

Abstract
Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) is a key enzyme of the purine recycling pathway that catalyzes the conversion of 5-phospho-ribosyl-α-1-pyrophosphate and guanine or hypoxanthine to guanosine monophosphate (GMP) or inosine monophosphate (IMP), respectively, and pyrophosphate (PPi). We report the first crystal structure of a fungal 6-oxopurine phosphoribosyltransferase, the Saccharomyces cerevisiae HGPRT (Sc-HGPRT) in complex with GMP. The crystal structures of full length protein with (WT1) or without (WT2) sulfate that mimics the phosphate group in the PPi binding site were solved by molecular replacement using the structure of a truncated version (Δ7) solved beforehand by multiwavelength anomalous diffusion. Sc-HGPRT is a dimer and adopts the overall structure of class I phosphoribosyltransferases (PRTs) with a smaller hood domain and a short two-stranded parallel β-sheet linking the N- to the C-terminal end. The catalytic loops in WT1 and WT2 are in an open form while in Δ7, due to an inter-subunit disulfide bridge, the catalytic loop is in either an open or closed form. The closure is concomitant with a peptide plane flipping in the PPi binding loop. Moreover, owing the flexibility of a GGGG motif conserved in fungi, all the peptide bonds of the phosphate binding loop are in trans conformation whereas in nonfungal 6-oxopurine PRTs, one cis-peptide bond is required for phosphate binding. Mutations affecting the enzyme activity or the previously characterized feedback inhibition by GMP are located at the nucleotide binding site and the dimer interface.
Keywords: crystal structure, phosphoribosyltransferase, HGPRT, cis/trans isomerization, peptide flipping, glycine residue
Introduction
Hypoxanthine-guanine phosphoribosyltransferase (HGPRT, Ec.2.4.2.8) catalyzes the synthesis of the purine nucleoside monophosphates, inosine 5′-monophosphate (IMP) and guanosine 5′-monophosphate (GMP), by transferring the phosphoribosyl moiety of 5-phosphoribosyl-α-1-pyrophosphate (PRPP) to either hypoxanthine or guanine.1 This reaction is Mg2+ dependant. Fungi have two specific 6-oxopurine phosphoribosyltransferase (PRT) enzymes that share at least 53% of identity. In Saccharomyces cerevisiae, one enzyme Sc-HGPRT (25.2 kDa, 221 residues) catalyzes the synthesis of IMP and GMP while Sc-XPRT preferentially recognizes xanthine and catalyzes its conversion to xanthosine 5′-monophosphate.2 PRTs are known to be active as dimers or tetramers3 and it has been shown that Sc-HGPRT is active in vivo as a dimer.4 Four loss-of-function mutations (G37D, R45K, R116C, and L47Q)5 have been identified in the HPT1 gene coding for Sc-HGPRT. Feedback inhibition by GMP has been documented and five mutations (F50S, K159R, K161R, V184I, and I212V) leading to a down-regulated enzyme have been characterized.6
The crystal structures of human, protozoan parasites, bacteria, and archaea 6-oxopurine PRTs either free or in complex with various substrates, products, analogs, or inhibitors have been solved (for a review, see Ref.7). Orotate, glutamine-amido, and 6-oxopurine PRTs belong to class I PRTs, which fold in two domains: a core with a conserved Rossman fold and a variable hood. The hood often contains a three- or four-stranded antiparallel β-sheet. Essential functions have been attributed to four connecting loops.1, 8 Loop I is involved in the binding of the inorganic pyrophosphate (PPi) and in most of the class I PRTs, an unusual nonproline cis peptide linkage allows the formation of a hydrogen bond between the terminal phosphate of PRPP or PPi and the nitrogen of the peptide bond. However, the trans peptide bond found in the structures of the human9–11 and Toxoplasma gondii enzymes12, 13 raised the question of a cis/trans isomerization requirement during catalysis.14, 15 Loop II is the catalytic loop and appears flexible because it can be “open” or “closed” upon transition state formation.3 Loop III is involved in the binding of the phosphoribosyl moiety of PRPP or of monophosphate nucleotides and magnesium. Loop IV, within the hood, interacts with the nucleobase and displays a β-strand and a coil connecting the hood to the core. Despite the fact that structures of 6-oxopurine PRTs are well documented, there was no structure of fungal 6-oxopurine PRTs.
Here, we present the first crystal structures of the S. cerevisiae HGPRT. The structures in complex with GMP and with or without sulfate show the relevance of four successive glycine residues in the PPi binding loop that are conserved in fungal purine PRTs. Conversely with nonfungal 6-oxopurine PRTs, all the peptide bonds of the PPi binding loop are in the trans conformation. Phylogenetic and structural analysis definitely places the Sc-HGPRT in an individualized sub-group of the purine PRT family.
Results
The S. cerevisiae HGPRT is a PRT of class I
The structure Δ7 of the truncated protein (residues 2–214) was first solved by multiwavelength anomalous diffusion (MAD) at 2.3 Å resolutions. The structures of the full length Sc-HGPRT (2-221) with (WT1) and without (WT2) sulfate in the PPi binding site, were afterwards solved by molecular replacement. In the three structures, HGPRT was a dimer with a GMP molecule bound in each binding site. In Δ7, there was a large deviation between chains A and B (RMSD 4.0 Å) while WT1 and WT2 contained more similar chains (RMSD < 0.33 Å), equivalent to the chain B of Δ7 (RMSD < 0.7 Å). The overall description will focus on WT2, the highest resolution structure (1.8 Å) (Fig. 1).
Figure 1.
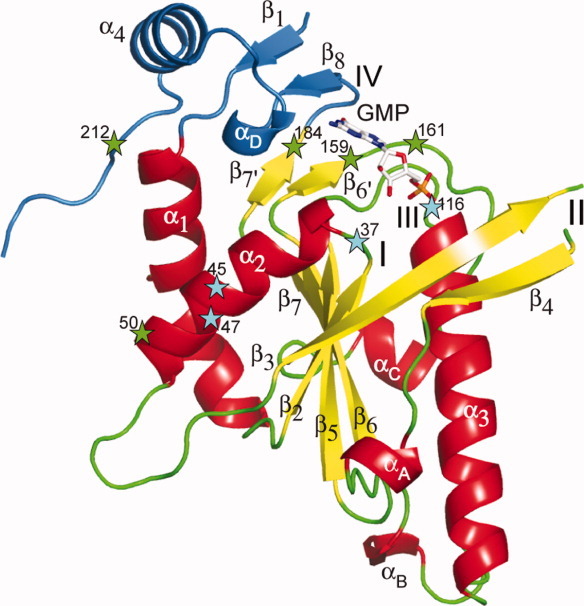
Crystal structure of the chain A of WT2 crystal form of Sc-HGPRT. The core domain is colored in red, yellow, and green according to secondary structure elements whereas the hood domain is colored in blue. The active site was marked by a bound GMP drawn as stick. Secondary structure elements and functional loops are numbered. Loss-of-function and down-regulation mutations are marked with a star colored in cyan and green, respectively. The central β-sheet [Nterm half of β3, β2, β5, β6, β7] is capped by a two-stranded β-sheet [β6′, β7′] and surrounded by three helices [α1, α2, α3]. The hood contains the strands β1 and β8 and the helix α4. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The monomer structure can be split into two domains, a central core and a small hood as for class I PRTs. The core domain (11–186) was a five-stranded twisted parallel β-sheet surrounded by three α-helices, α1 and α2 on one side, and α3 on the other side, and perpendicularly capped by a short two-stranded parallel β-sheet (Fig. 1). The interactions between the helices α1 and α2 and the central sheet were strongly hydrophobic. The replacement of the conserved leucine L47Q in this region (Supporting Information Fig. S1) resulted in a loss-of-function.6 The helix α3 that reaches 32 Å in length is a hallmark of Sc-HGPRT among PRTs. Moreover, the surface loops connecting β3–β5, α3–β6, and β6–β7 contain in Sc-HGPRT one-turn helices, αA (94–96), αB (145–147), and αC (169–173), respectively.
