

Abstract
Most equine herpesvirus 1 (EHV-1) strains, including the naturally occurring virulent RacL11 isolate, encode a large glycoprotein, gp2 (250 kDa), which is expressed from gene 71. Besides other alterations in the viral genome, the avirulent strain KyA harbors an in-frame deletion of 1,242 nucleotides in gene 71. To examine the contributions of gp2 variation to virus growth and virulence, mutant RacL11 and KyA viruses expressing full-length or truncated gp2 were generated. Western blot analyses demonstrated expression of a 250-kDa gp2 in cells infected with RacL11 virus or a mutant KyA virus harboring full-length gene 71, whereas a 75- to 80-kDa gp2 was detected in cells infected with KyA or mutant RacL11 virus expressing KyA gp2. The RacL11 gp2 precursor of 250 kDa in size and its truncated KyA counterpart of 80 kDa, as well as the 42-kDa carboxy-terminal gp2 subunit, were incorporated into virus particles. Absence of gp2 in RacL11 resulted in a 6-fold reduction of extracellular virus titers and a 13% reduction of plaque diameters, whereas gp2-negative KyA exhibited a 55% reduction in plaque diameter and a 51-fold decrease in extracellular virus titers. The massive growth defects of gp2-negative KyA could be restored by reinsertion of the truncated but not the full-length gp2 gene. The virulence of the generated gp2 mutant viruses was compared to the virulence of KyA and RacL11 in a murine infection model. RacL11 lacking gp2 was apathogenic for BALB/c mice, and insertion of the truncated KyA gp2 gene into RacL11 was unable to restore virulence. Similarly, replacement in the KyA genome of the truncated with the full-length RacL11 gene 71 did not result in the generation of virulent virus. From the results we conclude that full-length and truncated EHV-1 gp2 are not functionally equivalent and cannot compensate for the action of their homologues in allogeneic virus backgrounds.
Equine herpesvirus 1 (EHV-1), a member of Varicellovirus genus in the Alphaherpesvirinae subfamily, is one of the most prevalent viruses affecting equine populations worldwide. Infection with EHV-1 may result in rhinopneumonitis, abortion, and often fatal encephalomyelitis. The continuing threat imposed by EHV-1 results in ongoing efforts aiming at a better understanding of EHV-1 pathogenesis and at developing safe and effective vaccines (27).
Herpesviral glycoproteins are involved in different stages of virus infection and are major targets of the host's immune response. Envelope glycoproteins of EHV-1 are designated gB, gC, gD, gE, gG, gH, gI, gK, gL, and gM according their homologues in herpes simplex virus type 1 (HSV-1) (reviewed in reference 28). Compared to HSV-1 and other Alphaherpesvirinae, however, EHV-1 encodes two additional glycoproteins. One of the glycoproteins unique to EHV-1 was originally designated gp2 or gp300 (2, 45) and is expressed from gene 71 (EUS4) (38, 42). Homologues of gp2 are found in EHV-1, EHV-4, and asinine herpesvirus 3 (8) and are among the most-abundant and -immunogenic glycoproteins in EHV-1 and EHV-4 virions (1, 7). EHV-1 gp2 is rich in serine and threonine residues and is a heavily O-glycosylated protein with a molecular mass in the range of 192 to >400 kDa (38, 39, 45). In contrast to EHV-4 gp2, the EHV-1 glycoprotein was shown to be partially cleaved into two polypeptides in infected cells (19). Endoproteolytic cleavage occurs after each of two adjacent arginine (R) residues at positions 506 and 507 in the sequence HRGRAGGR506R507G, and results in a 42-kDa carboxy-terminal subunit, which contains the transmembrane anchor, and an N-terminal serine/threonine-rich component that is highly O-glycosylated (19, 43). In the absence of gp2, EHV-1 strains were shown to be impaired in virus egress, while secondary envelopment appears to occur with unaltered kinetics and efficiency (31, 32, 37). It was also reported that deletion of gene 71 in EHV-1 strain Ab4 resulted in attenuation, and the generated virus mutant used as an experimental vaccine conferred protection against pulmonary disease in mice after challenge with wild-type virus (21).
A number of EHV-1 strains with different biological properties have been described. While the RacL11 strain was isolated in the late 1950s from an aborted foal and exhibits high virulence in the natural host and laboratory animals, strain KyA, a candidate vaccine strain, is completely apathogenic for both mice and horses after serial passage in mouse L-M cells (6, 25, 26). As a result of a long history of passages in nonhomologous cells, KyA exhibits several genomic alterations, which predominantly affect the unique short (US) region of the genome. One of these alterations is a major deletion comprising genes 73 to 75, i.e., the gI and gE genes as well as a small open reading frame (ORF) unique to EHV-1 (15). An internal and substantial in-frame deletion of 1,242 bp in the gp2 ORF (gene 71) was also identified, which is predicted to lead to the expression of a truncated protein of only 383 amino acids in length (4, 5, 15) (Fig. 1E). Further, a frameshift mutation in gD (gene 72) results in the expression of a carboxy-terminally truncated version of this envelope constituent, which is involved in virus entry, egress, and cell-to-cell spread (12).
FIG.1.

(A) Schematic illustration of the EHV-1 RacL11 and KyA BAC clones (pRacL11 and pKyA) used for the construction of the gp2 revertant viruses (28). Shown are the overall organization of the circular pRacL11 and pKyA genomes and the BamHI map. Abbreviations: UL, unique long; IR, internal repeat; and TR, terminal repeat. (B) Organization of the unique short (US) region of pRacL11 from gene 69 (US3 homologue) through gene 76 (US9 homologue) and including the mini-F vector sequences derived from plasmid pHA2 and inserted in lieu of gene 71 encoding gp2 (28). (C) The modification of the KyA US region in comparison to the RacL11 US is shown. Arrows indicate transcriptional directions of the various ORFs. The KyA gE and gI genes as well as the unique gene 75 are deleted. The US9 homologue is not deleted in KyA, but its promoter is absent, and it therefore is not expressed (4, 5, 12). (D) PCR amplification using primers rev-1 and rev-2 (Table 1) resulted in a 2.8-kbp PCR product fragment from strain RacL11 and in a 1.6-kbp fragment from KyA DNA. Amplification products were cloned and termed p71L11 and p71KyA, respectively. Scales and restriction enzyme sites (B, BamHI; S, SphI) are given. (E) Comparison of the full-length (797-amino-acid) and truncated (383-amino-acid) gp2. The alignment was done using the MacVector software program (version 7.2; Oxford Molecular Group). Identical amino acids are in grey boxes; the arrows indicate the determined endoproteolytic cleavage sites that produce the carboxy-terminal 42-kDa subunit (43). The gp2 sequences were adapted from GenBank (M86664 for full-length and M87497 for truncated gp2).
The aim of the study presented here was to investigate the role in virus growth and virulence of the in-frame deletion present in KyA gene 71 (EUS4) that encodes gp2. To this end, recombinant viruses based on either RacL11 or KyA were generated and characterized, in which either the full-length or truncated form of gp2 was expressed. These studies showed that strain KyA indeed expresses a truncated gp2 but that this 80-kDa glycoprotein was not functionally equivalent to its full-length 250-kDa counterpart encoded by wild-type EHV-1.
MATERIALS AND METHODS
Plasmids.
The plasmids p71L11 and p71KyA were generated by insertion of a 2.8- and a 1.6-kbp PCR product into cloning vector pTZ18R (Pharmacia-Amersham). The inserted sequences comprise gene 71 of EHV-1 strain RacL11 and truncated gene 71 (EUS4) of EHV-1 strain KyA, respectively (Fig. 1). The PCR amplification products were obtained by performing a standard PCR (34) using primers rev-1 and rev-2, which contain BamHI and SphI restriction enzyme sites (32) (Table 1).
TABLE 1.
