

Abstract
Shwachman-Diamond syndrome (SDS) is an autosomal-recessive marrow failure syndrome with a predisposition to leukemia. SDS patients harbor biallelic mutations in the SBDS gene, resulting in low levels of SBDS protein. Data from nonhuman models demonstrate that the SBDS protein facilitates the release of eIF6, a factor that prevents ribosome joining. The complete abrogation of Sbds expression in these models results in severe cellular and lethal physiologic abnormalities that differ from the human disease phenotype. Because human SDS cells are characterized by partial rather than complete loss of SBDS expression, we interrogated SDS patient cells for defects in ribosomal assembly. SDS patient cells exhibit altered ribosomal profiles and impaired association of the 40S and 60S subunits. Introduction of a wild-type SBDS cDNA into SDS patient cells corrected the ribosomal association defect, while patient-derived SBDS point mutants only partially improved subunit association. Knockdown of eIF6 expression improved ribosomal subunit association but did not correct the hematopoietic defect of SBDS-deficient cells. In summary, we demonstrate an SBDS-dependent ribosome maturation defect in SDS patient cells. The role of ribosomal subunit joining in marrow failure warrants further investigation.
Introduction
Shwachman-Diamond syndrome (SDS) is characterized by impaired hematopoiesis and a predisposition to aplastic anemia, myelodysplasia, and myeloid leukemia.1,2 The clinical phenotype is not solely hematopoietic, with patients demonstrating multiorgan dysfunction, including pancreatic, skeletal, immunologic, and neurocognitive impairments.3,4 SDS is caused by biallelic mutations of the Shwachman-Bodian-Diamond syndrome (SBDS) gene, resulting in low levels of SBDS protein.5–7 Complete abrogation of SBDS expression appears to be lethal, since biallelic null mutations have not been found. This is consistent with the finding that Sbds deletion in mice is embryonic lethal.8
Hematopoietic progenitors from patients with SDS exhibit reduced colony formation even when cocultured with healthy control marrow stromal cells.2 Knockdown of Sbds by RNA interference (RNAi) in murine hematopoietic cells induces defects in granulocytic differentiation, myeloid progenitor generation, short-term hematopoietic engraftment, and B-lymphocyte numbers.9 SBDS knockdown impairs erythroid differentiation in vitro.10 Together, these data demonstrate a hematopoietic cell–intrinsic function for SBDS. In addition to defects in the myeloid hematopoietic lineage, the finding of marked abnormalities in lymphocyte numbers and function in SDS patients11 is consistent with a role for SBDS at the level of the hematopoietic stem cell or early uncommitted progenitor cell. Indeed, an increased risk of severe aplastic anemia is associated with SDS.
Loss of SBDS also has effects on blood development extrinsic to the hematopoietic stem cells. Dror and Freedman reported an impaired ability of marrow stroma from SDS patients to support hematopoiesis in long-term coculture with healthy control hematopoietic progenitor cells.2 In a murine model, targeted deletion in osteoprogenitor cells of Dicer1, an Rnase III endonuclease required for microRNA biogenesis and RNA processing, resulted in myelodysplasia and leukemia predisposition.12 Transcriptional profiling of the Dicer1-deleted osteoprogenitor cells revealed a significant reduction in Sbds expression. Targeted deletion of Sbds in osteoprogenitor cells similarly resulted in cytopenias and marrow dysplasia, reminiscent of the hematologic abnormalities in SDS. These studies demonstrate a role for Sbds in the marrow stroma to promote normal hematopoiesis.
The molecular mechanisms whereby depletion of SBDS protein impairs normal hematopoiesis remain poorly understood.13,14 Immunofluorescence studies demonstrate that the human SBDS protein is concentrated within the nucleolus of human cells6 as confirmed with multiple different antibodies and fixation conditions (S.A.C., Shimamura laboratory, unpublished data, 2010). Human SBDS associates with 60S ribosomal subunits but not with mature 80S subunits or polysomes.15 Multiple lines of evidence demonstrate an essential role for SBDS in the normal maturation of the ribosome.16–18 Additional effects of SBDS loss have also been described, including mitotic spindle destabilization,19 altered cellular stress responses,20 and impaired neutrophil chemotaxis.21,22 Data are currently lacking as to the mechanism whereby SBDS loss causes hematopoietic failure.13
Recent studies from animal models have demonstrated a role for SBDS orthologs in ribosome biogenesis. Biochemical studies demonstrate that Sbds is required for the elongation factor–like 1 (EFL1) GTPase-mediated release of eIF6 from the nascent 60S ribosomal subunit.17 Conditional deletion of Sbds in mouse liver results in fulminant liver failure with halfmer formation, consistent with a ribosomal-joining defect.17 Similar ribosomal profile disruption was seen in Dictyostelium discoideum, after acute loss of SBDS.18 Although increased 60S:80S ratios were reported in human SDS lymphoblasts, the dramatic ribosomal profile disruption observed in SBDS ortholog-deleted models were not found.18 Studies of SDS patient marrow fibroblasts did not demonstrate increased free ribosomal subunits or halfmers, compared with controls.15 The absence of halfmers in SDS patient cells, which retain some residual SBDS protein expression,19 raised the question of whether ribosome joining is impaired. We therefore investigated the ability of the 40S and 60S ribosomal subunits from SDS patient-derived cells to associate to form the 80S monomer.
Methods
Cell culture
Informed consent was obtained from patients with SDS per the Declaration of Helsinki in accordance with a human subjects' study protocol approved by the institutional review boards (IRBs) of the Seattle Children's Hospital and Fred Hutchinson Cancer Research Center. Control and SDS patient-derived lymphoblasts and bone marrow stromal cells were prepared and cultured as previously described.6 Anonymous healthy control bone marrow stromal cells were obtained from discarded bone marrow transplantation screens as approved by the Fred Hutchinson Cancer Research Center IRB.
CD34+ cells were purified from anonymous discarded full-term cord blood as previously described,23 using the CD34 Microbead kit (Miltenyi Biotec). Purified CD34+ cells were cryopreserved in 90% FBS/10% DMSO. CD34+ cells were cultured in SS FST6 (Stemspan SFEM; StemCell Technologies) supplemented with penicillin/streptomycin and 100 μg/mL SCF (Invitrogen), 100 μg/mL thrombopoietin (Tpo; PeproTech), 100 μg/mL Flt3 ligand (Invitrogen), and 100 μg/mL IL-6 (Invitrogen). All cells were grown in 5% CO2 humidified incubators at 37°C.
Plasmids
For patient-derived point mutants, full-length human SBDS was cloned into the pBABE vector6 with BamHI and EcoRI restriction enzymes. Clinically relevant point mutations (K33E, R126T, R169C) were introduced using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) and confirmed by sequencing. The SBDS cDNA inserts were cloned into the pHAGE-CMV-DsRed-IRES-ZsGreen lentiviral vectors as previously described.6
Cell transfection
Bone marrow stromal cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were harvested 72 hours after transfection. The following sequences were used to target eIF6 by RNAi: CAGATGTGCTCAAGGTGGAAGTCTT, CCAGGATGAGCTGTCCTCTCTTCTT, GATTCCCTCATTGACAGCCTCACCT. The scrambled control sequence was as follows: CAACAAGATGAAGAGCACCAA.