The catalytic loop II (64–102) that connected the corner of the central β-sheet to the strand β5 (Fig. 1) was not fully resolved. At the beginning of loop III, two acidic residues (110–111) conserved in 6-oxopurine PRTs (Supporting Information Fig. S1) were hydrogen bonded to the ribosyl 3′-OH (Fig. 2), directly (WT1 and Δ7) or via water molecules (WT2). A magnesium ion was coordinated to the ribosyl 2′-OH and 3′-OH in Δ7 chain A whereas it was coordinated to the D110/E111 carboxylate groups in the WT2 chain B (Fig. 2). At the end of loop III, the TRTT motif (115–118) tightly bound the GMP 5′-phosphate and the loss-of-function mutation R116C5 revealed the importance of the salt bridge interaction between Arg116 and Asp113 for positioning the loop.
Figure 2.
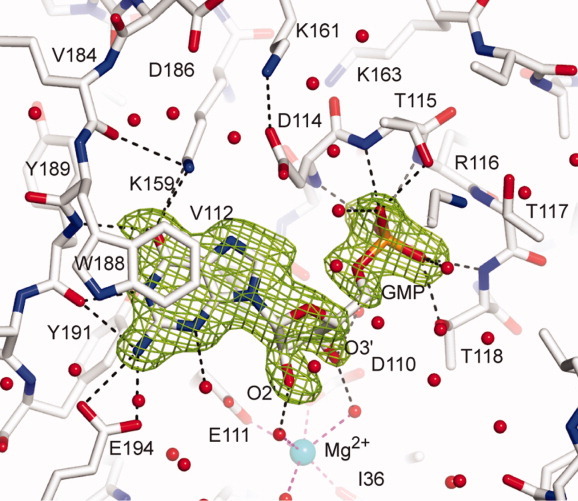
View of the active site. The 2Fo-Fc electron density maps are contoured at 1.5 σ around the GMP in the chain B of WT2 crystal form of Sc-HGPRT. The hydrogen bond network around the GMP molecule is drawn. The hexa-coordinated magnesium is drawn as a sphere colored in cyan. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The small hood domain (2–10 and 186–217) contained the helix α4, the one-turn 310-helix αD and a short parallel β-sheet formed by the strands β1 and β8 belonging to the amino- and the carboxy-termini, respectively. Loop IV (Fig. 1) was limited to the strand β8 (188–190) that bound the base (Fig. 2). The two-stranded hood and the WϕXXXW motif appear to be hallmarks of archaeal and fungal PRTs and bacterial XPRTs (Supporting Information Fig. S1).
The GMP nucleobase was sandwiched between the side-chains of Val112 from the core and the rings of Tyr191 and Trp188 from the hood (Fig. 2). Its recognition was achieved through six hydrogen bonds, two from O6-GMP to Nζ-Lys159 and N-Tyr189, one from N1-GMP to O-Tyr189 and three from N2-GMP to O-Tyr189, Oε1- and Oε2-Glu194. N3-GMP was hydrogen bonded to a water molecule whereas N7-GMP was not hydrogen bonded.
The conformation of the peptide bond Gly37-Gly38 in loop I of Sc-HGPRT
In Sc-HGPRT, loop I (36–40), defined as the PPi binding loop, contains a G/TGGG motif conserved in the fungal HGPRTs (Supporting Information Fig. S1). Successive glycine residues allow the backbone to bend outside the limits allowed with nonglycine residues.
A sulfate ion was found in the PPi binding sites of WT1 and Δ7 [Fig. 3(a–c)] but not in WT2 [Fig. 3(d)]. Superpositions of WT1 and Δ7 with PRT structures containing PPi or PRPP12, 14, 16–18 revealed that the sulfate ion occupied the site of PPi distal phosphate and to a lesser extent of PRPP β-phosphate. Thus sulfate ion binding mimics phosphate group binding. The Gly37-Gly38 peptide bond adopts clearly a trans conformation (Fig. 3) in all the structures with (Δ7 and WT1) or without (WT1) sulfate.
Figure 3.
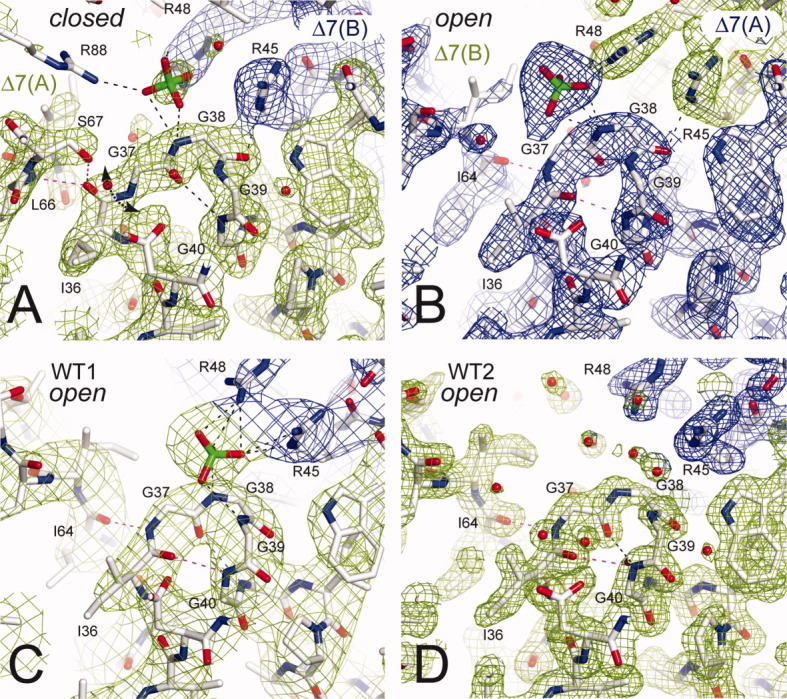
Views of the PPi binding loop. The 2Fo-Fc electron density maps are contoured at 1.5 σ around the PPi binding loop in Δ7 chain A (A) and chain B (B) at 2.3 Å resolution, in WT1 (C) at 3.4 Å resolution and WT2 (D) at 1.8 Å resolution. The Gly37-Gly38 peptide bond adopts clearly a trans conformation in all the structures with or without sulfate. The Ile36-Gly37 peptide bond plane was flipped in Δ7 between chains A and B (A, B). Maps are colored in green and blue for different chains. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In WT1, WT2, and the Δ7 chain B, the GGGG motif belongs to a 13-membered H-bonded turn [Fig. 3(b–d)] allowing a proper orientation of the NH-Gly38 to bind the sulfate [Fig. 3(b–c)] thus likely PPi. Among the four glycine residues, Gly37 is the only one located in the region of the Ramachandran plot with positive phi dihedral angle, which is unfavorable to nonglycine residues (Supporting Information Fig. S2). Hence, G37D is a loss-of-function mutation.5 In the Δ7 chain A, although the peptide plane 36–37 is flipped as described further, the peptide bond 37–38 is trans. Finally, owing to four successive glycine residues in the Sc-HGPRT loop I, the Gly37-Gly38 peptide bond clearly adopts a trans conformation whatever the presence of sulfate ion in the binding site (Fig. 3) thus we can expect the same changes with PPi or PRPP.