Primers used for generation of recombinant plasmids and detection of gp2 recombinant viruses
| Primer | Sequencea | Fragment (plasmid) generated |
|---|---|---|
| rev-1 | 5′-GCAGCATGCGAAATAGCCCCATACAAAAG-3′ | 2.8 kbp (p71L11) or 1.6 kbp (p71KyA) |
| rev-2 | 5′-GCAGGATCCATGTTTGTCTTAGCGATCAG-3′ | 2.8 kbp (p71L11) or 1.6 kbp (p71KyA) |
| gp2-1 | 5′-TTGGGATCCATGGGGTTCATCTATGCGCG-3′ | 2.4 or 1.14 kbp |
| gp2-2 | 5′-CGTGAATTCTTAATATACAGACGCTCGGG-3′ | 2.4 or 1.14 kbp |
Boldface type indicate restriction enzyme sites used for cloning into pTZ18R. Nucleotides in italics are random 5′ overhangs to facilitate restriction enzyme digestion and cloning.
Viruses and cells.
EHV-1 strains RacL11, KyA, and the avirulent modified live vaccine strain RacH, a derivative of strain RacL11 (17, 24, 25), were grown on rabbit kidney cells (RK13), which were propagated in Dulbecco's minimal essential medium supplemented with 10% fetal bovine serum. To generate recombinant viruses of strains RacL11 and KyA harboring either the full-length RacL11 or the truncated KyA gp2 gene, plasmid p71L11 or p71KyA was cotransfected with viral DNA cloned as bacterial artificial chromosomes (BAC) exactly as previously described (31) (Table 2). At day 5 after cotransfection, supernatants were harvested and transferred to fresh plates of RK13 cells, and recombinant nonfluorescing virus plaques were picked and purified to homogeneity by two rounds of plaque purification (32). At least two independent recombinant viruses were isolated from each of the cotransfections, and their growth properties in vitro and in vivo were compared. Because no differences between the individual viruses resulting from one transfection were observed, results for only one of the recombinant viruses are described (Table 2).
TABLE 2.
Viruses generated and used in this study
| Virus designation | Mode of virus generation | Virus and gp2 genotype |
|---|---|---|
| KyA | NAa | KyA, truncated gp2b |
| KyAΔgp2 | Transfection of pKyA | gp2-negative |
| KyARgp2Tc | Cotransfection of pKyA and p71KyA | KyA, truncated gp2b, revertant from KyAΔgp2 |
| KyARgp2Fc | Cotransfection of pKyA and p71L11 | KyA, full-length gp2d |
| RacL11 | NA | RacL11, full-length gp2d |
| L11Δgp2 | Transfection of BAC clone pRacL11 | gp2-negative |
| L11Rgp2Fc | Cotransfection of pRacL11 and p71L11 | RacL11, full-length gp2, revertant from L11Δgp2 |
| L11Rgp2Tc | Cotransfection of pRacL11 and p71KyA | RacL11, truncated gp2b |
| RacH | NA | RacH, full-length gp2 |
NA, not applicable.
Truncated gp2 as encoded by gene 71 (EUS4) of KyA.
Two independent isolates were purified to homogeneity in the case of KyARgpF, KyARgpT, L11RgpF, and L11RgpT, respectively. No differences in the in vitro or in vivo growth properties were observed; consequently, data for only one of the recombinant viruses each are presented.
Full-length gp2 as encoded by gene 71 (EUS4) of RacL11 or its avirulent derivative RacH.
DNA analysis.
Viral DNA isolated from eukaryotic cells was cleaved with restriction enzymes and separated by 0.8% agarose gel electrophoresis. Additionally, viral DNA was analyzed by PCR using primers gp2-1 and gp2-2 (Table 1). Using these primers, full-length gene 71 yields a 2.4-kbp fragment, whereas the truncated form of gene 71 of strain KyA results in an amplification product of 1.14 kbp (Fig. 2A).
FIG. 2.
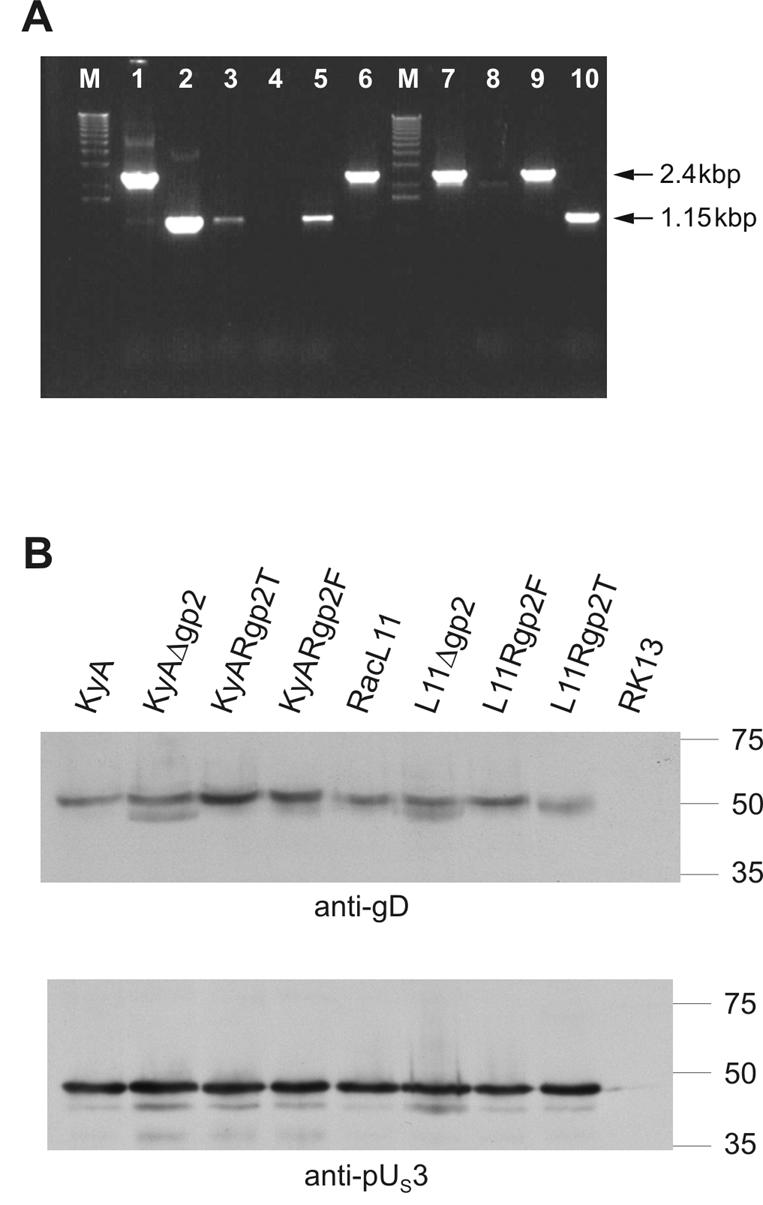
(A) PCR analysis of the generated RacL11 and KyA recombinant viruses using the primers gp2-1 and gp2-2. The full-length gp2 gene 71 yields a fragment of 2.4 kbp, whereas the truncated gene 71 results in a 1.14-kbp fragment. Lanes were loaded as follows: 1, p71L11; 2, p71KyA; 3, KyA; 4, KyAΔgp2; 5, KyARgp2T; 6, KyARgp2F; 7, RacL11; 8, L11Δgp2; 9, L11Rgp2F; 10, L11Rgp2T. The molecular size marker is the 1-kb ladder (Gibco-BRL). (B) Western blot analysis detecting expression of genes located up- or downstream of the pHA2 insertion present in the gp2-negative viruses. Infected-cell lysates were harvested 16 hpi, adjusted to identical protein concentrations, and separated by SDS-10% PAGE. Separated proteins were transferred to nitrocellulose and incubated with an anti-pUS3 or anti-gD-specific rabbit antiserum, which was used at a 1:1,000 dilution. The sizes of a prestained molecular weight marker (Precision Plus Protein; Bio-Rad) are given in thousands.
Antibodies.