Lentiviral infection
The Lentilox 3.7 system was used to introduce short hairpin RNAs (shRNA) into cells as previously described.24 Bone marrow stromal cells were infected with lentiviral vectors in the presence of 8 μg/mL hexadimethrine bromide (Sigma-Aldrich) as previously described.6 Cells were harvested 72 hours after the addition of lentivirus.
Antibodies and immunoblotting
Immunoblotting was carried out as previously described.15 Chicken anti-eIF6 (Abcam) was used at a 1/3000 dilution. Donkey anti–chicken IgG (Jackson ImmunoResearch Laboratories) was used at 1/20 000 dilution.
Sucrose density gradients
For polysome profiles, cells were extracted in polysome buffer (20mM HEPES [pH 7.4]), 100mM KCl, 10mM MgCl2, 1% NP-40, 1% sodium deoxycholate (DOC), 1mM DTT, 100 μg/mL cycloheximide, 200 μg/mL heparin, 1mM Pefabloc SC (Roche), 1.4μM Pepstatin A (Sigma-Aldrich), Complete Mini protease inhibitor EDTA free (Roche), 100 U/mL RNAseOut (Invitrogen) as previously described.15 Extracts were loaded onto either 7%-47% or 5%-50% sucrose density gradients (20mM HEPES, pH 7.4, 100mM KCl, 10mM MgCl2, 100 μg/mL cycloheximide, 200 μg/mL heparin) and centrifuged at 260 000g in a SW41 rotor for 2 hours at 4°C. Gradients were fractionated on a Teledyne Isco UA-6 with continuous UV monitoring at 254 nm.
Ribosome dissociation
Ribosomal subunit dissociation.
Cells were collected without cycloheximide pretreatment and extracted as previously described15 with 20mM HEPES (pH 7.4), 100mM KCl, 0.25mM MgCl2, 1mM DTT, 1% NP-40, 1% DOC, Complete Mini protease inhibitor EDTA free, 1mM Pefabloc SC, 1.4μM Pepstatin A, 100 U/mL RNAseOut. Extracts were loaded onto 5%-50% sucrose density gradients (20mM HEPES, pH 7.4, 100mM KCl, 0.25mM MgCl2, 200 μg/mL heparin) and analyzed as described.
Salt dissociation of ribosomal subunits.
Cells were collected without cycloheximide and extracted in polysome buffer minus cycloheximide as described in the previous paragraph. For salt dissociation, 3.5M KCl was added to a final concentration of 0.5M KCl. After a 5-minute incubation at room temperature, the extracts were loaded onto 5%-50% sucrose density gradients (20mM HEPES [pH 7.4], 500mM KCl, 10mM MgCl2, 200 μg/mL heparin) and analyzed as described.
Ribosomal subunit association assay
The ribosomal subunit association assay was modified from previously published protocols.25,26 Cells were extracted in assay buffer (20mM HEPES [pH 7.4], 100mM KCl, 0.25mM MgCl2, 1mM DTT, 1% NP-40, 1% DOC, Complete Mini protease inhibitor EDTA free, 1mM Pefabloc SC, 1.4μM Pepstatin A, 100 U/mL RNAseOut) for 10 minutes on ice. Extracts were clarified by centrifugation at 17 000g for 5 minutes at 4°C. The lower MgCl2 concentration dissociates ribosomes into 40S and 60S ribosomal subunits. To assay ribosomal subunit association, MgCl2 was added to an extract aliquot to achieve a final concentration of 10mM MgCl2 and incubated for 5 minutes at 37°C. Control extract aliquots were also incubated for 5 minutes at 37°C. Samples were cooled in ice water, loaded onto 5%-50% sucrose density gradients (20mM HEPES [pH 7.4], 100mM KCl, 0.25 or 10mM MgCl2, 200 μg/mL heparin), and analyzed as described.
The area under the curve for 40S and 60S ribosomal subunits and 80S ribosomes was determined by excising and weighing individual peaks from the polysome profiles. To determine the area under each peak, a baseline was manually drawn to best fit the curve (supplemental Figure 1A, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Each peak was cut out from an enlarged paper chart recording as shown. The mass of the 60S or 80S peak was compared with that of the 40S peak as an internal standard to determine their relative ratios. As a second approach, digitally captured A254nm absorbance readings (DATAQ Instruments) were also measured and used to quantitate the ribosomal peaks. Buffer alone was run through a control sucrose gradient in parallel to allow subtraction of the substantial buffer background from the polysome profiles (supplemental Figure 1B). With this methodology, the area under the curve was calculated from nadir to nadir for each 40S, 60S, and 80S peak by summing the digital measurements.
Recombinant SBDS protein
Wild-type (WT) and mutant SBDS cDNAs were cloned into the HAT bacterial expression vector pHAT10 (Clontech) and transformed into JM109 competent bacteria (Clontech). The recombinant protein was isolated over a TALON Metal Affinity Resin (Clontech) column per the manufacturer's instructions. To test the ability of recombinant SBDS proteins to associate with the 60S ribosomal subunit, each recombinant protein was added at a final concentration of 25nM to an SDS patient lymphoblast extract in assay buffer. The extracts were incubated for 15 minutes at room temperature followed by a 5-minute incubation at 37°C. After cooling in ice water, the extracts were centrifuged through sucrose gradients as described in the previous section. Proteins from each fraction were TCA precipitated as described in the next section and analyzed by immunoblotting as described in “Antibodies and immunoblotting.”
Protein analysis from sucrose gradients
Proteins were collected from sucrose density gradients as previously described.15 Briefly, fractions were precipitated with an equal volume of cold 20% TCA for 1 hour on ice. Precipitated proteins were pelleted and washed twice with cold acetone. Protein pellets were resuspended in sample buffer before immunoblotting as described.15
Methylcelluose colony formation
Cord blood–derived CD34+ cells were infected with lentiviral vectors expressing either a GFP marker together with an shRNA against SBDS or an mCherry marker together with an shRNA targeting eIF6. A scrambled shRNA was used as a control. Cells were infected with a virus-to-cell ratio of 10 in SS FST6 supplemented with 8 μg/mL hexadimethrine bromide at 37°C for 16 hours. After 2 washes in PBS, the cells were resuspended in fresh SS FST6 and incubated for an additional 24 hours. Cells positive for GFP and mCherry were sorted by flow cytometry. After an additional 24 hours of culture in SS FST6, 750 double-positive CD34+ cells were plated in MethoCult Classic (StemCell Technologies) in triplicate. Hematopoietic colonies were scored in a blinded fashion.
Results
Ribosomal profiles are altered in SDS patient cells
We have previously demonstrated that SBDS expression is high in marrow stromal cells and early hematopoietic precursors.27 A recent murine model demonstrated altered marrow stromal function after targeted Sbds depletion.12 We therefore first chose to examine the ribosomal profiles in SDS bone marrow stromal cells. Using physiologic salt conditions, SDS patient-derived marrow stromal cells demonstrated reduced relative levels of 80S ribosomes compared with healthy controls using the 40S peak to standardize across gradients (Figure 1A). This result was consistently seen for multiple SDS patients (DF259, FHCRC18, FHCRC30, FHCRC32, UW1) of different SBDS genotypes (supplemental Table 1). To test whether this reduction in the 80S subunit was also seen in hematopoietic cells, we compared polysome profiles from SDS patient lymphoblasts to those from healthy controls. Lymphocyte abnormalities are a common feature of SDS11 and were reported in a mouse model of SDS.9 The reduction in the 80S:40S ratio was also seen in the lymphoblasts from SDS patients (supplemental Figure 2).