In WT1, at the dimer interface, the sulfate ion was coordinated to the NH-Gly38 and NH-Gly39 from one subunit and to the Arg45 and Arg48 side-chains from the adjacent subunit [Fig. 3(c)]. Therefore, the PRPP binding relies on dimer formation. Arg45 and Arg48 are specifically conserved in fungal PRTs and the side-chain length is important as indicated by the R45K loss-of-function mutation.5
In Δ7, an inter-subunit disulfide bond stabilizes one loop II of the dimer in the closed form
In vivo, the Y1846 strain that lacked the HPT1 gene was complemented with the p3486 plasmid harboring the Hpt1Δ7 construct (Fig. 4). In agreement, in the full-length structures, the last seven residues of the C-terminal tail were not involved in the active site, the catalytic loop or the dimer interface (Figs. 1 and 5). Therefore, the shortening had no significant effect on activity or dimerization.
Figure 4.
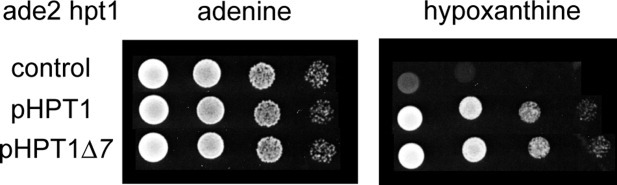
Functionality of the HPT1Δ7 construct. A serial dilution drop test was done on synthetic dextrose medium containing casaminoacids supplemented with either 0.3 mM adenine (left) or hypoxanthine (right). Gene HPT1Δ7 is sufficient to reestablish the growth of the strain in presence of hypoxanthine.
Figure 5.

Separated views of Sc-HGPRT dimers: (A) in WT2 with both catalytic loops in “open” form and 11 Å apart Cys97 sulfhydryl groups and (B) in Δ7 with the catalytic loop in “closed” and “open” forms in chains A and B, respectively with an inter-subunit Cys97-Cys97 disulfide bridge. Chains A and B are colored in salmon and magenta, respectively whereas catalytic loops are colored in blue (residues 65–100). Phe41 and Cys97 side-chains, GMP and sulfate are drawn as stick. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In the fungal 6-oxopurine PRTs, loop II sequences are about 15 residues longer than other PRTs and contain a conserved motif LSLYE (Supporting Information Fig. S1). In WT1 and WT2, loops II adopted the open form (Figs. 5 and 6). The Cterm half (64–69) of the strand β3 and the whole strand β4 formed a short anti-parallel β-sheet going away from the catalytic site. The extremity of the loop (70–84) was disordered. In this open form, the OH-Tyr69 of the LSLYE motif was at ∼16 Å apart from the Oγ-Thr117 of the TRTT motif. At the dimer interface, the Phe41 rings were stacked around the pseudo twofold axis [Fig. 5(a)] while the Cys97 residues were 11 Å far from each other since the crystals grew in the presence of DTT.
Figure 6.
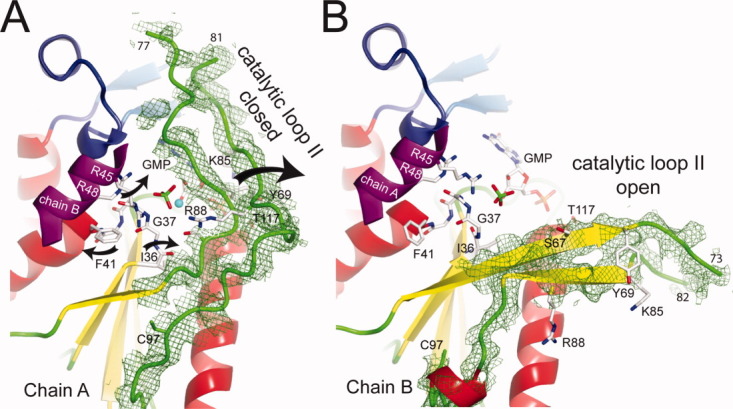
Peptide plane flipping. In the Δ7 crystal form of Sc-HGPRT, the Ile36-Gly37 peptide plane flips between the chain A in closed conformation (A) and the chain B in open conformation (B). Omit maps of electron density encompassing the residues 64–100 of chains A (A) and B (B) at 2.2 Å resolution were contoured at 1.5 σ. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In Δ7, the loop II of chain A became distorted as compared with the one of chain B or WT1 and WT2. This loop is better ordered in chain A where it covered the active site (closed form) than in chain B (open form). In the closed form, the OH-Tyr69 was hydrogen bound with Oγ-Thr117. At the dimer interface, the Cys97 shifted by 11 Å to form an inter-subunit disulfide bridge with the Cys97 of chain B [Fig. 5(b)] while the two Phe41 rings were no longer stacked [Fig. 5(b)]. Since, the crystal was obtained without DTT, the disulfide bridge along with crystal packing (Supporting Information Fig. S3) could be considered responsible for the closure.
In Δ7, one sulfate was coordinated to NH-Gly38 and Arg88 from chain A and Arg48 from chain B [Fig. 3(a)] whereas the other sulfate is simply coordinated to Arg48 from chain A [Fig. 3(b)]. The sulfate ions were similarly located in Δ7 chain A and WT1 but 2.3 Å apart in Δ7 chain B. The two sulfate binding patterns in Δ7 crystal can be attributed to the shift of the arginine side-chains at the dimer interface upon formation of the inter-subunit disulfide bridge. Indeed, superposition of WT1 and WT2 with Δ7 dimers reveals a 2.0 Å shift of the helices α2 of adjacent subunits and thus of the Arg45 and Arg48 side-chains.
Owing to the disulfide bridge, two states of the catalytic loop were revealed. The open form is identical to that found in WT1 and WT2 while the closed form is similar to that found in some PRT structures in complex with inhibitors and PRPP or PPi.14, 16, 19 These two forms could represent extreme conformations of loop II during catalysis.
