren MC, Arnebrant T. Structural stability effects on the adsorption and dodecyltrimethylammonium bromide-mediated elutability of bacteriophage-T4 lysozyme at silica surfaces. J Colloid Interface Sci. 1995;170:182–192. [Google Scholar]
- 15.Singla B, Krisdhasima V, McGuire J. Adsorption kinetics of wild type and two synthetic stability mutants of T4 phage lysozyme at silanized silica surfaces. J Colloid Interface Sci. 1996;182:292–296. [Google Scholar]
- 16.Apte JS, Gamble LJ, Castner DG, Campbell CT. Kinetics of leucine-lysine peptide adsorption and desorption at -CH3 and -COOH terminated alkylthiolate monolayers. Biointerphases. 2010;5:97–104. doi: 10.1116/1.3494080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weidner T, Samuel NT, McCrea K, Gamble LJ, Ward RS, Castner DG. Assembly and structure of alpha-helical peptide films on hydrophobic fluorocarbon surfaces. Biointerphases. 2010;5:9–16. doi: 10.1116/1.3317116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burkett SL, Read MJ. Adsorption-induced conformational changes of alpha-helical peptides. Langmuir. 2001;17:5059–5065. [Google Scholar]
- 19.Elbaum D, Harrington J, Roth EF, Nagel RL. Surface-activity of hemoglobin-S and other human hemoglobin variants. Biochim Biophys Acta. 1976;427:57–69. doi: 10.1016/0005-2795(76)90285-3. [DOI] [PubMed] [Google Scholar]
- 20.Kato A, Yutani K. Correlation of surface-properties with conformational stabilities of wild-type and 6 mutant tryptophan synthase alpha-subunits substituted in the same position. Protein Eng. 1988;2:153–156. doi: 10.1093/protein/2.2.153. [DOI] [PubMed] [Google Scholar]
- 21.Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell E, Wheeldon IR, Banta S. Broadening the cofactor specificity of a thermostable alcohol dehydrogenase using rational protein design introduces novel kinetic transient behavior. Biotechnol Bioeng. 2010;107:763–774. doi: 10.1002/bit.22869. [DOI] [PubMed] [Google Scholar]
- 23.Machielsen R, Uria AR, Kengen SWM, van der Oost J. Production and characterization of a thermostable alcohol dehydrogenase that belongs to the aldo-keto reductase superfamily. Appl Environ Microbiol. 2006;72:233–238. doi: 10.1128/AEM.72.1.233-238.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bohren KM, Page JL, Shankar R, Henry SP, Gabbay KH. Expression of human aldose and aldehyde reductases—site-directed mutagenesis of a critical lysine-262. J Biol Chem. 1991;266:24031–24037. [PubMed] [Google Scholar]
- 25.Norde W, Favier JP. Structure of adsorbed and desorbed proteins. Colloids Surf. 1992;64:87–93. [Google Scholar]
- 26.Giacomelli CE, Norde W. The adsorption-desorption cycle. Reversibility of the BSA-silica system. J Colloid Interface Sci. 2001;233:234–240. doi: 10.1006/jcis.2000.7219. [DOI] [PubMed] [Google Scholar]
- 27.Kondo A, Murakami F, Kawagoe M, Higashitani K. Kinetic and circular-dichroism studies of enzymes adsorbed on ultrafine silica particles. Appl Microbiol Biotechnol. 1993;39:726–731. doi: 10.1007/BF00164457. [DOI] [PubMed] [Google Scholar]
- 28.Evans JW. Random and cooperative sequential adsorption. Rev Mod Phys. 1993;65:1281–1329. [Google Scholar]
- 29.Bee JS, Chiu D, Sawicki S, Stevenson JL, Chatterjee K, Freund E, Carpenter JF, Randolph TW. Monoclonal antibody interactions with micro- and nanoparticles: adsorption, aggregation, and accelerated stress studies. J Pharm Sci. 2009;98:3218–3238. doi: 10.1002/jps.21768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tzannis ST, Hrushesky WJM, Wood PA, Przybycien TM. Adsorption of a formulated protein on a drug delivery device surface. J Colloid Interface Sci. 1997;189:216–228. [Google Scholar]