Figure 1.
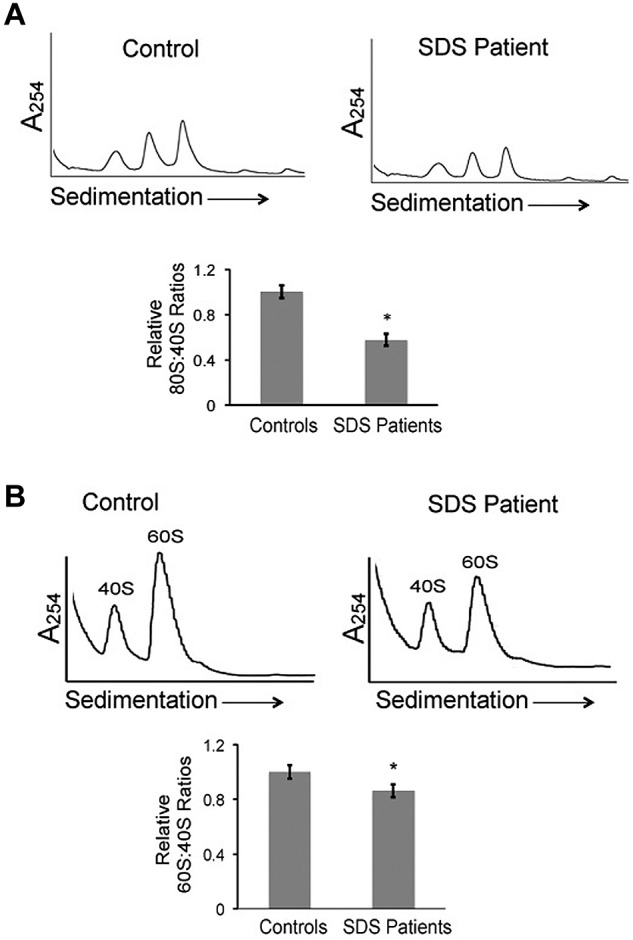
Ribosomal profiles are altered in SDS patient cells. (A) Bone marrow stromal cell lysates from healthy controls or SDS patients were fractionated through 7%-47% sucrose gradients by ultracentrifugation. Representative polysome profiles are shown. Relative 80S:40S ratios were quantified for 3 healthy controls versus 5 SDS patients (DF259, FHCRC18, FHCRC30, FHCRC32, UW1) of different SBDS genotypes (listed in supplemental Table 1). (B) Ribosomes were dissociated with 0.5M KCl or 0.25mM MgCl2 into 40S and 60S subunits, followed by sucrose density centrifugation. Representative polysome gradients are shown. 60S:40S ratios in 0.5M KCl were quantified for 3 healthy controls versus 3 SDS patients (DF259, FHCRC32, UW1) and the aggregate results are graphed; *P < .05. For 2 controls and 2 patients, the assay was repeated up to 3 times with consistent results (methods to quantitate the 40S, 60S, and 80S peaks are illustrated in supplemental Figure 1).
In yeast models, deletion of the SBDS ortholog SDO1 results in decreased 60S:40S ratios,16,28 though this was not observed after knockdown of SBDS orthologs in other model systems.17,18 The 40S and 60S ribosomal subunits from bone marrow stromal cells were dissociated with 0.5M KCl. The 40S and 60S ribosomal peaks were then evaluated after sucrose density sedimentation. SDS bone marrow stromal cells demonstrated a modest but consistent reduction in 60S:40S ratios, compared with controls (Figure 1B). Similar results were obtained when ribosomal subunits were dissociated by lowering the MgCl2 concentration (as shown below in Figure 5). Together, these data demonstrate that SDS patient cells have both reduced relative levels of 80S mature ribosomes and a reduction in the 60S:40S subunit ratio.
Figure 5.

Altered 60S:40S subunit ratios are SBDS-dependent. (A) SDS patient-derived bone marrow stromal cells were infected with lentiviral vectors expressing wild-type SBDS (WT) or SBDS point mutants (K33E, R126T, R169C). An empty vector was used as a control (Ctrl). Cells were lysed in 0.25mM MgCl2 to dissociate ribosomes into 40S and 60S subunits. Representative polysome profiles are shown for each condition. Profiles from WT SBDS (dotted lines) are overlaid for comparison with empty vector control or SBDS point mutants (solid lines). (B) 60S:40S ratio quantitation is reflective of 3 independent experiments; *P < .05.
To test whether the reduction of the 80S ribosome was SBDS-dependent, we introduced lentiviral vectors encoding shRNAs to knock down SBDS expression. To minimize off-target effects, we used 2 different shRNAs targeting different regions of SBDS (SBDS RNAi 1, SBDS RNAi 2). A scrambled shRNA was used as a control. To control for potential differences in cell culture/passage that might affect 80S levels, healthy bone marrow stromal cells from the same donor were divided into 3 equal aliquots before lentiviral infection. Seventy-two hours after lentiviral infection, bone marrow stromal cells were harvested and lysed for polysome profiling on sucrose gradients. Knockdown of SBDS protein was confirmed by Western blot, relative to tubulin as an internal standard (Figure 2A). SBDS-depleted bone marrow stromal cells demonstrated consistent reductions in 80S:40S ratios relative to scrambled controls (Figure 2B). Thus, knockdown of SBDS expression recapitulates the changes in the ribosomal profiles in SDS patient cells.
Figure 2.
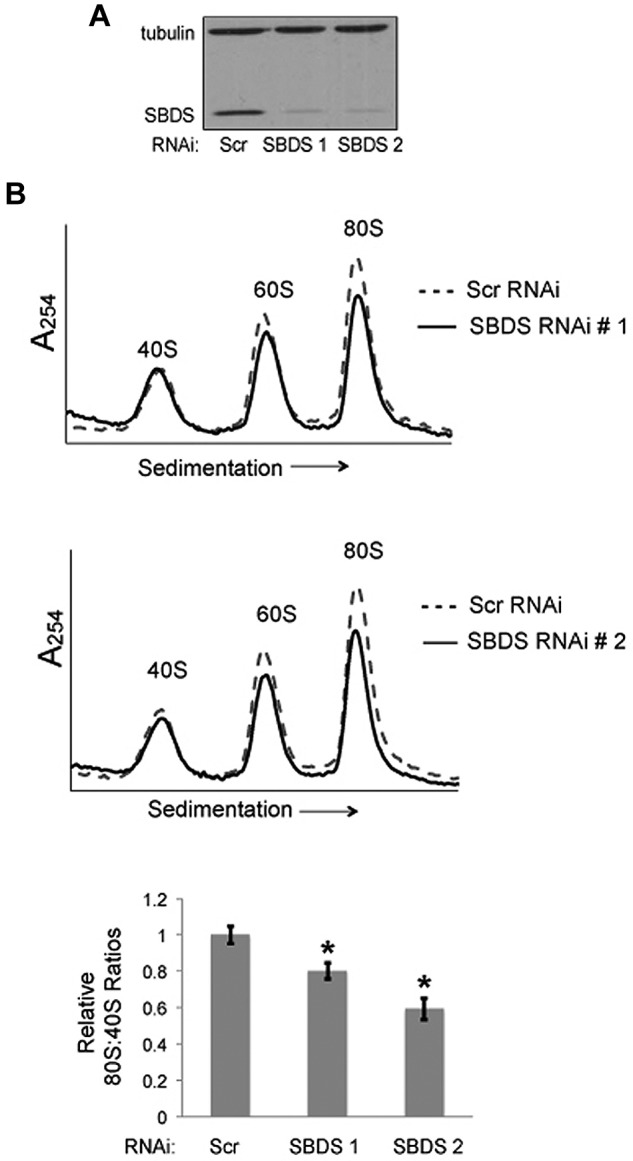
Knockdown of SBDS expression recapitulates ribosomal changes seen in SDS patients. (A) Healthy bone marrow stromal cells were infected with lentivirus encoding either a Scrambled (Scr) shRNA or an SBDS shRNA. Two different RNAi sequences targeting SBDS were assayed (no. 1, no. 2). SBDS knockdown was confirmed by immunoblot. Tubulin was assayed as a loading control. (B) The ribosomal profiles were analyzed by sucrose gradient centrifugation. Representative polysome profile overlays are shown for Scr versus SBDS RNAi experiments. 80S:40S ratios were quantitated for 3 independent experiments. 80S:40S ratios represent the average results for 3 independent experiments for each condition; *P < .05.