The flipping of the peptide plane Ile36-Gly37 stabilizes a closed active site
Superpositions of the open [Fig. 3(a)] and closed chains [Fig. 3(b–d)], using strand β2 and helix α2, revealed that the peptide plane Ile36-Gly37 was flipped and that minor adjustments of the backbone occurred between Ile36 and Gly38. Peptide-plane flipping is not a rare event in proteins and could be involved in substrate binding.20 In the closed form, Gly37 adopted a quasi fully extended conformation and the first turn of helix α2 was unwound [Fig. 3(a)]. The backbone dihedral angles ψIle36/φGly37 have values of 113/71° and −4°/−160° in WT2 and Δ7 chain A, respectively. A rotation about ψIle36 from 113° to −4° through 0° in concert with the rotation about φGly37 from 71° to −160° through 180° could follow favored area of Ramachandran plots without neighborhood hindrance (Supporting Information Fig. S2c). The rotation activation energy for peptide-plane flipping has been estimated to be 3 kcal/mol in a concerted mechanism.20 In the open form, O-Ile36 and NH-Gly37 were hydrogen bonded with NH-Gly40 and O-Ile64, respectively [Fig. 3(b–d)] whereas, in the closed form, O-Ile36 was hydrogen bonded with NH-Leu66 and Oγ-Ser67 from loop II and NH-Gly37 was free [Fig. 3(a)]. Hence, the flipping in loop I stabilizes the closed form of the catalytic loop II.
The disulfide bridge strongly impairs the enzyme activity
The kinetics parameters of the Sc-HGPRT enzyme were measured under reducing condition. The apparent KM for guanine and PRPP are 9.3 ± 1.8 μM and 70 ± 17 μM, respectively with a kcat value of about 30 s−1. The KM values are in the same order as those reported for a purified preparation of brewer's yeast, that is, 7.7 μM for guanine and 24 μM for PRPP21 and the kcat value is in the range of the values of 4–70 s−1 reported for other 6-oxopurine PRTs.12, 22, 23 Under oxidizing conditions, an inter-subunit disulfide bond, likely Cys97–Cys97, was shown by SDS-PAGE. Dimeric species formed the major band while a faint band of monomeric species was detected (data not shown). The oxidized enzyme had a low activity (ca. 10%). If the artifactual disulfide bridge totally impairs the enzyme activity, the residual activity could be inferred to residual reduced enzymes. Owing to the cytoplasmic localization of Sc-HGPRT and its reducing environment, the disulfide bond likely does not occur in vivo.
The yeast 6-oxopurine PRTs form a sub-group of PRTs
Eubacteria and ascomycetous yeasts possess two different 6-oxopurine PRTs: one xanthine-guanine specific (X(G)PRT) and one hypoxanthine specific, which can poorly use guanine (H(G)PRT). The eubacterial enzymes are clearly different (23% identity for E. coli PRTs), whereas the yeast ones are quite similar (56% identity for S. cerevisiae PRTs). In mammal and protozoan parasites, one enzyme has evolved in such a way that it uses the three 6-oxopurine substrates (HG(X)PRT). Both mammalian/protozoan and bacterial enzymes are very distant from the yeast ones (<12% identity).
Analysis of the quaternary architecture of known structures of 6-oxopurine PRTs indicates three dimer types (D1, D2, and D3) (Fig. 7). The three dimer interfaces involve (i) the Nterm part of the catalytic loop (ii) a part of the hood domain and (iii) the helix following the PPi binding loop (α2 in Sc-HGPRT). The inter-helical angles between these helices, which can be used to define dimer types, were −104°, −162°, and −67° for dimer D1, D2, and D3, respectively. Whatever the dimer type, the active sites are always made of residues from adjacent subunits.
Figure 7.
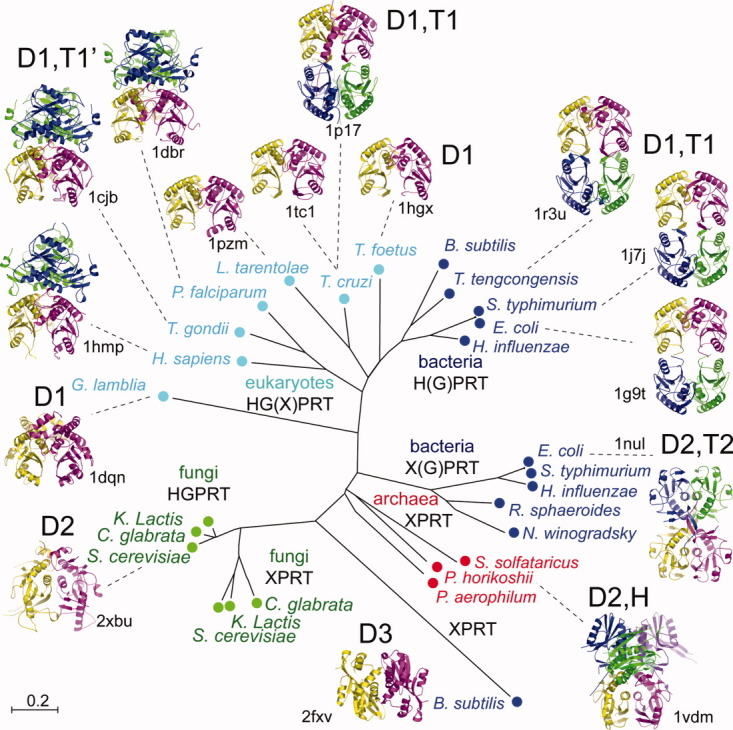
Phylogenetic tree along with quaternary structures of PRTs. In the upper part, HG(X)PRT from eukaryotes excluding fungi and XPRT from bacteria are made up of similar dimers D1 assembled or not as two different types of tetramers T1 and T1′. T. cruzi known to be functional as a dimer is found tetrameric in some crystal structures (1p17 and 1p19). On the lower part, fungi PRTs, bacteria XPRTs and archaea PRTs share another type of dimer D2 and are assembled as dimer, tetramer T2 or hexamer H. Dimer D3 of B. subtilis XPRT appears isolated in evolution and structure. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Dimer D1 is the most documented. The overall structures of their subunits are similar (RMSD 0.7–1.0 Å for 75% of Cα) except for G. lamblia GPRT (RMSD 1.4 Å for 50% of Cα).17 Frequently, dimer D1 assembles through the helices α3 or α1/α2 on the opposite face to form tetramers T1 or T1′, respectively. However, a few dimers D1 do not form higher oligomeric structure. For instance, in G. lamblia GPRT, an α-helix of the hood, which is equivalent to α4 in Sc-HGPRT, prevents the tetramer formation.17T. cruzi HGXPRT, which crystallized as tetramer T1′, is active as a dimer.14
Dimers D2 were found in the P. horikoshii PRT (Ph-PRT), Ec-XPRT, and Sc-HGPRT structures. The dimer interface is predominantly stabilized by hydrophobic interactions contributed by the loops II, the strands β3, the helices α2, and the hood residues 192–203 (Fig. 4). The dimer found in crystals of Sc-HGPRT is likely the active dimer in solution.4 Regarding the monomer, Sc-HGPRT is more similar to Ph-PRT (RMSD 2.2 Å for 50% of Cα)24 than to Ec-XPRT (RMSD 4.3 Å for 42% of Cα),18 and human HG(X)PRT (RMSD 5.5 Å for 46% of Cα).5 In Ph-PRT and Ec-XPRT, the interaction of the hoods leads to hexamer H or tetramer T2, respectively whereas in Sc-HGPRT, a longer helix α4 prevents it.
Dimer D3, which is common in orotate PRTs,25 was found in B. subtilis XPRT,26 which shares sequence similarities with adenine PRTs.