To detect EHV-1 gp2, we used monoclonal antibody (MAb) 8B6, which is directed against the terminal region of EHV-1 gp2 and recognizes the full-length as well as the 42-kDa cleavage product of gp2 (42, 43). MAb E2 directed against EHV-1 gM was kindly provided by Lindsey Day and Richard Killington, University of Leeds, Leeds, United Kingdom (9, 33, 36). For detection of gD and the EHV-1 gene 69 (US3) gene product, monospecific polyclonal rabbit antisera were used exactly as described earlier (3, 13, 14).
Virus titer and plaque diameter determinations.
Extracellular and cell-associated titers of the various viruses were determined after infection of 105 RK13 cells at a multiplicity of infection (MOI) of 3. Virus was allowed to attach for 2 h on ice, followed by a penetration period of 1 h at 37°C, after which residual input infectivity was neutralized by treatment with a citrate buffer (pH 3.0) (33). Supernatants of infected cells and infected-cell pellets were harvested 36 h after the temperature shift, and virus titers in both preparations were determined by culturing 10-fold dilutions on plates of RK13 cells. Plaque diameters were measured after plating of the viruses on RK13 cells and 3 days of incubation at 37°C under a 0.25% methylcellulose overlay (33). Cells were fixed with acetone and stained by indirect immunofluorescence using anti-gM MAb E2. For each virus, diameters of at least 50 plaques were determined using the ImageJ 1.28 software that is freely available from Wayne Rasband at the National Institutes of Mental Health (http://rsb.info.nih.gov/ij/docs/intro.html). Virus titers and plaque diameters were analyzed by performing an analysis of variance, followed by multiple comparisons of the groups. Therefore, P values were adjusted according to the method of Bonferroni as described previously (29). SAS software (version 8.2; SAS Institute, Cary, N.C.) was used for the statistical calculations.
Virus purification.
For virus purification, RK13 cells were infected with different viruses and incubated until complete cytopathic effect had developed. Cellular debris was removed by low-speed centrifugation (6,000 × g, 4°C, 15 min), and the virus-containing supernatants were centrifuged for 1 h at 4°C at 22,000 rpm in a TST-28 rotor (Beckman). The resulting pellet was resuspended in 200 μl of Tris-buffered saline (TBS) (200 mM NaCl, 2.6 mM KCl, 10 mM Tris-HCl [pH 7.5], 20 mM MgCl2, 1.8 mM CaCl2), and this preparation was layered onto a discontinuous sucrose gradient (30, 40, and 50% sucrose). After centrifugation for 2 h at 4°C and 20,000 rpm in a TST-28 rotor, virions were harvested by collecting the band at the boundary between 40 and 50% sucrose. Residual sucrose was removed from the virus-containing band by ultracentrifugation (1 h, 4°C, 20,000 rpm in a TST-28 rotor), and purified virions were finally resuspended in TBS. Virion preparations were checked for purity by negative staining. Formvar- and carbon-coated copper grids were pretreated with 5 μl of bacitracin, and then 5 μl of the virus suspension was added. After 5 min of incubation at 20°C, grids were dried and 10 μl of a 1:1 mixture of 2% uranyl acetate and bacitracin was added. Grids were analyzed using a Tecnai 12 (Philips) electron microscope.
Western blotting.
For Western blot analyses, cells were infected at an MOI of 2 with the various viruses, and cell lysates were prepared at 16 h after infection. Lysates of purified virions or infected cells were adjusted to equal protein concentrations determined by the bicinchoninic acid kit (Pierce) (30). Samples were separated by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis (SDS-10% PAGE) and transferred to nitrocellulose membranes (Schleicher & Schuell) by the semidry method (18). Free binding sites on the sheets were blocked by addition of 5% skim milk in phosphate-buffered saline containing 0.03% Tween (PBST) before the antibodies (suspended in PBST) were added. Bound antibodies were detected with anti-mouse or anti-rabbit immunoglobulin G peroxidase conjugates (Jackson Immunoresearch Laboratories) and visualized by enhanced chemiluminescence (ECL kit; Pharmacia-Amersham).
Animal experiments.
Animal experiments were conducted as described previously (17, 29). Briefly, 3- to 4-week old female BALB/c mice (10 mice per group) were infected with the various virus preparations by the intranasal route at 105 PFU. Virus was suspended in 20 μl of Dulbecco's minimal essential medium-10% fetal bovine serum. Two mice were euthanized on day 3 and three mice were euthanized on day 5 postinfection (p.i.). Lungs were homogenized and virus titers in lungs were determined by standard titration on RK13 cells. Also, individual weights of mice were determined on the day of infection (day 1) until day 13. Body weights were examined by an analysis of variance and multiple comparisons of the groups as described above. Due to the small number of samples, viral titers in lungs were compared by the nonparametric Kruskall-Wallis test using SAS software (version 8.2).
RESULTS
Generation of RacL11 and KyA mutant viruses harboring different gp2.
To generate recombinant viruses from EHV-1 strains RacL11 and KyA expressing either the full-length (gp2F, RacL11) or the truncated (gp2T, KyA) form of gp2, pRacL11 and pKyA DNAs were cotransfected with the recombinant plasmid p71L11 or p71KyA into RK13 cells. Because construction of the BAC clones of strains RacL11 and KyA resulted in the insertion of enhanced green fluorescent protein-encoding sequences, identification of gene 71-positive nonfluorescing virus plaques was readily possible (31). From day 1 after transfection, fluorescing foci resulting from expression of the enhanced green fluorescent protein by the reconstituted viruses were observed. Nonfluorescing recombinant virus plaques as the result of the insertion of gp2-encoding sequences were visible from day 2, and the recombinants were purified until a homogenous population of nonfluorescing virus progeny was obtained (Table 2). Recombinant viral DNA was extracted from infected cells; cleaved with restriction enzyme BamHI, HindIII, or EcoRI; and separated by 0.8% agarose gel electrophoresis. The resulting restriction enzyme patterns were compared with those of pRacL11, pKyA, and the parental EHV-1 strain RacL11 or KyA. Altered restriction fragment patterns were the consequence of the reinsertion of the full-length or truncated gp2 gene into the genomes of the two BAC clones and the concomitant loss of the mini-F vector sequences (31, 32). The observed restriction enzyme patterns obtained for the generated recombinant viruses exactly corresponded to the calculated patterns (Fig. 3). The correct insertion of the full-length or truncated gp2 gene was further confirmed by Southern blot analysis using labeled pHA2 or p71 as a probe (data not shown) and by PCR using primers gp2-1 and gp2-2 (Table 1; Fig. 2A). The full-length gp2 gene resulting in a PCR product of 2.4 kbp in size was detected in cells infected with RacL11, L11Rgp2F, and KyARgp2F, while the truncated gp2 gene resulting in a product of 1.14 kbp in size was amplified from cells infected with KyA, KyARgp2T, and L11Rgp2T (Table 2; Fig. 2A). In the case of pKyA-derived KyAΔgp2 and the pRacL11-derived L11Δgp2 virus, no amplification product was obtained. From the results of the restriction enzyme patterns, Southern blot experiments, and PCR assays, we concluded that the full-length or truncated form of gene 71 encoding gp2 was correctly inserted into the generated virus mutants (Table 2).
FIG. 3.