Ribosomal subunit association is impaired in SDS patient cells
A conserved role for SBDS in ribosome assembly has been demonstrated across diverse model systems.17,18 In these systems, complete abrogation of SBDS expression results in increased levels of free 40S and 60S subunits and the appearance of halfmer formation, consistent with a subunit-joining defect. Because these ribosomal features were not observed in SDS patient cells, it was possible that reduced 80S levels might reflect other processes, including decreased ribosomal protein stability29 and/or increased degradation of mature ribosomes.30,31 We therefore modified an in vitro subunit association assay25,26 that takes advantage of the magnesium-dependence of 80S formation from 40S and 60S subunits to directly test for ribosome subunit association defects in human SDS cells. In this assay, cell lysates were prepared using 0.25mM MgCl2 conditions to dissociate the 40S and 60S subunits. To assess whether the dissociated subunits maintained their structural integrity, the sedimentation profiles of the dissociated subunits were compared with that from parallel polysome profiles run in their native state. The positions of the 40S and 60S peaks were superimposable (supplemental Figure 3). We also confirmed that SBDS and eIF6 binding to the 60S ribosomal subunit were maintained under these low magnesium buffer conditions (supplemental Figure 4). Lysates were then divided into 2 equal aliquots. The first aliquot was maintained in 0.25mM MgCl2 and used to evaluate the dissociated 40S and 60S subunit levels. In the second aliquot, the MgCl2 concentration was raised to 10mM to assay the ability of the 40S and 60S ribosomal subunits to form the 80S monomer. The ribosomal profile for each aliquot was analyzed by sucrose gradient sedimentation. The ability of the subunits to form the 80S monomer was quantitated by measuring the area under each peak. The 40S peak provided an internal standard to compare relative changes in the 80S peak. Importantly, the 1:1 stoichiometry of the 40S to 60S subunits was maintained in the 80S monosome formed in vitro (supplemental Figure 5).
To assess whether the effect of ribosomal-joining factors was detected in this in vitro subunit association assay, we tested the effect of altering eIF6 expression levels. eIF6 is an anti–joining factor that associates with 60S ribosomal subunits to inhibit 40S and 60S subunit joining.25,26 eIF6 was of high interest because mutations in residues located along the 60S-binding site of the yeast eIF6 ortholog TIF6 are predicted to decrease Tif6 binding to the 60S subunit rescued the slow growth phenotype of the sdo1 mutant.16 Furthermore, SBDS orthologs function to couple the EFL1 GTPase activity with eIF6 release from the 60S subunit in mouse and D discoideum.17,18 Consistent with reports in other systems, knockdown of eIF6 expression in HeLa cells resulted in increased ribosomal subunit association in vitro (supplemental Figure 6A). Conversely, overexpression of eIF6 in HeLa cells resulted in reduced ribosome subunit association (supplemental Figure 6B). Thus, this in vitro assay recapitulates the effect of eIF6 on ribosomal subunit association.
We then examined ribosomal subunit association in bone marrow stromal cells from SDS patients compared with stromal cells from healthy controls. A statistically significant reduction in 80S monomer formation was observed in SDS patient cells relative to controls (Figure 3A). Results were consistent across independent SDS patient samples (DF259, FHCRC32, UW1, CH127) of different genotypes. Impaired ribosomal subunit association was also demonstrated by lymphoblasts from SDS patients (DF259, FHCRC18, FHCRC30, UW1) compared with control lymphoblasts (Figure 3B). Interestingly, ribosomal subunit association was diminished to a greater extent in lymphoblasts, a hematopoietic cell, relative to marrow stromal cells. Together, these data along with Figure 1A demonstrate reduced ribosomal subunit association in SDS cells.
Figure 3.

Ribosomal subunit association is impaired in SDS patient cells. (A) Bone marrow stromal cells or (B) lymphoblasts from healthy controls or SDS patients were lysed in 0.25mM MgCl2 to dissociate the 40S and 60S ribosomal subunits (left panels). MgCl2 was added (+Mg →) to an aliquot of each lysate to test the ability of the 40S and 60S ribosomal subunits to form 80S monomers (right panels). The resulting ribosomal profiles were analyzed by sucrose gradient sedimentation. Representative assays are shown. The resulting 80S:40S ratios were quantitated for 3 healthy controls versus 4 SDS patients of different SBDS genotypes as noted in the text; *P < .05.
Impaired ribosomal subunit association is SBDS-dependent
To determine whether the ribosomal subunit-joining defect is SBDS-dependent, we introduced WT SBDS cDNA or empty vector control into SDS patient-derived marrow stromal cells. SBDS protein expression was confirmed by immunoblot (Figure 4A). Subunit association was assayed as described in “Ribosomal subunit association assay.” Restoration of WT SBDS protein expression was sufficient to correct the defect in 80S formation in SDS patient cells (Figure 4B top left panel).
Figure 4.

Impaired ribosomal subunit association is SBDS-dependent. Ribosomal subunit association was assayed in SDS patient-derived bone marrow stromal cells infected with lentiviral vectors encoding WT or mutant (K33E, R126T, R169C) SBDS cDNA. Empty vector was used as a control (Ctrl). (A) SBDS protein expression was assayed by immunoblot for each condition. (B) Cells were lysed in 0.25mM MgCl2 to dissociate the ribosomal subunits. After the MgCl2 concentration was raised to 10mM, ribosomal subunit association was measured as described in Figure 3. Ribosomal profiles from WT SBDS (dotted lines) are overlaid for comparison with empty vector control or SBDS point mutants (solid lines). Representative polysome profiles are shown. (C) The 80S:40S ratio was quantitated for 3 independent experiments; *P < .05, **P < .01.
We then reasoned that if impaired ribosome joining contributes to disease pathogenesis, patient-derived SBDS mutations would be predicted to impair ribosomal subunit joining. Because human SBDS protein has 3 distinct structural domains32 that are highly conserved,33,34 we assayed the effects of patient-derived SBDS point mutants across different regions of SBDS: K33E, R126T, and R169C. WT SBDS, point mutant SBDS cDNAs, or empty vector controls were introduced into SDS patient-derived marrow stromal cells. SBDS protein expression was confirmed by immunoblot (Figure 4A). Cells were then lysed in 0.25mM MgCl2 to dissociate the 60S and 40S subunits. MgCl2 was added to each lysate to assess the capacity of the ribosomal subunits to associate. The resulting 80S:40S ratios were quantified after separation on sucrose gradients (Figure 4B). Whereas WT SBDS improved subunit association in SDS cells, both K33E and R126T mutants failed to improve 80S formation, compared with WT SBDS. The R169C point mutants also exhibited lower 80S:40S ratios compared with WT SBDS, though this did not reach statistical significance (Figure 4C).