The phylogenetic and structural analysis of available PRT sequences and structures show that fungal 6-oxopurine PRTs form a sub-group of PRTs more related to archaeal purine PRTs and eubacterial XPRTs than to eukaryotic and eubacterial HGPRT enzymes. Indeed, Sc-HGPRT has 21% identity with Ph-PRT and 12% with Ec-XPRT sequences (Supporting Information Fig. S1). The Sc-HGPRT structure can be considered as a paradigm for this sub-group.
Discussion
Mutations in Sc-HGPRT affect feedback inhibition by GMP
Five mutations have been selected and characterized making Sc-HGPRT more or less sensitive to feedback inhibition by GMP.6 K159R, V184I, and K161R are in the vicinity of the purine binding site whereas I212V and F50S are located at the dimeric interface. It is to note that all substitutions selected by reduced feedback inhibition affect also the catalytic activity to different extents.
The conserved Lys159 binds directly the O6-GMP. Therefore its replacement might affect both catalytic activity in the destabilization of the guanine substrate and the feedback inhibition by a lack of binding of GMP. Among the five known down regulation mutations, K159R is the one that affects the most the HGPRT activity (6% of WT activity) and feedback inhibition.
In fungi, Val184 is conserved or replaced by a threonine and does not bind directly to the GMP. In Sc-HGPRT, Val184 and Tyr189 side chains are in close contact and are constrained to lie in a unique orientation. The Tyr189 backbone amino- and carbonyl-groups are both hydrogen bonded to the purine. Therefore, a slight shift of Tyr189 resulting from the bulkier substitution V184I could affect the purine binding, the HGPRT activity (63% of WT activity) and the feedback inhibition. Accordingly, in other species, the substitution of Val184 by isoleucine goes with substitution of Tyr189 by a less bulky side chain (Ile, Leu, or Val).
Lys161 is conserved in fungi 6-oxopurine PRTs and replaced in other species by arginine or tryptophane. In Sc-HGPRT, Lys161 forms a salt bridge with the conserved Asp114 in a way similar as in other PRT structures in complex with GMP or IMP. In human HG(X)PRT, it has been evidenced that the homolog Asp137 could act as a general base for N7-purine deprotonation toward its activation as a nucleophile,27 and in the structure of Ec-XPRT in complex with guanine and cPRPP, the homolog Asp92 interacts with the likely protonated N7-guanine.18 The substitution K161R could affect the interaction with Asp114 and thus the HGPRT activity (51% of WT activity). Unexpectedly, a further mutation I212V restores partially the HGPRT activity (63% of WT activity). The decrease of HGPRT activity (31%) for F50S mutant reinforces the evidence of functional dimer and these results suggest that dimeric organization may be also important for the feedback inhibition. Finally, the selection of mutants more or less sensitive to feedback inhibition appears to be a powerful tool to identify residues indirectly involved in the catalytic mechanism.
The GGGG motif replaces the cis nonproline peptide plane required in the PPi binding loop of PRTs
Generally, a nonproline cis peptide bond (37–38) is required in the loop I of PRTs to allow the backbone amide of the residue in position 38 to point into the PPi binding site. During folding, this peptide bond could adopt the favorable trans conformation then, during dimerization, the energetically unfavorable (∼20 kcal/mol) cis conformation could be stabilized through several inter- and intra-subunit polar interactions involving the side-chain of the residue 38 (R/K/T) in the presence of the conserved Gly39 (Supporting Information Fig. S1).
Occasionally, a trans peptide bond was found in some oligomeric structures of human and T. gondii HG(X)PRTs9–13 and orotate PRTs.25, 28 As a consequence, the backbone amide pointed opposite to the PPi binding site and locally loop I displayed large distortions. Moreover, both conformations have been found in one structure of uracil PRT.29 Finally, trans conformation observed in Tg-HGPRT crystal12 and in the GTP-activated S. solfataricus UPRT30 have been considered a consequence of crystal packing.14 Although a rotational activation energy of about 20 kcal/mol must result in a rather slow process,15 it was suggested that a cis/trans isomerization could occur during catalysis.31 Biological relevance of cis/trans isomerization in PRTs still remains controversial.10, 14
Nevertheless, fungi have evolved a distinctive strategy to overcome this energetic barrier, in selecting a GGGG motif in the PPi binding loop of 6-oxopurine PRTs allowing the backbone amide at residue 38 to point into the binding site although the Gly37–Gly38 peptide bond is in a trans conformation.
Sc-HGPRT adopts an open and a closed conformation in the dimer
In the crystal structures of purine PRTs, loop II adopts an “open” form and a “closed” form in inhibited complex structures. We found a similar behavior with the two forms of Sc-HGPRT structures. Although the length of loop II is variable from 15 to 35 residues, a tyrosine (Y69 in Sc-HGPRT) is conserved in fungi (Supporting Information Fig. S1).16, 32 In the open form, this tyrosine was far away from the nucleotide binding site whereas in the closed form the tyrosine hydroxyl was H-bonded to the nucleotide 5′-phosphate.14, 16, 19 The overall similarity of fold between the closed forms infers the relevance of the conformation of loop II brought to the artifactual disulfide bond.
Across the dimer interface, the stacking/unstacking of the Phe41 rings that seems coupled with the conformational changes of loop I could be related to cooperativity between the two binding sites. In Homo sapiens HG(X)PRT, despite a different dimer interface, it was already argued that the rotation of Phe74 side chain (equivalent to yeast Phe41) could assist the stabilization of the PPi loop upon GMP binding.10
In conclusion, regarding the PPi binding loop, the 6-oxopurine PRT sequences of fungi harbor a stretch of four glycine residues that adopt a trans conformation in the current X-ray structure of S. cerevisiae HGPRT. This differs from other PRT structures where a cis nonproline peptide plane was required suggesting that during catalysis this particular peptide bond could undergo a cis/trans rearrangement. Such a conformational change is not required in 6-oxopurine PRTs of fungi.
Materials and Methods
Yeast strains
S. cerevisiae strain Y1846 (MATa, ade2::KanMX4, hpt1::KanMX4, his3, leu2, ura3) was transformed with the empty plasmid (CEN and URA3) as control (p605) or containing either the wild type HPT1 gene (pHPT1) or the truncated version (pHPT1Δ7) where 21 base pairs had been removed at the 3′ end before the stop codon.
Functionality tests of the HPT1Δ7 construct have been realized with a serial dilution drop test. The experiment was done on synthetic dextrose medium containing casaminoacids supplemented with either 0.3 mM adenine or hypoxanthine.
Over-expression and purification
Expression and purification of Sc-HGPRT were performed as described elsewhere.6 The Δ7 recombinant enzyme, a Sc-HGPRT variant lacking the last seven residues, was prepared as part of a strategy to improve the crystallization process. The HPT1 gene was modified by PCR using the oligonucleotides GAGATTCCATATGTCGGCAAACGATAAGCAA and CGACCTGCTCAGCTCAAATAAAGATGTCATTGCCCTG. The PCR product was inserted into the EcoRV site of the Bluescript SK vector. The plasmid was restricted with Bpu1102 and NdeI, and ligated in the same restriction sites of pET3a (Novagen). C41(DE3) cells were transformed with the plasmid. Δ7 was purified as described for Sc-HGPRT.6 The Δ7 protein was expressed in the E. coli Met-auxotrophic strain B834 in the presence of l-selenomethionine (SeMet). After 10–12 h induction, the SeMet-labeled protein (SeMet-Δ7) was purified as described for Sc-HGPRT.6 The SeMet substitution of the three Met sites and the initial methionine residue removal were confirmed by mass spectrometry. Protein preparations were dialyzed against 20 mM Tris-HCl pH 8.0 buffer and concentrated to 12 mg/mL. Before crystallization, 4 mM guanosine 5′-monophosphate disodium salt (GMP) and 4 mM MgCl2, were added to the protein solution.