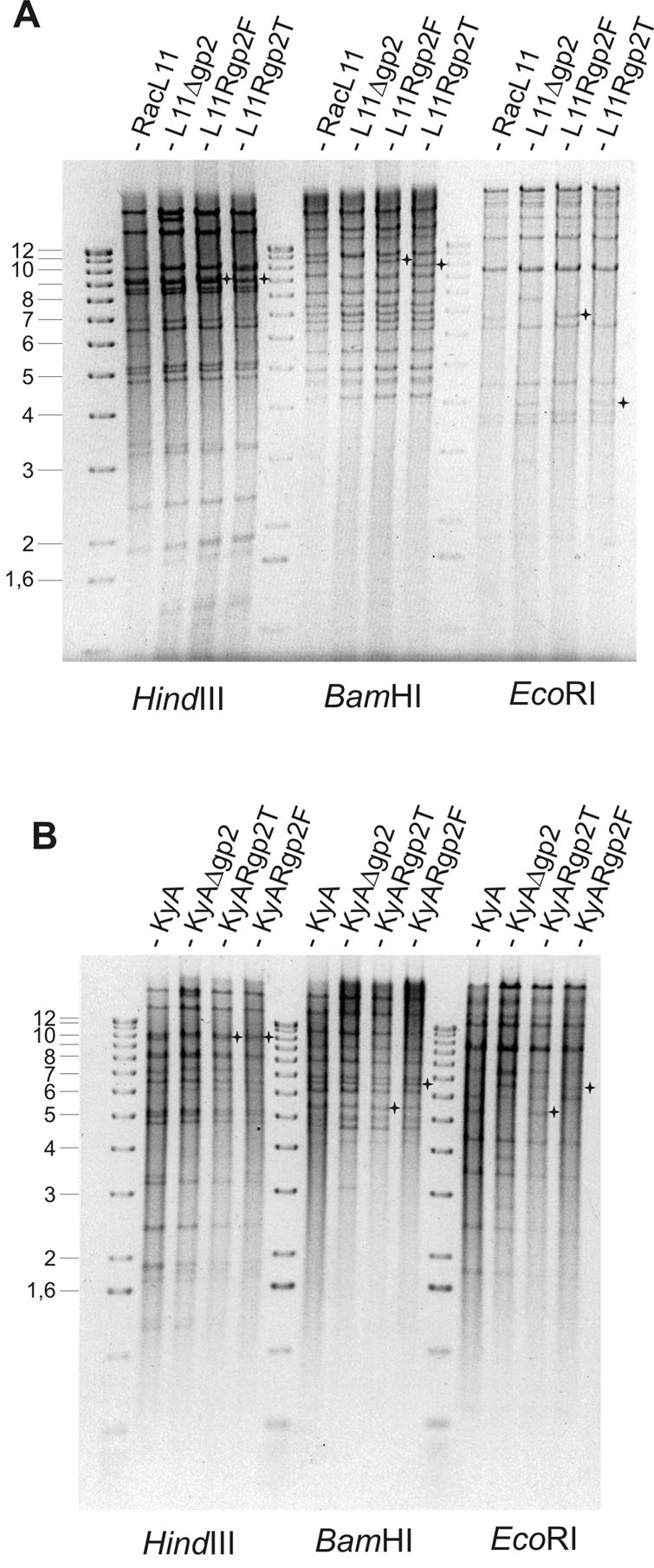
Agarose gel electrophoresis of L11Rgp2F, L11Rgp2T, parental RacL11, and L11Δgp2 DNA (A) and of KyARgp2T, KyARgp2F, KyA, and KyAΔgp2 DNA (B). Viral DNA was isolated from infected RK13 cells; digested with the restriction enzyme BamHI, EcoRI, or HindIII; and separated in 0.8% agarose gels. Asterisks mark fragments in L11Rgp2F, L11Rgp2T, KyARgp2T, and KyARgp2F DNA, which resulted from insertion of the different gene 71 sequences. The molecular size marker is the 1-kb ladder (Gibco-BRL), and sizes are given in kbp. Gel images were scanned and color inverted using Adobe Photoshop 7.0.
Although based on the transcriptional organization of the EHV-1 US region, down regulation of gene expression up- or downstream of the pHA2 insertion sites in L11Δgp2 and KyAΔgp2 was not expected (4), expression of the US3-encoded protein kinase (pUS3) located upstream of the mini-F vector insertion and of gD located immediately downstream of the vector sequences was verified (Fig. 1). Western blot analyses performed on infected-cell lysates harvested at 16 h p.i. (hpi) demonstrated that expression of both pUS3 and gD was virtually identical in all viruses used in the study (Fig. 2B). From the results we concluded that the presence of the pHA2 sequences in L11Δgp2 and KyAΔgp2 did not have detrimental effects on gene expression in the US region of mutant viruses.
Truncated gp2 encoded by strain KyA is indeed expressed in infected cells.
In a series of experiments, the expression of the heavily O-glycosylated protein gp2 in virus-infected cells and purified virions was analyzed. Previous attempts to demonstrate expression of truncated KyA gp2 had been unsuccessful, because the available gp2-specific MAbs recognized only the uncleaved full-length precursor and no gp2-specific signals could be detected by Western blotting, indirect immunofluorescence, or immunoprecipitation analyses of cells infected with EHV-1 strain KyA (31). Lysates of RK13 cells infected with RacL11, KyA, or the constructed recombinant viruses harboring either the full-length or the truncated form of the gp2 gene (Table 2) were prepared and analyzed by Western blotting using the gp2-specific MAb 8B6 that recognizes the 42-kDa carboxy-terminal cleavage product of the glycoprotein (19, 42). In cells infected with RacL11 or mutant viruses harboring the full-length gp2 (L11Rgp2F and KyARgp2F), the 42-kDa carboxy-terminal cleavage product of gp2 and the 250-kDa uncleaved glycoprotein were detected by MAb 8B6 (Fig. 4). In contrast, in cells infected with strain KyA or mutant viruses harboring the KyA gp2 gene (KyARgp2T and L11Rgp2T), a 75- to 80-kDa uncleaved glycoprotein was specifically detected by MAb 8B6. As described for the RacL11 gp2 and for a variety of other wild-type viruses, a 42-kDa cleavage product was detected in cells expressing the truncated form of gp2 encoded by KyA (Fig. 4), indicating that a truncated form of gp2 encoded by KyA gene 71 is indeed expressed as a 75- to 80-kDa uncleaved glycoprotein. Truncated gp2 is—similar to full-length gp2—partially cleaved and the cleavage results in the generation of a carboxy-terminal subunit with an Mr of 42,000.
FIG. 4.

Western blot analysis of gp2 in strains RacL11 (A) and KyA (B) harboring either truncated or full-length gp2. Infected-cell lysates were separated by SDS-10% PAGE, transferred to nitrocellulose, and incubated with anti-gp2 MAb 8B6 at a 1:2,000 dilution. MAb 8B6 recognizes the 42-kDa carboxy-terminal cleavage product of gp2 (solid arrowhead) and the uncleaved full-length (open arrowhead) as well as truncated (circle) gp2. The sizes of a prestained molecular weight marker (Sea Blue; Novex) are given in thousands.
Full-length and truncated gp2 are incorporated into extracellular virions.
To analyze incorporation of full-length and truncated gp2 into the viral envelope, extracellular virions of RacL11, KyA, or the different recombinant viruses were purified by sucrose-gradient centrifugation. Purified virus preparations were separated by SDS-10% PAGE, and proteins were transferred to nitrocellulose sheets. By using MAb 8B6, both the full-length uncleaved gp2 and the 42-kDa carboxy-terminal cleavage product were detected in purified virions of the wild-type RacL11 strain or strain RacH, which also expresses full-length gp2 (32) (Fig. 5). Incorporation of the uncleaved gp2 and the carboxy-terminal cleavage product was also observed in the case of purified KyA virions, because both the 42-kDa cleavage product-specific band and the 75- to 80-kDa-protein band representing uncleaved truncated KyA gp2 were detected in purified virion preparations (Fig. 5A).
FIG. 5.

Purified RacL11, RacH, and KyA virions (A) or recombinant KyA and RacL11 viruses expressing either full-length or truncated gp2 (B) were separated by SDS-10% PAGE, transferred to nitrocellulose, and incubated with either anti-gp2 MAb 8B6 (left panels) or anti-gM MAb E2 (right panels). The 42-kDa carboxy-terminal cleavage product of gp2 and the uncleaved full-length gp2 are indicated by solid and open arrowheads, respectively. Uncleaved truncated gp2 is marked with a circle. The sizes of a prestained molecular weight marker (Sea Blue [Novex] [A] or Precision Plus Protein [Bio-Rad] [B]) are given in thousands.
It was noticed that signal intensities of both the uncleaved 75- to 80-kDa gp2 and the carboxy-terminal 42-kDa protein were approximately 10-fold higher in purified KyA virions than in both RacL11 or RacH virions (Fig. 5A), although identical amounts of protein were loaded in each lane. To confirm the specificity of the enhanced incorporation of gp2 in the case of KyA, an identical nitrocellulose membrane containing separated virion proteins of RacL11, RacH, and KyA was incubated with the gM-specific MAb E2. The 50- to 55-kDa gM-specific band was detected in comparable intensities in each of the virus-infected cell lysates, confirming that truncated KyA gp2 appeared to be incorporated more efficiently into purified virions (Fig. 5A).