Because we had observed reduced levels of the 60S subunit in SDS patient cells (Figure 1B), we also examined the effects of WT and mutant SBDS cDNAs on the 60S:40S ratio. SDS patient-derived bone marrow stromal cells were infected with lentiviral vectors harboring WT SBDS, SBDS point mutants (K33E, R126T, or R169C), or empty vector controls for 72 hours, followed by lysis in 0.25mM MgCl2. 40S and 60S ribosomal subunits were then separated through a sucrose density gradient. The introduction of WT SBDS resulted in an increased 60S peak, while the 40S peak remained unchanged (Figure 5A). Both the R126T and R169C point mutants improved 60S:40S ratios to a similar extent as that observed with WT SBDS. In contrast, the K33E point mutant did not significantly improve 60S:40S levels in SDS cells compared with WT SBDS (Figure 5B). Thus, an increase in 60S levels was not sufficient to improve 80S formation from the dissociated subunits. Together, these results demonstrate SBDS-dependent impairments in the association of the 40S and 60S subunits and 60S subunit levels in SDS cells.
We have previously shown that SBDS protein associates with the 60S subunit.15 To investigate why the K33E mutation, which resides in the amino-terminal domain of SBDS, was unable to correct the deficiency of 60S subunit levels, we generated cDNAs that progressively deleted 5, 10, or 15 amino acids from the SBDS amino terminus. These SBDS cDNAs were introduced into marrow stromal cells from SDS patients and expression of the SBDS deletion mutants was confirmed by Western blot (Figure 6A). Progressive deletion of the amino-terminal amino acids successively weakened the ability of SBDS to associate with the 60S subunit (Figure 6B). Thus, the amino terminus of SBDS is required for its association with 60S subunits and may account, at least in part, for the inability of the K33E mutant to correct the 60S subunit deficiency.
Figure 6.
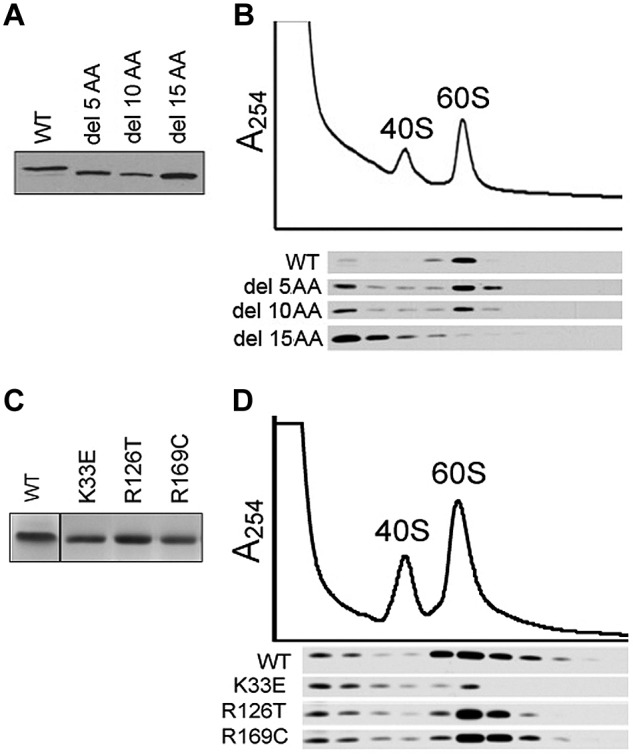
Association of SBDS point mutants with the 60S ribosomal subunit. (A) Marrow stromal cells from SDS patients were infected with FLAG-tagged wild-type SBDS (WT) or SBDS N-terminal deletion mutants lacking the first 5, 10, or 15 amino acids (del 5 AA to del 15 AA). Protein expression was determined with an anti-FLAG Western blot of whole-cell lysates. (B) Cells harboring the indicated SBDS cDNAs were lysed in 0.25mM MgCl2, followed by sucrose density fractionation. Proteins were precipitated from each gradient fraction and immunoblotted with an anti-FLAG antibody (bottom panel). (C) A Coomassie stain of the indicated purified recombinant SBDS proteins is shown. (D) Equimolar quantities of recombinant SBDS proteins were added to SDS lymphoblast lysates prepared in 0.25mM MgCl2 to dissociate the ribosomal subunits. Lysates were fractionated by sucrose density sedimentation to identify 40S and 60S subunit peaks. Proteins were precipitated from equal aliquots of each gradient fraction and immunoblotted for SBDS.
To further investigate this hypothesis, we compared the ability of the different SBDS missense mutants to associate with the 60S subunit. Equimolar quantities of WT or mutant SBDS recombinant protein (Figure 6C) were added to lysates from SDS patient lymphoblasts in 0.25mM MgCl2 buffer to dissociate the ribosomal subunits. After sedimentation of the lysates through sucrose gradients to isolate the 60S subunits, gradient fractions were immunoblotted for SBDS protein. Only the K33E SBDS mutant protein showed a consistent reduction in 60S subunit association compared with WT SBDS or the R126T or R169C mutants (Figure 6D). These results suggest that a subset of SBDS mutations diminish SBDS association with the 60S subunit, while other mutations affect SBDS-mediated subunit association despite intact 60S binding.
Knockdown of eIF6 improves subunit association but not hematopoiesis in SBDS-deficient cells
eIF6 is a ribosome assembly factor that functions to promote 60S subunit maturation,35,36 as well as to prevent ribosomal subunit joining.26 Mutations in yeast TIF6 that impair its binding to the 60S subunit improve the slow-growth phenotype associated with deletion of the SBDS ortholog SDO1. To determine whether eIF6 association with the 60S subunit is altered in SDS patients, lysates from primary marrow stromal cells of controls versus SDS patients were sedimented through sucrose gradients. Proteins from each gradient fraction were blotted for eIF6. The 60S ribosomal protein RPL3 was also blotted as an internal standard to allow comparison of relative eIF6 levels between blots from different gradients. To control for potential differences in the efficiency of protein recovery from each fraction, each gradient fraction was spiked with equal quantities of recombinant purified luciferase protein before protein extraction and analyzed by Western blot with an anti-luciferase antibody. No significant differences in the steady-state levels of 60S-associated eIF6 were noted in patients versus healthy controls (Figure 7A). To further pursue this question, the effect of SBDS overexpression in SDS patient cells was also assessed. As shown in supplemental Figure 7, no SBDS-dependent change was noted in the steady-state levels of 60S-associated eIF6, normalized to RPL3 to control for SBDS-dependent changes in 60S subunit levels. Finally, we performed stable isotype labeling by amino acids in cell culture (SILAC) analysis of 2 different pairs of control cells versus SDS patient lymphoblast cells. Proteins in the 60S subunit fractions were extracted and analyzed by tandem mass spectrometry. The relative ratio of 60S-associated eIF6 in each control versus patient sample pair was similar (supplemental Figure 8), consistent with the results of the Western blots across the gradients in Figure 7A. Taken together with prior published reports, these studies suggest that in patient cells, SBDS might function during the displacement of eIF6 when ribosomal subunits are joining without preventing steady-state binding of eIF6 to pre-60S subunits.