- 31.Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of protein pharmaceuticals: an update. Pharm Res. 2010;27:544–575. doi: 10.1007/s11095-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 32.Chi EY, Krishnan S, Randolph TW, Carpenter JF. Physical stability of proteins in aqueous solution: mechanism and driving forces in nonnative protein aggregation. Pharm Res. 2003;20:1325–1336. doi: 10.1023/a:1025771421906. [DOI] [PubMed] [Google Scholar]
- 33.Kelly SM, Price NC. The application of circular dichroism to studies of protein folding and unfolding. Biochim Biophys Acta. 1997;1338:161–185. doi: 10.1016/s0167-4838(96)00190-2. [DOI] [PubMed] [Google Scholar]
- 34.Kelly SM, Price NC. Circular dichroism to study protein interactions. Curr Protoc Protein Sci. 2006;20:10. doi: 10.1002/0471140864.ps2010s46. [DOI] [PubMed] [Google Scholar]
- 35.van Mierlo CPM, Steensma E. Protein folding and stability investigated by fluorescence, circular dichroism (CD), and nuclear magnetic resonance (NMR) spectroscopy: the flavodoxin story. J Biotechnol. 2000;79:281–298. doi: 10.1016/s0168-1656(00)00244-3. [DOI] [PubMed] [Google Scholar]
- 36.Rezwan K, Meier L, Gauckler L. Lysozyme and bovine serum albumin adsorption on uncoated silica and A1OOH coated silica. Biomaterials. 2005;26:4351–4357. doi: 10.1016/j.biomaterials.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 37.Felsovalyi F, Maniagalli P, Bureau C, Kumar S, Banta S. The reversibility of lysozyme on silica. Langmuir. 2011;27:11873–11882. doi: 10.1021/la202585r. [DOI] [PubMed] [Google Scholar]
- 38.Zoungrana T, Findenegg GH, Norde W. Structure, stability, and activity of adsorbed enzymes. J Colloid Interface Sci. 1997;190:437–448. doi: 10.1006/jcis.1997.4895. [DOI] [PubMed] [Google Scholar]
- 39.Norde W, MacRitchie F, Nowicka G, Lyklema J. Protein adsorption at solid-liquid interfaces: reversibility and conformation aspects. J Colloid Interface Sci. 1986;112:447–456. [Google Scholar]
- 40.Pellenc D, Gallet O, Berry H. Adsorption-induced conformational changes in protein diffusion-aggregation surface assemblies. Phys Rev E Stat Nonlin Soft Matter Phys. 2005;72:051904. doi: 10.1103/PhysRevE.72.051904. [DOI] [PubMed] [Google Scholar]
- 41.Bagchi P, Birnbaum SM. Effect of pH on the adsorption of immunoglobulin G on anionic poly(vinyltoluene) model latex particles. J Colloid Interface Sci. 1981;83:460–478. [Google Scholar]
- 42.Tilton RD, Gast AP, Robertson CR. Surface diffusion of interacting proteins—effect of concentration on the lateral mobility of adsorbed bovine serum-albumin. Biophys J. 1990;58:1321–1326. doi: 10.1016/S0006-3495(90)82473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Billsten P, Carlsson U, Jonsson BH, Olofsson G, Hook F, Elwing H. Conformation of human carbonic anhydrase II variants adsorbed to silica nanoparticles. Langmuir. 1999;15:6395–6399. [Google Scholar]
- 44.Sethuraman A, Vedantham G, Imoto T, Przybycien T, Belfort G. Protein unfolding at interfaces: slow dynamics of alpha-helix to beta-sheet transition. Proteins. 2004;56:669–678. doi: 10.1002/prot.20183. [DOI] [PubMed] [Google Scholar]
- 45.Buijs J, Hlady V. Adsorption kinetics, conformation, and mobility of the growth hormone and lysozyme on solid surfaces, studied with TIRF. J Colloid Interface Sci. 1997;190:171–181. doi: 10.1006/jcis.1997.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knotts TA, Rathore N, de Pablo JJ. Structure and stability of a model three-helix-bundle protein on tailored surfaces. Proteins. 2005;61:385–397. doi: 10.1002/prot.20581. [DOI] [PubMed] [Google Scholar]
- 47.Sreerama N. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.