

Abstract
Background
Tri- and tetra-nucleotide repeats in mammalian genomes can induce formation of alternative non-B DNA structures such as triplexes and guanine (G)-quadruplexes. These structures can induce mutagenesis, chromosomal translocations and genomic instability. We wanted to determine if proteins that bind triplex DNA structures are quantitatively or qualitatively different between colorectal tumor and adjacent normal tissue and if this binding activity correlates with patient clinical characteristics.
Methods
Extracts from 63 human colorectal tumor and adjacent normal tissues were examined by gel shifts (EMSA) for triplex DNA-binding proteins, which were correlated with clinicopathological tumor characteristics using the Mann-Whitney U, Spearman’s rho, Kaplan-Meier and Mantel-Cox log-rank tests. Biotinylated triplex DNA and streptavidin agarose affinity binding were used to purify triplex-binding proteins in RKO cells. Western blotting and reverse-phase protein array were used to measure protein expression in tissue extracts.
Results
Increased triplex DNA-binding activity in tumor extracts correlated significantly with lymphatic disease, metastasis, and reduced overall survival. We identified three multifunctional splicing factors with biotinylated triplex DNA affinity: U2AF65 in cytoplasmic extracts, and PSF and p54nrb in nuclear extracts. Super-shift EMSA with anti-U2AF65 antibodies produced a shifted band of the major EMSA H3 complex, identifying U2AF65 as the protein present in the major EMSA band. U2AF65 expression correlated significantly with EMSA H3 values in all extracts and was higher in extracts from Stage III/IV vs. Stage I/II colon tumors (p = 0.024). EMSA H3 values and U2AF65 expression also correlated significantly with GSK3 beta, beta-catenin, and NF- B p65 expression, whereas p54nrb and PSF expression correlated with c-Myc, cyclin D1, and CDK4. EMSA values and expression of all three splicing factors correlated with ErbB1, mTOR, PTEN, and Stat5. Western blots confirmed that full-length and truncated beta-catenin expression correlated with U2AF65 expression in tumor extracts.
Conclusions
Increased triplex DNA-binding activity in vitro correlates with lymph node disease, metastasis, and reduced overall survival in colorectal cancer, and increased U2AF65 expression is associated with total and truncated beta-catenin expression in high-stage colorectal tumors.
Background
DNA and RNA are dynamic molecules that adopt several different secondary and tertiary structures. DNA can form a stable triple helix in which a purine- or pyrimidine-rich third strand forms sequence-specific H-bonds (Hoogsteen and reverse-Hoogsteen) with a purine-rich strand in the major groove of the Watson-Crick duplex in polypyrimidine-polypurine repeat sequences [1]. Guanine (G)-rich DNA and RNA can also form G-quadruplexes that also use Hoogsteen and reverse Hoogsteen G*G bonds in a non-canonical four-stranded topology. G-quadruplexes specifically have been implicated at DNA telomere ends, the purine-rich DNA strands of oncogenic promoters, and in RNA 5’-untranslated regions (UTR) near translation start sites [2]. For example, a nuclease-sensitive element in the human c-MYC promoter that can form either a DNA triplex or G-quadruplex interferes with DNA transcription [3]. Transient Hoogsteen base pairs have been detected in DNA duplexes bound to transcription factors and in damaged DNA, suggesting that the DNA double helix can resonate and form excited-state Hoogsteen base pairs that can expand its structural complexity [4].
Genomic instability in association with carcinogenesis is well established and promotes multiple hallmarks of cancer [5]. Repetitive DNA, such as tri- and tetranucleotide sequences, is genetically unstable, and expansions of such DNA repeats are associated with numerous hereditary neurological diseases including Fragile X syndrome, myotonic dystrophy, and Friedreich’s ataxia [6,7]. Many of these DNA repeat sequences can exist in at least two different conformations, and at least 10 non-B DNA conformations can form, perhaps transiently, at specific sequences due to negative supercoiling generated by DNA replication, transcription, protein binding, or during DNA repair [8]. Non-B DNA structures such as cruciforms, triplexes and G-quadruplexes can cause mutations such as deletions, expansions, and translocations [9,10]. Bacolla et al. found that genes containing long polypyrimidine-polypurine sequences are more susceptible to chromosomal translocations than genes that do not contain these sequences [11]. Researchers have located “hotspot” regions of the genome at or near sequences with the potential to form non-B DNA structures, including the region in the promoter of the human c-MYC gene capable of forming triplex or G-quadruplex DNA that overlaps with one of the major breakpoint hotspots in c-MYC-induced lymphomas and leukemias [12,13]. The recently created Non-B Database ( http://nonb.abcc.ncifcrf.gov) can be used to predict the capability of a DNA sequence in mammalian genomes to form any of a variety of non-B structures [14].