Figure 7.

Knockdown of eIF6 expression improves ribosome association in SDS patient cells but does not rescue the marrow failure phenotype. (A) Lysates from healthy controls versus SDS patients were sedimented through sucrose gradients under native conditions. Exogenous recombinant luciferase protein was added in equal amounts to each fraction before protein extraction to control for potential variations in protein recovery between fractions. Proteins from each fraction were immunoblotted for eIF6, RPL3, and luciferase and quantitated by densitometry. Levels of eIF6 in the 60S fractions were normalized to the 60S subunit protein RPL3 in the 60S fractions as an internal control to allow comparison between different gradient blots and the results of 3 experiments are graphed. The fraction of free eIF6 versus 60S-associated eIF6 was also calculated as a percentage of total eIF6. No statistically significant difference was found between patients versus controls (P = .4). (B) SDS patient-derived marrow stromal cells were transfected with a scrambled RNAi sequence (Scr) or RNAi targeting eIF6. Knockdown of eIF6 in SDS patient cells was confirmed by Western blot with tubulin used as a loading control. Cells were lysed in the presence of 0.25mM MgCl2 to dissociate the ribosomal subunits and assayed in “Ribosomal subunit association assay.” Representative results for cells transfected with the indicated RNAi target sequences are shown. 80S formation relative to the 40S peak was quantitated for 3 independent experiments with cells from SDS patients (FHCRC18, FHCRC30, UW1) of different SBDS genotypes (*P < .05). (C) CD34+ cells were infected with the indicated lentiviral vectors (+) encoding an RNAi sequence targeting SBDS, eIF6, or scrambled control (Scr). Hematopoietic progenitor colony formation in methylcellulose was quantitated in a blinded fashion in triplicate for each condition.
Based on data showing that TIF6 mutations interfering with its ribosomal binding resulted in rescue of the slow growth phenotype of Δsdo1 yeast,16 we next examined whether knockdown of eIF6 expression improved ribosomal subunit association in SDS patient cells. Knockdown of eIF6 protein levels in SDS patient-derived marrow stromal cells was confirmed by Western blot (Figure 7B). Bone marrow stromal cells from each condition were lysed in 0.25mM MgCl2 for testing in the subunit association assay. Knockdown of eIF6 expression rescued the ribosomal subunit association defect in SDS cells compared with scrambled controls (Figure 7B). Similar results were observed in 3 independent SDS patient samples (FHCRC18, FHCRC30, UW1), demonstrating that modulation of eIF6 expression improves ribosomal subunit association across different SBDS mutant genotypes.
Because eIF6 knockdown corrected the ribosomal subunit association defect, we explored the effect of eIF6 knockdown on the hematopoietic failure in SBDS-deficient cells. We constructed bicistronic lentiviral vectors expressing either a GFP marker together with an shRNA against SBDS or an mCherry marker together with an shRNA-targeting eIF6. To minimize off-target effects, 2 different shRNAs targeting different regions of the eIF6 gene were assayed. Healthy human CD34+ cells were infected with lentiviral vectors encoding scrambled control shRNAs, or shRNAs targeting SBDS or eIF6. Seventy-two hours after infection, cells were positively sorted for fluorescent markers (GFP, mCherry) and plated in methylcellulose. Hematopoietic progenitor colony formation was quantified in triplicate in a blinded fashion for each condition (Figure 7C, supplemental Figure 9). Knockdown of SBDS expression in human CD34+ cells consistently decreased the number of myeloid colonies, compared with scrambled controls. The effect on erythroid colony formation was variable after SBDS knockdown, with some experiments showing more pronounced reduction in erythroid colonies compared with controls (Figure 7C vs supplemental Figure 9). eIF6 knockdown decreased formation of both myeloid and erythroid colonies, with a more pronounced effect on the erythroid lineage compared with SBDS shRNA. Knockdown of eIF6 did not improve hematopoietic colony formation in SBDS-deficient CD34+ cells. Instead, hematopoietic colony formation was further reduced after eIF6 knockdown in SBDS-deficient CD34+ cells. Thus, while eIF6 knockdown improves ribosomal subunit association, it does not rescue SBDS-dependent marrow failure.
Discussion
Ribosome assembly is a complex and highly regulated process.37–39 Initial assembly steps of the 40S and 60S ribosomal subunits are coordinated with the generation of a precursor rRNA transcript which is then cleaved to form the nascent pre-40S and pre-60S ribosomal subunits. Each subunit undergoes further nuclear and cytoplasmic maturation via additional assembly factors37 before finally joining together into the mature 80S ribosome to begin protein translation. We demonstrate that 40S and 60S ribosomal subunit association is impaired in SDS patient cells. Ribosomal subunit association is improved after restoration of wild-type SBDS expression. In support of a central role for ribosome joining in SDS, pathogenic patient-derived SBDS missense mutations compromised the ability of SBDS to promote ribosomal subunit association.
Prior evidence for the role of SBDS in ribosome joining was based on its biochemical and genetic interactions with eIF6 and EFL1, factors which regulate ribosome joining; however, ribosomal joining was not directly assessed. We used an in vitro assay based on the studies of Ceci et al to test whether SBDS affects the association of the 40S and 60S ribosomal subunits.26 This assay interrogates the ability of the 40S and 60S subunits to associate together into the 80S monomer. It is important to note, however, that this assay does not model translation initiation, which is a complex multistep energy-driven process.40 Nonetheless, the assay models the effects of previously characterized ribosome assembly factors such as eIF6 on ribosome joining26 and is a useful tool for further analyses of the ability of 40S and 60S subunits, assembled in the presence or absence of SBDS, to associate together.
Prior reports demonstrated that SBDS promotes eIF6 release from isolated 60S subunits.17 These experiments were conducted with eIF6-loaded pre-60S subunits isolated from the livers of Sbds-deleted mice which rapidly die of severe liver failure.17 Increased eIF6 binding was also reported after SBDS knockdown in the TF-1 leukemia cell line.41 Our studies here examined ribosomes isolated from human patients. Cells from SDS patients exhibited lower 60S and 80S levels and impaired subunit association while eIF6 binding to 60S subunits at steady state was not affected. Introduction of WT SBDS failed to reduce steady-state levels of 60S-associated eIF6. One possibility is that perhaps in some cell systems, SBDS does not prevent binding of eIF6 to pre-60S subunits, although SBDS contributes to the displacement of eIF6 during subunit association. Further studies are needed to investigate this process to better understand the role of SBDS in disease pathogenesis.