Two possibilities were investigated that could account for the more-efficient incorporation of truncated gp2 into purified virions. First, the absence of a major envelope constituent, the gE-gI complex, in the case of KyA could result in a “compensatory” incorporation of larger amounts of gp2. Second, truncated gp2 by itself might be incorporated more efficiently. Virions were purified from cells infected with RacL11 or KyA recombinants expressing either full-length or truncated gp2. Western blot analyses revealed that signal intensities of all purified virions with the anti-gD antibody (data not shown) and the E2 anti-gM antibody were virtually identical, similar to the situation described for the parental RacL11 and KyA viruses (Fig. 5B). Signal intensities of gp2 detected with MAb 8B6, however, varied considerably, and efficient incorporation of gp2 into virions could be observed in the case of L11Rgp2T and KyARgp2T, but not in L11Rgp2F or KyARgp2F virions, where the signals of the 250-kDa precursor and especially the 42-kDa cleavage product were barely visible (Fig. 5B). These results indicated that the efficiency of gp2 incorporation into purified virions is regulated independently of the absence or presence of other major US glycoproteins but is dependent on the KyA-specific deletion in the amino-terminal portion of the envelope constituent (Fig. 1E).
Growth properties of L11Rgp2F, L11Rgp2T, KyARgp2T, and KyARgp2F.
The growth properties of the generated recombinant viruses on RK13 cells were analyzed. First, plaque diameters of L11Rgp2F and L11Rgp2T (Table 2) were compared to those of parental RacL11 and the gp2-negative viruses L11Δgp2 and KyAΔgp2. It could be shown that the approximately 13% reduction in plaque diameters of L11Δgp2 was virtually fully restored after introduction of the full-length RacL11 gp2 as well as the truncated KyA gp2 into the gp2-negative L11Δgp2, and no statistically significant differences between the plaque sizes of RacL11 and L11Rgp2F were observed (Fig. 6A). Similarly, the highly significant reduction in plaque diameters of the gp2-negative KyAΔgp2 of approximately 55% (P < 0.001) could be restored completely by reinsertion of KyA gene 71 (EUS4) encoding the truncated glycoprotein (Table 2), and plaque diameters observed after plating of KyAgp2T were very similar to those of the parental KyA (Fig. 6B). In contrast, the replacement of truncated with full-length RacL11 gp2-encoding gene 71 in the KyARgp2F mutant (Table 2) was not able to fully restore plaque sizes, and only 78% of the plaque diameters of the KyA virus were observed (Fig. 6B). The >20% difference between plaque sizes observed for KyARgp2F and KyA or KyARgp2T was highly significant (P < 0.001).
FIG. 6.

Plaque sizes and end-point titers of RacL11 (A) and KyA (B) recombinant viruses harboring the full-length or truncated form of gp2. For plaque size determinations, 100 PFU of each virus (Table 2) per well was plated on RK13 cells seeded in six-well plates. Shown are means and standard deviations of diameters of 50 plaques measured for each virus at 3 days p.i. Plaque diameters of RacL11 or KyA were set at 100%. Error bars indicate standard deviations. Cell-associated (open bars) and extracellular (black bars) end-point titers (36 hpi) of the generated RacL11 and KyA recombinant viruses were determined by infection of 105 RK13 cells at an MOI of 3 and titrations of cell pellets or infected-cell supernatants on RK13 cells. Asterisks indicate statistically significant (P < 0.001 in panels on the left; P < 0.0125 in panels on the right) differences. Letters (a or b) indicate that annotated groups were not different from each other.
In a second set of experiments, virus titers of the various recombinant viruses were determined by infecting 105 RK13 cells at an MOI of 3. Mutant virus growth properties were compared to those of the parental and the gp2-negative viruses, respectively. Extracellular and cell-associated virus titers determined at 36 hpi are summarized for all viruses in Fig. 6. The gp2 revertant viruses—i.e., L11Rgp2F, L11Rgp2T, KyARgp2F, and KyARgp2T—exhibited extracellular and cell-associated titers that were virtually identical to those determined for parental strain RacL11 or KyA, respectively. End-point extracellular titers determined at 36 hpi ranged from 1.4 × 106 to 2.7 × 106 PFU/ml, irrespective of whether the full-length or truncated gp2 was reintroduced in either the RacL11 or KyA background. In the case of RacL11, cell-associated and extracellular titers of gp2 revertant viruses were between two- and sixfold higher than those of the respective gp2-negative viruses (Fig. 6C). The extracellular virus titers of KyA viruses harboring the authentic truncated or the RacL11 full-length gp2 were significantly higher (between 27- and 51-fold, respectively; P < 0.001) than those determined for the gp2-negative virus (Fig. 6A). The reduction in titers of cell-associated virus titers was comparable to that observed in the case of the RacL11 mutant viruses and ranged from five- to ninefold (Fig. 6B).
These results are in perfect agreement with those of earlier studies and indicated that both the full-length RacL11 gp2 and the truncated KyA gp2 are able to restore the defects of the gp2 deletion in their parental strains or in allogeneic virus backgrounds with regard to virus egress. However, the inability of full-length gp2 to restore the plaque sizes of the gp2-negative KyAΔgp2 virus to those induced by the parental KyA virus indicated that full-length and truncated gp2 are functionally different with regard to cell-to-cell spread in different virus backgrounds.
Animal experiments.
The pathogenicities of the generated gp2 revertant viruses L11Rgp2F, L11Rgp2T, KyARgp2T, and KyARgp2F were analyzed in a murine model of EHV-1 infection and compared to those of their parental or the respective gp2-negative viruses. Mice were divided into groups of 10 animals and infected intranasally with 105 PFU per mouse. Animals were monitored for 13 days after infection, and the body weights, the virus titers in lungs, and the behavior of each individual mouse were recorded daily. The results of the experiment are summarized in Fig. 7. Mice infected with KyARgp2T, KyARgp2F, KyAΔgp2, or KyA did not exhibit a significant reduction of body weights after infection; rather, mean body weights remained constant or increased until day 13 after infection. In contrast, RacL11- or L11Rgp2F-infected mice exhibited a massive reduction in body weights of up to 28%, and clinical symptoms indicative of EHV-1 infection were observed from days 2 to 8 after infection. In addition, mice inoculated with these viruses did not reach preinfection weights until the end of the observation period, and one mouse in the L11Rgp2F group died as a consequence of infection on day 2 p.i.
FIG. 7.

(A and B) Development of mean body weights after infection of mice with EHV-1. Mice were in groups of 10, and each mouse was infected intranasally with 105 PFU of the indicated virus. Mean body weights were determined on the day of infection (day 1) and on the following 12 days. Mean body weights and standard deviations (error bars) are shown. (A) Mice infected with RacL11, L11Δgp2, L11Rgp2F, or L11Rgp2T; (B) mice infected with KyA, KyAΔgp2, KyARgp2T, or KyARgp2F. (C and D) Virus titers in lungs were determined in two mice of each group on day 3 and from three mice of each group on day 5 after infection. Titers in lungs and standard deviations (error bars) are shown. (C) Mice infected with RacL11, L11Δgp2, L11Rgp2F, or L11Rgp2T; (D) mice infected with KyA, KyAΔgp2, KyARgp2T, or KyARgp2F. Asterisks indicate statistically significant differences (P < 0.001) between RacL11/L11Rgp2F- and KyA-inoculated mice; mean body weights in the L11Δgp2 group were also different from those of the KyA group on days 5 and 8 to 13 (P < 0.001).