While the existence of triplex or G-quadruplex nucleic acids in vivo has yet to achieve mainstream acceptance, eukaryotic proteins that recognize and bind to these alternative structures do exist. For example, the Fragile X mental retardation protein (FMRP) binds an intramolecular G-quartet in target mRNAs, and loss of function of this protein causes the Fragile X mental retardation syndrome [15]. We have studied proteins in Saccharomyces cerevisiae and HeLa carcinoma cells that bind specifically to a purine-motif triplex DNA probe in gel shifts (EMSA) where the third strand is G-rich and photo-crosslinked with a psoralen group (Ps~) [16-18]. Stm1, the major purine-motif triplex DNA-binding protein in S. cerevisiae, also binds to G-quartet DNA and RNA in vitro[19]. Using Southwestern blotting where HeLa nuclear extracts were separated by SDS-PAGE, blotted and probed with the same radio-labeled purine triplex DNA used in EMSA, we found that 100-, 60-, and 15-kDa bands were hybridized with the triplex DNA probe, whereas only the 100-kDa band was also hybridized with the parent duplex DNA probe [16]. RecQ-family helicases, including the WRN helicase, have been shown to preferentially bind to and unwind aberrant DNA structures such as triplex and G-quadruplex DNAs, which are believed to exist in vivo as intermediates in DNA replication, recombination, and repair. The WRN helicase is deficient in patients with Werner syndrome, an autosomal recessive disease causing premature aging that is associated with numerous age-related phenotypes, including a high predisposition to cancer [20]. Others have examined specific aspects of WRN expression in colorectal cancer, such as the presence of allelic variants and colorectal cancer risk and WRN promoter methylation as it correlates with a CpG island methylation phenotype (CIMP)-high diagnosis [21,22]. These studies led us to question whether triplex DNA-binding proteins and WRN helicase expression are quantitatively and/or qualitatively different in human colorectal tumors and corresponding normal tissues, if there is any correlation with clinical prognosis, and identify purine-motif triplex DNA-binding proteins in human cells.
Numerous genetic, cytogenetic, and epigenetic aberrations act at specific stages in colorectal cancer initiation and progression and influence response to therapy, such as inactivation of tumor suppressor APC as an initiating event and KRAS or BRAF mutations as markers of non-response to EGFR-targeted therapy [23]. High-throughput studies have suggested the existence of additional undiscovered cancer genes that may promote colorectal cancer development [24-26]. Colorectal cancer is also one of the more genetically unstable cancers, with about 65% of sporadic adenomas and cancers being characterized by chromosomal instability (CIN), 10-15% characterized by microsatellite instability (MSI), and approximately 20% having a CIMP phenotype, with some overlap among these characteristics.
We have found higher triplex DNA-binding activity in vitro in colorectal tumor extracts than in corresponding normal tissue extracts using EMSA, and that this increased binding activity correlated significantly with the spread of cancer to the lymph nodes, metastasis, and reduced overall survival. We also found that expression of the triplex/G-quadruplex-unwinding helicase WRN correlated significantly with total triplex DNA-binding activity in EMSAs in both normal and tumor tissue extracts. Biotin purine-motif triplex DNA affinity identified three multifunctional splicing factors: U2AF65, PSF, and p54nrb, and an anti-U2AF65 antibody produced a super-shifted EMSA band. High U2AF65 expression was associated with advanced colon tumor stages and with p54nrb and PSF expression in tumors. U2AF65 expression also correlated significantly with both total and truncated beta-catenin, as well as NF- B p65, PCNA, EGFR, mTOR, PTEN, and Stat5 in colorectal tumors.