Knockdown of SBDS in human CD34+ cells resulted in reduced numbers of myeloid and erythroid colonies, with a more pronounced effect on the myeloid lineage. These results are consistent with SBDS depletion in other model systems, which have demonstrated defects in granulocytic differentiation and myeloid progenitor generation, as well as erythroid differentiation.9,10 Mutations in TIF6, the yeast ortholog of eIF6, that are predicted to impair the association of Tif6 with the 60S ribosomal subunit, result in correction of the slow growth phenotype in sdo1 mutants.16 We found that knockdown of eIF6 expression was sufficient to correct the ribosomal subunit association defect in SDS patient cells but failed to correct the hematopoietic failure associated with SDS. Loss of SBDS expression results in multiple cellular abnormalities, including mitotic spindle destabilization,19 impaired chemotaxis,21 increased p53 expression,42,43 accelerated apoptosis,44 increased abundance of reactive oxygen species,45 and altered cellular stress responses.20 While some of these functions may be secondary downstream effects resulting from disruption of ribosome biogenesis, the demonstration of microtubule stabilization with purified SBDS protein in vitro supports a direct role for SBDS in the mitotic spindle.19 This raises the possibility that improvement in ribosome joining is insufficient to rescue marrow failure because other SBDS functions are also coordinately required for normal hematopoiesis. In addition to its role in ribosome joining, eIF6 also plays an important role in 60S subunit maturation.34,35 Thus, it is also possible that specific inhibition of the ribosome-joining function of eIF6, without affecting eIF6's function in 60S maturation, might ameliorate marrow failure in SDS. Further studies are needed to understand the precise molecular function of SBDS in marrow failure.
Ribosomal abnormalities have emerged as a common molecular theme underlying additional marrow failure syndromes such as Diamond-Blackfan anemia and dyskeratosis congenita. Different steps in ribosome biogenesis are targeted in each syndrome and each syndrome exhibits distinct clinical phenotypes.46,47 How the specific ribosomal abnormalities in each syndrome ultimately contribute to different disease phenotypes remains an area of active investigation. Consistent with predictions from model systems, this current study demonstrates that patients with SDS exhibit impaired ribosomal subunit association and abnormalities in 60S ribosomal subunit homeostasis. Altered availability of 60S subunits is predicted to affect the efficiency of joining to the 40S subunit during translation initiation and may provide an additional mechanism for regulation of gene expression.48–50 The molecular mechanisms regulating the dissociation of the ribosomal subunits after completion of protein translation to allow reinitiation of translation remain poorly understood. Regulation of ribosome joining may also affect the formation of active mRNA-associated translation-initiating ribosomes versus inactive nontranslating monomers.
Finally, the ribosomal subunit association assay we describe here may now be explored as a potential functional screen for ribosomal abnormalities in patients presenting with marrow failure of unknown etiology. In addition, approximately 10% of patients who meet the clinical diagnostic criteria for SDS lack identifiable mutations in the SBDS gene. It is unclear whether these patients harbor mutations elsewhere in the SBDS molecular pathway. Potential clinical applications of the ribosome association assay are currently under investigation.
Supplementary Material
Acknowledgments
The authors thank Dr Colleen Delaney (Fred Hutchinson Cancer Research Center) for input regarding the cord blood studies, Michelle Brault (Seattle Children's Research Institute) for virus preparation, Dr David Morris (University of Washington) and Dr Adam Geballe (Fred Hutchinson Cancer Research Center) for helpful discussions, and Dr Vivian MacKay for advice and critical reading of the manuscript.
This work was supported by grants from the National Heart, Lung, and Blood Institute (NHLBI; A.S.). N.B. was supported by a T32 Research Training Grant in Hematology, University of Washington.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: N.B., S.A.C., and T.N. performed the experiments and analyzed the data; N.B. and A.S. wrote the manuscript; S.A.C. and T.N. reviewed the manuscript; and A.S. oversaw the design and analysis of this project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Akiko Shimamura, MD, PhD, Pediatric Oncology, D2-100, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Seattle, WA 98109; e-mail: ashimamu@fhcrc.org.
References
- 1.Smith OP, Hann IM, Chessells JM, Reeves BR, Milla P. Haematological abnormalities in Shwachman-Diamond syndrome. Br J Haematol. 1996;94(2):279–284. doi: 10.1046/j.1365-2141.1996.d01-1788.x. [DOI] [PubMed] [Google Scholar]
- 2.Dror Y, Freedman MH. Shwachman-Diamond syndrome: an inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood. 1999;94(9):3048–3054. [PubMed] [Google Scholar]
- 3.Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009;23(2):233–248. doi: 10.1016/j.hoc.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dror Y, Donadieu J, Koglmeier J, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci. 2011;1242(1):40–55. doi: 10.1111/j.1749-6632.2011.06349.x. [DOI] [PubMed] [Google Scholar]
- 5.Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33(1):97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 6.Austin KM, Leary RJ, Shimamura A. The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood. 2005;106(4):1253–1258. doi: 10.1182/blood-2005-02-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woloszynek JR, Rothbaum RJ, Rawls AS, et al. Mutations of the SBDS gene are present in most patients with Shwachman-Diamond syndrome. Blood. 2004;104(12):3588–3590. doi: 10.1182/blood-2004-04-1516. [DOI] [PubMed] [Google Scholar]
- 8.Zhang S, Shi M, Hui CC, Rommens JM. Loss of the mouse ortholog of the Shwachman-Diamond syndrome gene (sbds) results in early embryonic lethality. Mol Cell Biol. 2006;26(17):6656–6663. doi: 10.1128/MCB.00091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rawls AS, Gregory AD, Woloszynek JR, Liu F, Link DC. Lentiviral-mediated RNAi inhibition of sbds in murine hematopoietic progenitors impairs their hematopoietic potential. Blood. 2007;110(7):2414–2422. doi: 10.1182/blood-2006-03-007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sen S, Wang H, Nghiem CL, et al. The ribosome-related protein, SBDS, is critical for normal erythropoiesis. Blood. 2011;118(24):6407–6417. doi: 10.1182/blood-2011-02-335190. [DOI] [PubMed] [Google Scholar]
- 11.Dror Y, Ginzberg H, Dalal I, et al. Immune function in patients with Shwachman-Diamond syndrome. Br J Haematol. 2001;114(3):712–717. doi: 10.1046/j.1365-2141.2001.02996.x. [DOI] [PubMed] [Google Scholar]
- 12.Raaijmakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson AW, Ellis SR. Of blood, bones and ribosomes: is Shwachman-Diamond syndrome a ribosomopathy? Genes Dev. 2011;25(9):898–900. doi: 10.1101/gad.2053011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burwick N, Shimamura A, Liu JM. Non-Diamond Blackfan anemia disorders of ribosome function: Shwachman Diamond syndrome and 5q- syndrome. Semin Hematol. 2011;48(2):136–143. doi: 10.1053/j.seminhematol.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganapathi KA, Austin KM, Lee CS, et al. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007;110(5):1458–1465. doi: 10.1182/blood-2007-02-075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menne TF, Goyenechea B, Sanchez-Puig N, et al. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39(4):486–495. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- 17.Finch AJ, Hilcenko C, Basse N, et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011;25(9):917–929. doi: 10.1101/gad.623011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong CC, Traynor D, Basse N, Kay RR, Warren AJ. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood. 2011;118(16):4305–4312. doi: 10.1182/blood-2011-06-353938. [DOI] [PubMed] [Google Scholar]