Kinetics and activity studies
The enzymatic synthesis of GMP was monitored using the spectrophotometric method33 with a Varian Cary 4000 spectrophotometer equipped with an external temperature controller. Initial velocities were determined at 28°C in 100 mM Tris-HCl pH 8.0, 10 mM MgCl2. The change in extinction coefficient at 258 nm for the conversion of guanine into GMP was found to be 3000 ± 168 M−1 cm−1 in this condition. To determine the Km, increasing concentrations of guanine (2–100 μM) or PRPP (10–900 μM) were used in excess of PRPP (700 μM) or guanine (80 μM), respectively. After 20 min incubation with 2 mM DTT or 2 mM CuCl2, the reaction was started by addition of 0.1 μg of the protein.
Crystallization
Sc-HGPRT wild type protein crystallizes in two different forms WT1 and WT2. Crystals appeared in less than 2 days at 20°C, by the sitting drop vapor diffusion method, after mixing 2 μL of WT protein solution containing 4 mM DTT with 2 μL of reservoir solution. For WT1 crystals, the reservoir solution contained 1.6 M ammonium sulfate, 4% PEG 400, 0.2 M Na-K tartrate, 50 mM Na citrate, 50 mM MES, pH 5.8 while for WT2 crystals, it contained 0.2 M ammonium acetate, 30% PEG 4000, 0.1 M tri-sodium citrate and pH 5.6. Crystallization of Δ7 and SeMet-Δ7 proteins were carried out at 20°C using microbatch technique. Crystals appeared in 48 h after mixing 2 μL of truncated protein solution without DTT with 2 μL of the precipitant solution used for WT2 crystals. Crystals were cryo-protected with mother liquor containing 15% (v/v) glycerol and flash-frozen in liquid nitrogen.
Data collection
WT1 crystals belonging to the orthorhombic space group P21212 diffracted to 3.45 Å at the beamline BM30a (ESRF, Grenoble). The low resolution of the dataset was related to a high 74% solvent content. WT2 crystals belonged to the monoclinic space group P21 and diffracted to 1.8 Å resolution (ID29, ESRF). Δ7 crystals belonging to the tetragonal space group I4 diffracted to 2.3 Å resolution at the beamline BW7A (DESY, Hamburg) and ID23-1 (ESRF). The SeMet-Δ7 dataset were collected at 3.1 Å resolution at three wavelengths (ID29, ESRF) from a single frozen crystal. It was isomorphous to the unlabeled Δ7 crystals. All data were processed with MOSFLM.34 Data treatment statistics are given in Table I.
Table I.
Summary of Data Collection Statistics
| SeMet-Δ7 | |||||
|---|---|---|---|---|---|
| Dataset | WT1 | WT2 | Peak | Edge | Δ7 |
| Space group | P21212 | P21 | I4 | ||
| Cell dimensions (Å°) | a = 118.08 | a = 51.53 | a = 129.63 | a = 128.44 | |
| b = 176.36 | b = 77.54 | c = 56.52 | c = 56.13 | ||
| c = 91.11 | c = 56.63 | ||||
| β = 95.18 | |||||
| Wavelength (Å) | 0.9800 | 0.9340 | 0.97915 | 0.97933 | 1.1000 |
| Resolution (Å) | 32.94–3.45 | 30.00–1.80 | 41–3.10 | 50.0–2.30 | |
| Highest shell (Å) | 3.64–3.45 | 1.90–1.80 | 3.21–3.10 | 2.38–2.30 | |
| Rsyma | 0.09 (0.65) | 0.06 (0.37) | 0.11 (0.54) | 0.12 (0.63) | 0.06 (0.34) |
| Ranoma | 0.05 (0.19) | 0.05 (0.23) | |||
| <I/σ(I)>a | 10.5 (1.9) | 15.3 (3.0) | 11.5 (7.4) | 10.1 (3.3) | 14.94 (2.9) |
| Completenessa (%) | 99.4 (100.0) | 96.6 (98.4) | 99.9 (100.0) | 99.9 (99.7) | 98.3 (96.5) |
| Redundancya | 3.3 (3.3) | 4.3 (3.6) | 6.7 (5.8) | 6.7 (5.3) | 7.1 (7.2) |
| Wilson Bfactor (Å2) | 120 | 22 | 97 | 97 | 58 |
Value in parentheses are for the highest resolution shells.
Crystal structure determination and refinement
Whatever the PRT structure used as search model and taking into account or not the noncrystallographic symmetries (NCS) present in the crystals, molecular replacement failed to solve both Sc-HGPRT and Δ7 structures. The Δ7 structure was first solved by MAD using the 3.3 Å anomalous scattering signal from the SeMet-Δ7 dataset using SOLVE.35 Experimental phases were improved by density modification techniques and 45% of the model was built using RESOLVE.35
Then, using 3.0 Å resolution data of the Δ7 dataset, the model was 66% complete with chain A (168 residues) better defined than chain B (114 residues). The resolution was progressively increased to 2.3 Å. Several rounds of crystallographic refinement and side-chains rebuilding were carried out using CNS36 and XTALVIEW,37 respectively. Finally, TLS refinement with groups including either the catalytic loop or the remaining of the chain was carried out using REFMAC.38 The final model contains residues 5–77 and 81–214 of chain A and residues 5–73 and 82–214 of chain B. One GMP molecule and one sulfate ion were found in each active site. The occupancy of the sulfate ions was refined then fixed to 0.5. A residual electron density peak observed in the active site of chain A was assigned to a magnesium ion based on ligand distances shorter than 2.5 Å.
Afterwards, WT1 and WT2 structures were solved by molecular replacement with MOLREP39 using the coordinates of the Δ7 chain B as search model. WT1 and WT2 structures were refined using CNS and PHENIX,40 respectively. In WT1, the four chains A–D contain residues 5–71 and 84–217. NCS restraints were applied through all stages of refinement. Between two symmetry related Lys182 residues, a 6 σ residual density blob was not assigned. In WT2, chain A contained residues 5–69 and 84–217 while chain B contained residues 5–72 and 84–217. The refinement statistics were reported in Table II. The figures were drawn using PYMOL.41
Table II.