Materials and methods
Preparation of cytoplasmic and nuclear extracts of tissue and cell lines. Tissue samples of tumor and adjacent normal mucosa were collected after surgical resections after informed consent, verification by a pathologist, and snap-frozen in liquid nitrogen. The patients had not previously received any chemotherapy, therefore the tissues are chemotherapy naïve. Frozen tissue samples were prepared as described by Asangani et al. [27]. The samples were pulverized with a Sartorius Mikrodismembrator, then extracted for 30 min on ice with Schaffner lysis buffer A (10 mM HEPES-Na + pH 7.9, 10 mM KCl, 0.1 mM EDTA pH 8.0, 0.1 mM EGTA pH 8, 1 mM dithiothreitol, 0.5% Triton X-100, Sigma phosphatase inhibitor cocktail 2, and Roche Complete Mini protease inhibitor) and centrifuged at 13,000 rpm, 4°C in a microcentrifuge to produce cytoplasmic extracts. The nuclear pellet was extracted for 30 min on ice with Schaffner buffer C (20 mM HEPES-Na + pH 7.9, 0.4 M NaCl, 0.1 mM EDTA pH 8.0, 0.1 mM EGTA pH 8.0, 1 mM dithiothreitol, 20% glycerol, with phosphatase and protease inhibitors) and centrifuged at 13,000 rpm, 4°C in a microcentrifuge to produce nuclear extracts [28]. Total protein concentrations were determined using the Pierce BCA Protein Assay kit. Colorectal cancer cell lines and HeLa cytoplasmic or nuclear extracts were similarly prepared using Schaffner buffers A and C, respectively.
Purine-motif triplex DNA formation and 33P-labeling
Purine triplex DNA oligonucleotide sequences and probe formation were as previously described [16,17]. The parent duplex oligonucleotides are PuGA: 5’ – AATTCCTAAGGGAGGGGAGGGGAGGGTAGCT – 3’ and complementary strand PuCT: 5’ – AGCTACCCTCCCCTCCCCTCCCTTAGG – 3’. The parent duplex DNA was made by annealing equimolar (0.1 mM) concentrations of the PuGA and PuCT oligonucleotides at room temperature after boiling for 2 min in 40 mM Tris-HCl pH 8.0, 10 mM MgCl2, 0.01% NP-40. The purine-motif triplex-forming oligonucleotide (TFO) contained a 4’-(hydroxymethyl)-4,5’,8-trimethylpsoralen-hexyl (Ps~) moiety at the 5’-terminus (Eurogentec): 5’ – Ps ~ GGG TGG GGT GGG GTG GGT -3’. To form triplex DNA, the parent duplex DNA and a 10-fold molar excess of TFO were incubated for 4 h at 30°C in 40 mM Tris HCl pH 8.0, 100 mM MgCl2, 0.01% NP-40. Psoralenated TFO was then cross-inked with the parent DNA duplex with a 366 nm UV transilluminator for 10 min on ice. Purine triplex DNA (1 x 10-7 M) was 3’ end-labeled with T4 kinase (New England Biolabs) and γ-33P dATP for 1 h at 37°C. Unincorporated labeling dATP was removed from the reaction by centrifuging the reaction mixture with an equal volume of 10 mM Tris-HCl pH 8.0, 10 mM MgCl2, 0.05% Triton X-100 through a G25 Microspin column (GE Healthcare).
Electrophoretic mobility shift assay (EMSA) and super-shift EMSA
Gel shifts were also done as previously described [16,17]. In this study 5 μg total protein from tissue extracts or 1.5 μg HeLa or colorectal cancer cell line cytoplasmic or nuclear extracts were mixed with 1 nM 33P-labeled purine triplex DNA and 2 μg poly (dIdC) carrier DNA in binding buffer (25 mM HEPES-Na + pH 7.9, 50 mM KCl, 10% glycerol, 0.5 mM dithiothreitol, 2 mM MgCl2) for 30 min at room temperature. Protein-triplex DNA probe complexes were resolved by nondenaturing PAGE at 7 V/cm for 90 min through a 5% acrylamide/0.25% bisacrylamide gel containing 22 mM Tris borate, 0.5 mM EDTA, and 5% glycerol. Protein-probe complexes were visualized using autoradiography and quantitated with a Storm 840 PhosphorImager (Molecular Dynamics). Major EMSA H3 bands from each tissue sample were normalized by dividing by the H3 band value of HeLa nuclear extract present in each gel. For super-shift EMSA, protein extracts were incubated in the same binding buffer with purine triplex DNA probe for 30 min at room temperature, then 400 ng of anti-U2AF65 MC3 antibody or mouse IgG antibody as a negative control (Santa Cruz) were added to the reaction and incubated for 1 h at room temperature. PAGE gels were run as for regular EMSA with the addition of a circulating cooling water bath to the gel apparatus.