- 19.Austin KM, Gupta ML, Coats SA, et al. Mitotic spindle destabilization and genomic instability in Shwachman-Diamond syndrome. J Clin Invest. 2008;118(4):1511–1518. doi: 10.1172/JCI33764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ball HL, Zhang B, Riches JJ, et al. Shwachman-Bodian Diamond syndrome is a multi-functional protein implicated in cellular stress responses. Hum Mol Genet. 2009;18(19):3684–3695. doi: 10.1093/hmg/ddp316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wessels D, Srikantha T, Yi S, Kuhl S, Aravind L, Soll DR. The Shwachman-Bodian-Diamond syndrome gene encodes an RNA-binding protein that localizes to the pseudopod of dictyostelium amoebae during chemotaxis. J Cell Sci. 2006;119(Pt 2):370–379. doi: 10.1242/jcs.02753. [DOI] [PubMed] [Google Scholar]
- 22.Orelio C, Kuijpers TW. Shwachman-Diamond syndrome neutrophils have altered chemoattractant-induced F-actin polymerization and polarization characteristics. Haematologica. 2009;94(3):409–413. doi: 10.3324/haematol.13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delaney C, Varnum-Finney B, Aoyama K, Brashem-Stein C, Bernstein ID. Dose-dependent effects of the notch ligand Delta1 on ex vivo differentiation and in vivo marrow repopulating ability of cord blood cells. Blood. 2005;106(8):2693–2699. doi: 10.1182/blood-2005-03-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tulpule A, Lensch MW, Miller JD, et al. Knockdown of Fanconi anemia genes in human embryonic stem cells reveals early developmental defects in the hematopoietic lineage. Blood. 2010;115(17):3453–3462. doi: 10.1182/blood-2009-10-246694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valenzuela DM, Chaudhuri A, Maitra U. Eukaryotic ribosomal subunit anti-association activity of calf liver is contained in a single polypeptide chain protein of mr = 25,500 (eukaryotic initiation factor 6). J Biol Chem. 1982;257(13):7712–7719. [PubMed] [Google Scholar]
- 26.Ceci M, Gaviraghi C, Gorrini C, et al. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature. 2003;426(6966):579–584. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 27.Wong TE, Calicchio ML, Fleming MD, Shimamura A, Harris MH. SBDS protein expression patterns in the bone marrow. Pediatr Blood Cancer. 2010;55(3):546–549. doi: 10.1002/pbc.22573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore JB, 4th, Farrar JE, Arceci RJ, Liu JM, Ellis SR. Distinct ribosome maturation defects in yeast models of Diamond-Blackfan anemia and Shwachman-Diamond syndrome. Haematologica. 2010;95(1):57–64. doi: 10.3324/haematol.2009.012450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorenstein C, Warner JR. Synthesis and turnover of ribosomal proteins in the absence of 60S subunit assembly in Saccharomyces cerevisiae. Mol Gen Genet. 1977;157(3):327–332. doi: 10.1007/BF00268670. [DOI] [PubMed] [Google Scholar]
- 30.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10(5):602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- 31.Luzzatto L, Banks J, Marks PA. Protein synthesis in erythroid cells. 3. Monoribosome and polyribosome function in the cell-free system. Biochim Biophys Acta. 1965;108(3):434–446. doi: 10.1016/0005-2787(65)90035-3. [DOI] [PubMed] [Google Scholar]
- 32.de Oliveira JF, Sforca ML, Blumenschein TM, et al. Structure, dynamics, and RNA interaction analysis of the human SBDS protein. J Mol Biol. 2010;396(4):1053–1069. doi: 10.1016/j.jmb.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 33.Shammas C, Menne TF, Hilcenko C, et al. Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome. J Biol Chem. 2005;280(19):19221–19229. doi: 10.1074/jbc.M414656200. [DOI] [PubMed] [Google Scholar]
- 34.Savchenko A, Krogan N, Cort JR, et al. The Shwachman-Bodian-Diamond syndrome protein family is involved in RNA metabolism. J Biol Chem. 2005;280(19):19213–19220. doi: 10.1074/jbc.M414421200. [DOI] [PubMed] [Google Scholar]
- 35.Sanvito F, Piatti S, Villa A, et al. The beta4 integrin interactor p27(BBP/eIF6) is an essential nuclear matrix protein involved in 60S ribosomal subunit assembly. J Cell Biol. 1999;144(5):823–837. doi: 10.1083/jcb.144.5.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basu U, Si K, Warner JR, Maitra U. The Saccharomyces cerevisiae TIF6 gene encoding translation initiation factor 6 is required for 60S ribosomal subunit biogenesis. Mol Cell Biol. 2001;21(5):1453–1462. doi: 10.1128/MCB.21.5.1453-1462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kressler D, Hurt E, Bassler J. Driving ribosome assembly. Biochim Biophys Acta. 2010;1803(6):673–683. doi: 10.1016/j.bbamcr.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Panse VG, Johnson AW. Maturation of eukaryotic ribosomes: acquisition of functionality. Trends Biochem Sci. 2010;35(5):260–266. doi: 10.1016/j.tibs.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Acker MG, Lorsch JR. Mechanism of ribosomal subunit joining during eukaryotic translation initiation. Biochem Soc Trans. 2008;(Pt4):653–657. doi: 10.1042/BST0360653. [DOI] [PubMed] [Google Scholar]
- 40.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11(2):113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sezgin G, Henson AL, Nihrane A, et al. Impaired growth, hematopoietic colony formation, and ribosome maturation in human cells depleted of Shwachman-Diamond syndrome protein SBDS [published online ahead of print September 19, 2012]. Pediatr Blood Cancer. doi: 10.1002/pbc.24300. doi: 10.1002/pbc.24300. [DOI] [PubMed] [Google Scholar]
- 42.Elghetany MT, Alter BP. p53 protein overexpression in bone marrow biopsies of patients with Shwachman-Diamond syndrome has a prevalence similar to that of patients with refractory anemia. Arch Pathol Lab Med. 2002;126(4):452–455. doi: 10.5858/2002-126-0452-PPOIBM. [DOI] [PubMed] [Google Scholar]
- 43.Dror Y. P53 protein overexpression in Shwachman-Diamond syndrome. Arch Pathol Lab Med. 2002;126(10):1157–1158. doi: 10.5858/2002-126-1157b-PPOISS. [DOI] [PubMed] [Google Scholar]
- 44.Rujkijyanont P, Watanabe K, Ambekar C, et al. SBDS-deficient cells undergo accelerated apoptosis through the fas-pathway. Haematologica. 2008;93(3):363–371. doi: 10.3324/haematol.11579. [DOI] [PubMed] [Google Scholar]
- 45.Ambekar C, Das B, Yeger H, Dror Y. SBDS-deficiency results in deregulation of reactive oxygen species leading to increased cell death and decreased cell growth. Pediatr Blood Cancer. 2010;55(6):1138–1144. doi: 10.1002/pbc.22700. [DOI] [PubMed] [Google Scholar]
- 46.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–122. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luzzatto L, Karadimitris A. Dyskeratosis and ribosomal rebellion. Nat Genet. 1998;19(1):6–7. doi: 10.1038/ng0598-6. [DOI] [PubMed] [Google Scholar]
- 48.Martin-Marcos P, Hinnebusch AG, Tamame M. Ribosomal protein L33 is required for ribosome biogenesis, subunit joining, and repression of GCN4 translation. Mol Cell Biol. 2007;27(17):5968–5985. doi: 10.1128/MCB.00019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foiani M, Cigan AM, Paddon CJ, Harashima S, Hinnebusch AG. GCD2, a translational repressor of the GCN4 gene, has a general function in the initiation of protein synthesis in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11(6):3203–3216. doi: 10.1128/mcb.11.6.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steffen KK, MacKay VL, Kerr EO, et al. Yeast life span extension by depletion of 60S ribosomal subunits is mediated by Gcn4. Cell. 2008;133(2):292–302. doi: 10.1016/j.cell.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.