Summary of Crystallographic Refinement Statistics
| WT1 (2jkz) | WT2 (2xbu) | Δ7 (2jky) | |
|---|---|---|---|
| Solvent content (%) | 74 | 45 | 47 |
| Resolutiona (Å) | 32.94–3.45 | 28.21–1.80 | 30.27–2.30 |
| 3.67–3.45 | 1.85–1.80 | 2.36–2.30 | |
| No. reflections | 25,547 (4006) | 39,543 (2734) | 19,335 (1405) |
| Rwork | 0.221 (0.347) | 0.167 (0.220) | 0.204 (0.263) |
| Rfree | 0.234 (0.350) | 0.206 (0.247) | 0.235 (0.305) |
| No. of chains | 4 | 2 | 2 |
| No. of atoms | |||
| Protein | 6508 | 3249 | 3300 |
| Water | none | 475 | 95 |
| Ligands | 116 | 57 | 59 |
| Occupancy-factors | |||
| SO4 | 1/1/1/1 | 0/0 | 0.5/0.5 |
| Mg | 0/0/0/0 | 0/1 | 1/0 |
| B-factors | |||
| Protein (Å2) | 116. | 25.6 | 63. |
| Water (Å2) | n.a. | 40.1 | 66. |
| R.M.S deviations | |||
| Bond lengths (Å) | 0.008 | 0.004 | 0.007 |
| Bond angles (°) | 1.30 | 0.91 | 1.05 |
| Ramachandran (%)b | 82.4/17.6/0.0 | 95.8/3.7/0.5 | 91.2/8.8/0.0 |
Highest resolution shell is shown in parenthesis.
% of residues in most favored regions/allowed regions/disfavored regions.
Phylogenetic analysis
A rooted phylogenetic tree for various 6-oxopurine PRTs was constructed using the MEGA software on a multialignment calculated with CLUSTALW.42
Broader Audience Summary
We report the first X-ray structure of a yeast 6-oxopurine phosphoribosyltransferase. The phosphate binding is possible through a loop containing four successive glycine residues. Such a short sequence is often incorporated as flexible linker in fusion proteins without considering that this peptide chain can adopt specific conformations.
Accession number
The atomic coordinates and structure factors of WT1, WT2, and Δ7 crystal structures have been deposited in the Protein Data Bank with accession numbers 2jkz, 2xbu, and 2jky, respectively.
Acknowledgments
The authors wish to thank staffs of beamlines of ESRF (Grenoble, France) and DESY (Hamburg, Germany) for their support. Thanks to Dr Vern L. Schramm for inhibitor gift and to Dr Naoki Kunishima for providing unpublished results about P. horikoshii PRT structure.
Glossary
Abbreviations
- DTT
di-thiothreitol
- Ec
Escherichia coli
- GMP
guanosine 5′ -monophosphate
- GPRT
guanine PRT
- HGPRT
hypoxanthine-guanine PRT
- HPT1
gene coding for Sc-HGPRT
- Hs
Homo sapiens
- IMP
inosine 5′ -monophosphate
- MAD
multiwavelength anomalous diffusion
- NCS
noncrystallographic symmetry
- Ph
Pyrococcus horikoshii
- PPi
pyrophosphate
- PRPP
5-phosphoribosyl-1-α -pyrophosphate
- PRT
phosphoribosyltransferase
- Sc
Saccharomyces cerevisiae
- SeMet
selenomethionine
- WT1
orthorhombic crystal structure of Sc-HGPRT in complex with GMP and sulfate (PDB ID: 2jkz)
- WT2
monoclinic crystal structure of Sc-HGPRT in complex with GMP without sulfate (PDB ID: 2xbu)
- XPRT
xanthine PRT
- Δ 7
crystal structure of the truncated version of Sc-HGPRT in complex with GMP and sulfate (PDB ID: 2jky)
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Schramm VL, Grubmeyer C. Phosphoribosyltransferase mechanisms and roles in nucleic acid metabolism. Prog Nucleic Acid Res Mol Biol. 2004;78:261–304. doi: 10.1016/S0079-6603(04)78007-1. [DOI] [PubMed] [Google Scholar]
- 2.Guetsova ML, Crother TR, Taylor MW, Daignan-Fornier B. Isolation and characterization of the Saccharomyces cerevisiae XPT1 gene encoding xanthine phosphoribosyl transferase. J Bacteriol. 1999;181:2984–2986. doi: 10.1128/jb.181.9.2984-2986.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Subbayya INS, Balaram H. A point mutation at the subunit interface of hypoxanthine-guanine-xanthine phosphoribosyltransferase impairs activity: role of oligomerization in catalysis. FEBS Lett. 2002;521:72–76. doi: 10.1016/s0014-5793(02)02826-0. [DOI] [PubMed] [Google Scholar]
- 4.Nussbaum RL, Caskey CT. Purification and characterization of hypoxanthine-guanine phosphoribosyltransferase from Saccharomyces cerevisiae. Biochemistry. 1981;20:4584–4590. doi: 10.1021/bi00519a011. [DOI] [PubMed] [Google Scholar]
- 5.Lecoq K. 2000. Régulation de l'expression des gènes de biosynthèse de l'AMP et recyclage des composés puriques chez la levure Saccharomyces cerevisiae PhD Thesis N 766, Bordeaux 2 University.
- 6.Breton A, Pinson B, Coulpier F, Giraud M-F, Dautant A, Daignan-Fornier B. Lethal accumulation of guanylic nucleotides in Saccharomyces cerevisiae HPT1-deregulated mutants. Genetics. 2008;178:815–824. doi: 10.1534/genetics.107.083295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monzani PS, Trapani S, Thiemann OH, Oliva G. Crystal structure of Leishmania tarentolae hypoxanthine-guanine phosphoribosyltransferase. BMC Struct Biol. 2007;7:59. doi: 10.1186/1472-6807-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craig SP, III, Eakin AE. Purine phosphoribosyltransferases. J Biol Chem. 2000;275:20231–20234. doi: 10.1074/jbc.R000002200. [DOI] [PubMed] [Google Scholar]
- 9.Eads JC, Scapin G, Xu Y, Grubmeyer C, Sacchettini JC. The crystal structure of human hypoxanthine-guanine phosphoribosyltransferase with bound GMP. Cell. 1994;78:325–334. doi: 10.1016/0092-8674(94)90301-8. [DOI] [PubMed] [Google Scholar]
- 10.Keough DT, Brereton IM, de Jersey J, Guddat LW. The crystal structure of free human hypoxanthine-guanine phosphoribosyltransferase reveals extensive conformational plasticity throughout the catalytic cycle. J Mol Biol. 2005;351:170–181. doi: 10.1016/j.jmb.2005.05.061. [DOI] [PubMed] [Google Scholar]
- 11.Keough DT, Hockova D, Holy A, Naesens LM, Skinner-Adams TS, Jersey J, Guddat LW. Inhibition of hypoxanthine-guanine phosphoribosyltransferase by acyclic nucleoside phosphonates: a new class of antimalarial therapeutics. J Med Chem. 2009;52:4391–4399. doi: 10.1021/jm900267n. [DOI] [PubMed] [Google Scholar]
- 12.Héroux A, White EL, Ross LJ, Borhani DW. Crystal structures of the Toxoplasma gondii hypoxanthine-guanine phosphoribosyltransferase-GMP and -IMP complexes: comparison of purine binding interactions with the XMP complex. Biochemistry. 1999;38:14485–14494. doi: 10.1021/bi990507q. [DOI] [PubMed] [Google Scholar]