Mice infected with the gp2-negative L11Δgp2 mutant derived from the virulent RacL11 virus clearly exhibited a loss of body weight, especially compared to mice infected with avirulent KyA or its derivatives (Fig. 7). The reduction in mean body weight induced by L11Δgp2 was significantly different from that of KyA (P < 0.001) and is in contradiction to previous results obtained after mutagenesis of strain Ab4, in which the deletion of gp2 resulted in complete attenuation (21). Surprisingly, however, mice infected with L11Rgp2T that expresses the truncated KyA gp2 exhibited a reduction in mean body weight only for the first 3 days after infection; thereafter, mean body weights increased and reached preinfection weights. This may indicate that in vivo replication of a RacL11 virus expressing truncated gp2 is even more restricted than that of a gp2-negative RacL11 (Fig. 7A).
The determination of the titers in mouse lungs determined on days 3 (two mice per group) and 5 (three mice per group) after infection corroborated the results of the mean body weight determinations, although the determined differences in virus titers between individual groups were not statistically significant. Titers of RacL11 and the L11Rgp2F revertant virus were almost identical on days 3 and 5 p.i., whereas deletion of gene 71 in the virulent RacL11 (L11Δgp2) resulted in an approximately threefold reduction in lung virus titers. The reduction in lung virus titers could not be restored to RacL11 levels by the insertion of the KyA gene 71 (Fig. 7C). Titers in lungs of mice inoculated with KyA and its various derivatives were approximately 10-fold lower compared to the RacL11 counterparts on both day 3 and 5 after infection (Fig. 7D). Similar to the findings described above, replacement of KyA gene 71 with the RacL11 counterpart did not result in enhanced virus replication in murine lungs.
From the results of the animal experiment, we concluded that strain KyA encodes a gp2 that is functionally different from that expressed by strain RacL11. The animal experiment also showed that full-length gp2 alone is not able to restore virulence of vaccine strain KyA, although the in vitro growth properties of KyA expressing the authentic truncated or the full-length gp2 were similar. Likewise, expression of truncated instead of authentic full-length gp2 in a RacL11 background resulted in complete attenuation of a virulent wild-type virus.
DISCUSSION
In this study, experiments were conducted to elucidate the function of the in-frame deletion of gene 71, which leads to the expression of a truncated gp2 from the EHV-1 strain KyA. The salient findings reported here are that we were able to demonstrate that strain KyA indeed expresses a truncated gp2 with an Mr of 75,000 to 80,000, which is partially cleaved into two subunits. The full-length gp2 and the carboxy-terminal subunit are incorporated into purified virions similar to full-length RacL11 gp2. Furthermore, truncated gp2 is not functionally equivalent to the full-length version of the envelope constituent. This conclusion is drawn from the differences of the growth properties in cultured cells of several gp2 revertant viruses based on RacL11 or KyA. In addition, an animal experiment involving the generated mutant viruses demonstrated that truncated gp2 is not able to fully restore the virulent phenotype when introduced into RacL11 and that the full-length gene gp2 alone cannot render vaccine virus KyA pathogenic for mice when inserted instead of the truncated ORF.
Previous studies have demonstrated that the effects of a deletion of gene 71 (EUS4) encoding gp2 with respect to the virus growth properties are strain specific to some extent, which is reflected by the differences in virus growth restrictions reported for gp2-negative Ab4, RacL11, RacH, and KyA. In any virus strain analyzed so far, however, gp2 was shown to be dispensable for growth in cultured cells but was shown to be involved in virus egress at a step after secondary envelopment as well as in direct cell-to-cell spread (31, 32, 37). It was also suggested that the unique glycoprotein functionally interacts with another viral protein(s), as deduced from the demonstration of massive growth defects of a gp2-gM double deletion mutant (32). The nature of the putative interaction of gp2 with other viral protein(s) has yet to be identified, but it is clear that gp2 function is partially dependent on the overall organization of the US region of the genome, more specifically the absence or presence of gE, gI, and the US9 homologous protein, as well as a carboxy-terminal mutation of the gD ORF (12, 39). This interpretation is supported by the fact that gp2-negative viruses of the EHV-1 strains RacL11 and KyA (L11Δgp2 and KyAΔgp2) exhibited significantly different growth properties. In both gp2-negative viruses, the cell-associated virus titers were reduced by 5- to 9-fold, but only in the case of KyAΔgp2 were plaque diameters (approximately by 55%) and extracellular virus titers (51-fold) significantly reduced (31). Previous data and the results reported here show that both the full-length RacL11 gp2 and the truncated KyA gp2 are involved in direct cell-to-cell spread in cell culture, although gp2 function is more prominent in strain KyA, which also lacks the gE and gI genes (15). It has been shown in all Alphaherpesvirinae analyzed so far—and also in the case of EHV-1—that the gE-gI complex facilitates cell-to-cell spread, because gE-gI-negative viruses induce only small plaques compared to those of their parental viruses (10, 20, 22, 35, 40, 46). Whereas Marek's disease virus and varicella-zoster virus mutants devoid of gE or gI are replication incompetent or significantly impaired in direct cell-to-cell spread, their only means of replication in cultured cells, deletion of the glycoprotein complex, leads to a small-plaque phenotype in HSV-1, pseudorabies virus, bovine herpesvirus 1, and EHV-1 wild-type strains (16, 22). In stark contrast, EHV-1 strain KyA efficiently replicates in cultured cells and plaque formation appears normal despite the absence of the gE-gI complex. It is tempting to speculate that KyA has compensated for the absence of gE and gI by a concomitant truncation of gp2 and has thereby overcome its growth restriction in cultured cells (15, 39). This hypothesis is supported by the failure of a complete restoration of the plaque sizes by the introduction of full-length gp2 into a KyA genomic background, clearly demonstrating that full-length RacL11 gp2 exerts functions different from truncated gp2 encoded by KyA.
It has been shown that gp2 contains both N-linked and high levels of O-linked oligosaccharides (42, 45). A very striking feature of the gene 71 product is a region of repeated serine and threonine residues (Fig. 1E), which are extensively modified by the addition of O-glycans, resulting in the high molecular mass of gp2 (42). In virus-infected cells, full-length gp2 is cleaved into two domains, the 42-kDa carboxy-terminal component and an amino-terminal subunit of approximately 200 kDa containing the serine and threonine residues (44). In addition to the incorporation of uncleaved gp2 into virions (41), the 42-kDa carboxy-terminal cleavage product was also shown to be a virion component (this report). The incorporation of a glycoprotein precursor into purified virions was unexpected but apparently was not caused by contamination of virus preparations with infected cells. Electron microscopy and control reactions of purified virion lysates with an anti-gM antibody, which detected only the fully processed form of the glycoprotein and not its precursors in a Western blot, clearly showed the purity of virion preparations (Fig. 5). This finding suggests an incomplete processing of gp2 in infected cells and incorporation of processed and unprocessed gp2 into finally enveloped virions, the mechanism of which is currently under investigation.
Using a MAb recognizing the 42-kDa carboxy-terminal subunit, we were able to unequivocally demonstrate expression of the truncated form of gp2 encoded by KyA for the first time. As observed for the full-length 250-kDa gp2 encoded by strain RacL11, the truncated 75- to 80-kDa glycoprotein is cleaved, and the same 42-kDa carboxy-terminal subunit that is observed in cells infected with wild-type EHV-1 is generated. In addition, both the precursor 75- to 80-kDa and the 42-kDa gp2 moieties are incorporated into KyA virions, indicating a similar processing of truncated and full-length gp2. It was very notable that the incorporation of truncated KyA gp2 into purified virions appeared to occur with approximately 10-fold-higher efficiency compared to that of RacL11 gp2. This enhanced incorporation was probably caused by a more efficient incorporation of the truncated gp2 per se, and not by the absence of other major constituents of the virus envelope such as gE and gI in the case of KyA. This conclusion was based on recombinant RacL11 viruses (that harbor gE and gI) expressing truncated gp2, which also incorporated significantly more of the glycoprotein than did revertant or wild-type RacL11 virus expressing full-length gp2. The higher levels of truncated gp2 in purified virions may be accounted for by the large in-frame deletion, which results in the loss of the majority of the serine/threonine-rich domain and, consequently, the O-glycosylation sites in the amino-terminal part of the glycoprotein (5). The carboxy-terminal domain and the cleavage site, however, remain unaffected by the deletion in KyA gp2 (Fig. 1E). In addition, we think that it is unlikely that the different reactivities of truncated and full-length gp2 with the 8B6 antibody are caused by different affinities to the truncated or full-length form of the glycoprotein, because signal intensities with the 42-kDa carboxy-terminal subunit, which is identical between the RacL11 and KyA versions of the envelope constituent, were also reduced in purified virions harboring full-length gp2.