- 13.Schumacher MA, Carter D, Ross DS, Ullman B, Brennan RG. Crystal structures of Toxoplasma gondii HGXPRTase reveal the catalytic role of a long flexible loop. Nat Struct Biol. 1996;3:881–887. doi: 10.1038/nsb1096-881. [DOI] [PubMed] [Google Scholar]
- 14.Canyuk B, Medrano FJ, Wenck MA, Focia PJ, Eakin AE, Craig SP., III Interactions at the dimer interface influence the relative efficiencies for purine nucleotide synthesis and pyrophosphorolysis in a phosphoribosyltransferase. J Mol Biol. 2004;335:905–921. doi: 10.1016/j.jmb.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 15.Jabs A, Weiss MS, Hilgenfeld R. Non-proline cis peptide bonds in proteins. J Mol Biol. 1999;286:291–304. doi: 10.1006/jmbi.1998.2459. [DOI] [PubMed] [Google Scholar]
- 16.Shi W, Li CM, Tyler PC, Furneaux RH, Cahill SM, Girvin ME, Grubmeyer C, Schramm VL, Almo SC. The 2.0 Å structure of malarial purine phosphoribosyltransferase in complex with a transition-state analogue inhibitor. Biochemistry. 1999;38:9872–9880. doi: 10.1021/bi990664p. [DOI] [PubMed] [Google Scholar]
- 17.Shi W, Munagala NR, Wang CC, Li CM, Tyler PC, Furneaux RH, Grubmeyer C, Schramm VL, Almo SC. Crystal structures of Giardia lamblia guanine phosphoribosyltransferase at 1.75 Å. Biochemistry. 2000;39:6781–6790. doi: 10.1021/bi000128t. [DOI] [PubMed] [Google Scholar]
- 18.Vos S, Parry RJ, Burns MR, de Jersey J, Martin JL. Structures of free and complexed forms of Escherichia coli xanthine-guanine phosphoribosyltransferase. J Mol Biol. 1998;282:875–889. doi: 10.1006/jmbi.1998.2051. [DOI] [PubMed] [Google Scholar]
- 19.Balendiran GK, Molina JA, Xu Y, Torres-Martinez J, Stevens R, Focia PJ, Eakin AE, Sacchettini JC, Craig SP., III Ternary complex structure of human HGPRTase, PRPP, Mg2+, and the inhibitor HPP reveals the involvement of the flexible loop in substrate binding. Protein Sci. 1999;8:1023–1031. doi: 10.1110/ps.8.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunasekaran K, Gomathi L, Ramakrishnan C, Chandrasekhar J, Balaram P. Conformational interconversions in peptide beta-turns: analysis of turns in proteins and computational estimates of barriers. J Mol Biol. 1998;284:1505–1516. doi: 10.1006/jmbi.1998.2154. [DOI] [PubMed] [Google Scholar]
- 21.Miller RL, Bieber AL. Purification and properties of inosine monophosphate: pyrophosphate phosphoribosyltransferase (EC 2.4.2.8) from brewers yeast. Biochemistry. 1968;7:1420–1426. doi: 10.1021/bi00844a026. [DOI] [PubMed] [Google Scholar]
- 22.Focia PJ, Craig SP, III, Eakin AE. Approaching the transition state in the crystal structure of a phosphoribsyltransferase. Biochemistry. 1998;37:17120–17127. doi: 10.1021/bi9821465. [DOI] [PubMed] [Google Scholar]
- 23.Lee CC, Craig SP, III, Eakin AE. A single amino acid substitution in the human and a bacterial hypoxanthine phosphoribosyltransferase modulates specificity for the binding of guanine. Biochemistry. 1998;37:3491–3498. doi: 10.1021/bi9720179. [DOI] [PubMed] [Google Scholar]
- 24.Sugahara M, Kunishima N. 2004. Crystal structure of purine phosphoribosyltransferase from Pyrococcus horikoshii OT3. doi: 10.2210/pdb1vdm/pdb.
- 25.Scapin G, Grubmeyer C, Sacchettini JC. Crystal structure of orotate phosphoribosyltransferase. Biochemistry. 1994;33:1287–1294. doi: 10.1021/bi00172a001. [DOI] [PubMed] [Google Scholar]
- 26.Arent S, Kadziola A, Larsen S, Neuhard J, Jensen KF. The extraordinary specificity of xanthine phosphoribosyltransferase from Bacillus subtilis elucidated by reaction kinetics, ligand binding, and crystallography. Biochemistry. 2006;45:6615–6627. doi: 10.1021/bi060287y. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Grubmeyer C. Catalysis in human hypoxanthine-guanine phosphoribosyltransferase: Asp 137 acts as a general acid/base. Biochemistry. 1998;37:4114–4124. doi: 10.1021/bi972519m. [DOI] [PubMed] [Google Scholar]
- 28.Chang C, Li H, Clancy S, Joachimiak A. 2007. Crystal structure of phosphoribosyltransferase from Corynebacterium diphtheriae. doi: 10.2210/pdb2p1z/pdb.
- 29.Arent S, Harris P, Jensen KF, Larsen S. Allosteric regulation and communication between subunits in uracil phosphoribosyltransferase from Sulfolobus solfataricus. Biochemistry. 2005;44:883–892. doi: 10.1021/bi048041l. [DOI] [PubMed] [Google Scholar]
- 30.Christoffersen S, Kadziola A, Johansson E, Rasmussen M, Willemoës M, Jensen KF. Structural and kinetic studies of the allosteric transition in Sulfolobus solfataricus uracil phosphoribosyltransferase: permanent activation by engineering of the C-terminus. J Mol Biol. 2009;393:464–477. doi: 10.1016/j.jmb.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 31.Héroux A, White EL, Ross LJ, Davis RL, Borhani DW. Crystal structure of Toxoplasma gondii hypoxanthine-guanine phosphoribosyltransferase with XMP, pyrophosphate, and two Mg2+ ions bound: insights into the catalytic mechanism. Biochemistry. 1999;38:14495–14506. doi: 10.1021/bi990508i. [DOI] [PubMed] [Google Scholar]
- 32.Medrano FJ, Wenck MA, Eakin AE, Craig SPIII. Functional roles for amino acids in active site loop II of a hypoxanthine phosphoribosyltransferase. Biochim Biophys Acta. 2003;1650:105–116. doi: 10.1016/s1570-9639(03)00206-1. [DOI] [PubMed] [Google Scholar]
- 33.Carter CE. Reaction of 6-mercaptopurine with inosine- and guanosine-5′-phosphate pyrophosphorylase purified from E. coli. Biochem Pharmacol. 1959;2:105–111. doi: 10.1016/0006-2952(59)90077-2. [DOI] [PubMed] [Google Scholar]
- 34.Leslie A. Joint CCP4 + ESF-EAMCB Newsletter on Protein. Crystallography. 1992;26:1781–1802. [Google Scholar]
- 35.Terwilliger TC. Automated side-chain model building and sequence assignment by template matching. Acta Crystallogr D Biol Crystallogr. 2003;59:45–49. doi: 10.1107/S0907444902018048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 37.McRee DE. XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 38.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 39.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 40.Afonine PV, Grosse-Kunstleve RW, Adams PD. The Phenix refinement framework. CCP4 Newsletter. 2005;42 contribution 8. [Google Scholar]
- 41.DeLano W. The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 42.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.