It has been described for EHV-1 and closely related Alphaherpesvirinae that mutations in the US region, where as many as five (glyco)proteins are clustered in the case of EHV-1, result in reduced replication and or attenuation of mutant viruses in vivo (11, 21-23). Similarly, it could be shown for EHV-1 that deletion of the unique gp2 gene or the deletion of the gE and gI encoding genes from virulent strains resulted in the attenuation of the virus (16, 21). Recently, we were able to demonstrate that reinsertion of gE and gI into the KyA genome is sufficient to rescue a virulent phenotype, although virulence was not completely restored to that of RacL11 (16). It is shown here that expression of full-length gp2 derived from the virulent strain RacL11 by strain KyA was insufficient to restore virulence in the BALB/c mouse model of infection. On the other hand, deletion of gp2 in the virulent strain RacL11 resulted in a recombinant virus that clearly exhibited residual virulence, because a significant loss of body weight was observed in animals injected with gp2-negative RacL11. The latter observation is in conflict with previous results reported for strain Ab4, in which deletion of gene 71 resulted in complete attenuation (21). This discrepancy between virulent strains RacL11 and Ab4 may be explained by strain-specific differences but may also be caused by the fact that the Ab4 mutant was obtained by conventional marker insertion of the LacZ gene using homologous recombination in eukaryotic cells, which required multiple rounds of plaque purification and during which compensatory mutations may have arisen (21, 37). In this context it is interesting that reinsertion of the truncated KyA gene 71 (EUS4) in a RacL11 genomic background not only was unable to restore virulence but also appeared to have an attenuating effect that was greater than deletion of the authentic full-length ORF (16).
Taken together, the findings reported here on the growth properties in cultured cells of RacL11 and KyA viruses expressing the two different forms of the unique gp2 as well as the animal studies indicate that gp2 expressed from strain RacL11 is functionally different from the truncated variant gp2 encoded by the attenuated strain KyA. Future studies will concentrate on the elucidation of the suspected coevolution of the US genes expressed by virulent RacL11 and the avirulent KyA strain by exchanging individual US genes between the two strains. These experiments should shed light on the putative functional interdependence of gp2 truncation and the absence of the gE-gI complex.
Acknowledgments
The technical assistance of Jennifer L. Gardell is gratefully acknowledged, and we thank Daniel Schumacher for performing the negative staining of virion preparations.
This work was supported by funds of the Harry M. Zweig Memorial Fund for Equine Research granted to N.O. and by NIH grants AI-22001 and P20-RR018724 to D.J.O.
REFERENCES
- 1.Allen, G. P., and J. T. Bryans. 1986. Molecular epizootiology, pathogenesis, and prophylaxis of equine herpesvirus-1 infections. Prog. Vet. Microbiol. Immunol. 2:78-144. [PubMed] [Google Scholar]
- 2.Allen, G. P., and M. R. Yeargan. 1987. Use of lambda gt11 and monoclonal antibodies to map the genes for the six major glycoproteins of equine herpesvirus 1. J. Virol. 61:2454-2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colle, C. F., and D. J. O'Callaghan. 1996. Localization of the US protein kinase of equine herpesvirus type 1 is affected by the cytoplasmic structures formed by the novel IR6 protein. Virology 220:424-435. [DOI] [PubMed] [Google Scholar]
- 4.Colle, C. F., III, and D. J. O'Callaghan. 1995. Transcriptional analyses of the unique short segment of EHV-1 strain Kentucky A. Virus Genes 9:257-268. [DOI] [PubMed] [Google Scholar]
- 5.Colle, C. F., III, C. C. Flowers, and D. J. O'Callaghan. 1992. Open reading frames encoding a protein kinase, homolog of glycoprotein gX of pseudorabies virus, and a novel glycoprotein map within the unique short segment of equine herpesvirus type 1. Virology 188:545-557. [DOI] [PubMed] [Google Scholar]
- 6.Colle, C. F., III, E. B. Tarbet, W. D. Grafton, S. R. Jennings, and D. J. O'Callaghan. 1996. Equine herpesvirus-1 strain KyA, a candidate vaccine strain, reduces viral titers in mice challenged with a pathogenic strain, RacL. Virus Res. 43:111-124. [DOI] [PubMed] [Google Scholar]
- 7.Crabb, B. S., G. P. Allen, and M. J. Studdert. 1991. Characterization of the major glycoproteins of equine herpesviruses 4 and 1 and asinine herpesvirus 3 using monoclonal antibodies. J. Gen. Virol. 72:2075-2082. [DOI] [PubMed] [Google Scholar]
- 8.Crabb, B. S., and M. J. Studdert. 1990. Comparative studies of the proteins of equine herpesviruses 4 and 1 and asinine herpesvirus 3: antibody response of the natural hosts. J. Gen. Virol. 71:2033-2041. [DOI] [PubMed] [Google Scholar]
- 9.Day, L. 1999. Characterisation of selected glycoproteins of equine herpesvirus-1. Ph.D. thesis. University of Leeds, Leeds, United Kingdom.
- 10.Dingwell, K. S., C. R. Brunetti, R. L. Hendricks, Q. Tang, M. Tang, A. J. Rainbow, and D. C. Johnson. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68:834-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzmaurice, T., C. Walker, A. Kukreja, Y. Sun, S. M. Brown, and H. J. Field. 1997. The pathogenesis of ED71, a defined deletion mutant of equine herpesvirus-1, in a murine intranasal infection model for equine abortion. J. Gen. Virol. 78:2167-2169. [DOI] [PubMed] [Google Scholar]
- 12.Flowers, C. C., E. M. Eastman, and D. J. O'Callaghan. 1991. Sequence analysis of a glycoprotein D gene homolog within the unique short segment of the EHV-1 genome. Virology 180:175-184. [DOI] [PubMed] [Google Scholar]
- 13.Flowers, C. C., S. P. Flowers, S. R. Jennings, and D. J. O'Callaghan. 1995. Synthesis and processing of equine herpesvirus 1 glycoprotein D. Virology 208:9-18. [DOI] [PubMed] [Google Scholar]
- 14.Flowers, C. C., and D. J. O'Callaghan. 1992. Equine herpesvirus 1 glycoprotein D: mapping of the transcript and a neutralization epitope. J. Virol. 66:6451-6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flowers, C. C., and D. J. O'Callaghan. 1992. The equine herpesvirus type 1 (EHV-1) homolog of herpes simplex virus type 1 US9 and the nature of a major deletion within the unique short segment of the EHV-1 KyA strain genome. Virology 190:307-315. [DOI] [PubMed] [Google Scholar]
- 16.Frampton, A. R., Jr., P. M. Smith, Y. Zhang, T. Matsumura, N. Osterrieder, and D. J. O'Callaghan. 2002. Contribution of gene products encoded within the unique short segment of equine herpesvirus 1 to virulence in a murine model. Virus Res. 90:287-301. [DOI] [PubMed] [Google Scholar]
- 17.Hübert, P. H., S. Birkenmaier, H. J. Rziha, and N. Osterrieder. 1996. Alterations in the equine herpesvirus type-1 (EHV-1) strain RacH during attenuation. Zentbl. Vetmed. B 43:1-14. [DOI] [PubMed] [Google Scholar]
- 18.Kyhse-Andersen, J. 1984. Electroblotting of multiple gels: a simple apparatus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J. Biochem. Biophys. Methods 10:203-209. [DOI] [PubMed] [Google Scholar]
- 19.Learmonth, G. S., D. N. Love, J. E. Wellington, J. R. Gilkerson, and J. M. Whalley. 2002. The carboxy-terminal regions of the envelope glycoprotein gp2 of equine herpesviruses 1 and 4 are antigenically distinct. Arch. Virol. 147:607-615. [DOI] [PubMed] [Google Scholar]
- 20.Mallory, S., M. Sommer, and A. M. Arvin. 1998. Analysis of the glycoproteins I and E of varicella-zoster virus (VZV) using deletional mutations of VZV cosmids. J. Infect. Dis. 178(Suppl. 1):S22-S26. [DOI] [PubMed] [Google Scholar]
- 21.Marshall, K. R., Y. Sun, S. M. Brown, and H. J. Field. 1997. An equine herpesvirus-1 gene 71 deletant is attenuated and elicits a protective immune response in mice. Virology 231:20-27. [DOI] [PubMed] [Google Scholar]
- 22.Matsumura, T., T. Kondo, S. Sugita, A. M. Damiani, D. J. O'Callaghan, and H. Imagawa. 1998. An equine herpesvirus type 1 recombinant with a deletion in the gE and gI genes is avirulent in young horses. Virology 242:68-79. [DOI] [PubMed] [Google Scholar]
- 23.Matsumura, T., D. J. O'Callaghan, T. Kondo, and M. Kamada. 1996. Lack of virulence of the murine fibroblast adapted strain, Kentucky A (KyA), of equine herpesvirus type 1 (EHV-1) in young horses. Vet. Microbiol. 48:353-365. [DOI] [PubMed] [Google Scholar]
- 24.Mayr, A., J. Pette, K. Petzoldt, and K. Wagener. 1968. Studies on the development of a live vaccine against rhinopneumonitis (mare abortion) of horses. Zentbl. Vetmed. B 15:406-418. [PubMed] [Google Scholar]
- 25.O'Callaghan, D. J., W. P. Cheevers, G. A. Gentry, and C. C. Randall. 1968. Kinetics of cellular and viral DNA synthesis in equine abortion (herpes) virus infection of L-M cells. Virology 36:104-114. [DOI] [PubMed] [Google Scholar]
- 26.O'Callaghan, D. J., J. M. Hyde, G. A. Gentry, and C. C. Randall. 1968. Kinetics of viral deoxyribonucleic acid, protein, and infectious particle production and alterations in host macromolecular syntheses in equine abortion (herpes) virus-infected cells. J. Virol. 2:793-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Callaghan, D. J., and N. Osterrieder. 1999. The equine herpesviruses, p. 508-515. In R. G. Webster and A. Granoff (ed.), Encyclopedia of virology. Academic Press, Harcourt Brace & Co., San Diego, Calif.
- 28.Osterrieder, N., O. R. Kaaden, and A. Neubauer. 1999. Structure and function of equine herpesvirus glycoproteins: a review, p. 111-118. In Proceedings of the 8th International Conference on Equine Infectious Diseases. R&W Publications, Newmarket, United Kingdom.
- 29.Osterrieder, N., A. Neubauer, C. Brandmüller, O. R. Kaaden, and D. J. O'Callaghan. 1996. The equine herpesvirus 1 IR6 protein influences virus growth at elevated temperature and is a major determinant of virulence. Virology 226:243-251. [DOI] [PubMed] [Google Scholar]
- 30.Osterrieder, N., A. Neubauer, B. Fakler, C. Brandmüller, C. Seyboldt, O. R. Kaaden, and J. D. Baines. 1997. Synthesis and processing of the equine herpesvirus 1 glycoprotein M. Virology 232:230-239. [DOI] [PubMed] [Google Scholar]
- 31.Rudolph, J., D. J. O'Callaghan, and N. Osterrieder. 2002. Cloning of the genomes of equine herpesvirus type 1 (EHV-1) strains KyA and racL11 as bacterial artificial chromosomes (BAC). J. Vet. Med. B Infect. Dis. Vet. Public Health 49:31-36. [DOI] [PubMed] [Google Scholar]
- 32.Rudolph, J., and N. Osterrieder. 2002. Equine herpesvirus type 1 devoid of gM and gp2 is severely impaired in virus egress but not direct cell-to-cell spread. Virology 293:356-367. [DOI] [PubMed] [Google Scholar]
- 33.Rudolph, J., C. Seyboldt, H. Granzow, and N. Osterrieder. 2002. The gene 10 (UL49.5) product of equine herpesvirus 1 is necessary and sufficient for functional processing of glycoprotein M. J. Virol. 76:2952-2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schumacher, D., B. K. Tischer, W. Fuchs, and N. Osterrieder. 2000. Reconstitution of Marek's disease virus serotype 1 (MDV-1) from DNA cloned as a bacterial artificial chromosome and characterization of a glycoprotein B-negative MDV-1 mutant. J. Virol. 74:11088-11098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher, D., B. K. Tischer, S. M. Reddy, and N. Osterrieder. 2001. Glycoproteins E and I of Marek's disease virus serotype 1 are essential for virus growth in cultured cells. J. Virol. 75:11307-11318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seyboldt, C., H. Granzow, and N. Osterrieder. 2000. Equine herpesvirus 1 (EHV-1) glycoprotein M: effect of deletions of transmembrane domains. Virology 278:477-489. [DOI] [PubMed] [Google Scholar]
- 37.Sun, Y., A. R. MacLean, J. D. Aitken, and S. M. Brown. 1996. The role of the gene 71 product in the life cycle of equine herpesvirus 1. J. Gen. Virol. 77:493-500. [DOI] [PubMed] [Google Scholar]
- 38.Sun, Y., A. R. MacLean, D. Dargan, and S. M. Brown. 1994. Identification and characterization of the protein product of gene 71 in equine herpesvirus 1. J. Gen. Virol. 75:3117-3126. [DOI] [PubMed] [Google Scholar]
- 39.Telford, E. A., M. S. Watson, K. McBride, and A. J. Davison. 1992. The DNA sequence of equine herpesvirus-1. Virology 189:304-316. [DOI] [PubMed] [Google Scholar]
- 40.Trapp, S., N. Osterrieder, G. M. Keil, and M. Beer. 2003. Mutagenesis of a bovine herpesvirus type 1 genome cloned as an infectious bacterial artificial chromosome: analysis of glycoprotein E and G double deletion mutants. J. Gen. Virol. 84:301-306. [DOI] [PubMed] [Google Scholar]
- 41.Turtinen, L. W., and G. P. Allen. 1982. Identification of the envelope surface glycoproteins of equine herpesvirus type 1. J. Gen. Virol. 63:481-485. [DOI] [PubMed] [Google Scholar]
- 42.Wellington, J. E., G. P. Allen, A. A. Gooley, D. N. Love, N. H. Packer, J. X. Yan, and J. M. Whalley. 1996. The highly O-glycosylated glycoprotein gp2 of equine herpesvirus 1 is encoded by gene 71. J. Virol. 70:8195-8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wellington, J. E., A. A. Gooley, D. N. Love, and J. M. Whalley. 1996. N-terminal sequence analysis of equine herpesvirus 1 glycoproteins D and B and evidence for internal cleavage of the gene 71 product. J. Gen. Virol. 77:75-82. [DOI] [PubMed] [Google Scholar]
- 44.Wellington, J. E., D. N. Love, and J. M. Whalley. 1996. Evidence for involvement of equine herpesvirus 1 glycoprotein B in cell-cell fusion. Arch. Virol. 141:167-175. [DOI] [PubMed] [Google Scholar]
- 45.Whittaker, G. R., L. A. Wheldon, L. E. Giles, J. M. Stocks, I. W. Halliburton, R. A. Killington, and D. M. Meredith. 1990. Characterization of the high Mr glycoprotein (gP300) of equine herpesvirus type 1 as a novel glycoprotein with extensive O-linked carbohydrate. J. Gen. Virol. 71:2407-2416. [DOI] [PubMed] [Google Scholar]
- 46.Zuckermann, F. A., T. C. Mettenleiter, C. Schreurs, N. Sugg, and T. Ben-Porat. 1988. Complex between glycoproteins gI and gp63 of pseudorabies virus: its effect on virus replication. J. Virol. 62:4622-4626. [DOI] [PMC free article] [PubMed] [Google Scholar]