Statistical correlations
The Wilcoxon Sign Rank Test was used to compare the level of the major EMSA H3 complex and WRN expression in total, cytoplasmic, and nuclear extracts of colorectal tumors and corresponding normal tissues. The Mann-Whitney U test was used with SPSS version 13.0 to compare quantitative variables in two independent groups. Spearman correlations among continuous variables were computed. Chi square (Bonferroni-corrected) were used for grouped/dichotomized variables. Survival was estimated using Kaplan-Meier analysis, and differences were calculated using Mantel-Cox log-rank statistics; primary endpoints were tumor-related death (disease-specific survival), death (overall survival), and tumor recurrence (recurrence-free survival, R0-patients only). The following variables were dichotomized according to the median value: protein levels in nuclear and total extracts (cytoplasm and nucleus) ratios (tumor/normal) as high levels in tumor (values above the median) vs. low levels in tumor (values below the median) as compared with normal tissue, involved lymph nodes as pN0 vs. pN1-3, distant metastasis as M0 vs. M1, surgical curability as curative vs. non-curative resection (R0 vs. R1/2).
Purification of triplex DNA-binding proteins using biotin/streptavidin affinity
Biotinylated purine triplex DNA was formed using a 3’ biotinylated PuCT oligonucleotide (Eurogentec): 5’ – AGCTACCCTCCCCTCCCCTCCCTTAGGAATTTT-biotin-3’ annealed to the PuGA complementary strand, then annealed and crosslinked with the Ps ~ TFO as described above. Purification of DNA-binding proteins using biotin/streptavidin affinity systems, as described in Current Protocols in Molecular Biology [29], was performed in separate 2 ml reactions containing either 800 μg RKO colorectal cancer cell nuclear extract or 1085 μg RKO cytoplasmic extract, EMSA binding buffer (25 mM HEPES-Na + pH 7.9, 50 mM KCl, 10% glycerol, 0.5 mM dithiothreitol, 2 mM MgCl2), 600 μg poly (dIdC), 1 nM biotinylated purine triplex DNA, and 150 μl pretreated streptavidin agarose (Fluka) while rotating for 2 hr at room temperature. Streptavidin agarose was gently pelleted and washed three times with binding buffer. Laemmli buffer was added directly to the agarose pellet and boiled for 5 min to elute bound protein(s). Proteins were separated using 10% SDS-PAGE and stained with Coomassie blue. Two bands (100 and 60 kDa) from the nuclear extract reaction and one band (65 kDa) from the cytoplasmic extract reaction were excised from the gel and submitted to the German Cancer Research Center (DKFZ) Functional Proteome Analysis laboratory for sequencing and analysis using nano-HPLC ESI-MS-MS and identified using MASCOT database searches.
Western blotting
Western blot analysis was performed using standard procedures as described in Current Protocols in Molecular Biology [27]. 25 μg total protein from tissue or cell line cytoplasmic or nuclear extract was separated by 10% SDS-PAGE, then electro-transferred to nitrocellulose membranes in 25 mM Tris, 190 mM glycine with 20% methanol. After blocking in 5% milk in Tris-buffered saline with 0.2% Tween-20 (TBST) for 1 hr at room temperature, membranes were incubated with antibodies against WRN (H-300 Santa Cruz sc-5629, 1:500), U2AF65 (MC3 Santa Cruz sc-53942, 1:2000), PSF (39-1 Santa Cruz sc-101137, 1:2000), p54nrb (H-85 Santa Cruz sc-67016, 1:2000) in 5% milk-TBST for 1 hr at room temperature, or beta-catenin (L87A12 Cell Signaling CS-2698, 1:1000) or actin (Sigma A2066, 1:1000) in 5% milk in TBST overnight at 4°C. Blots were washed with TBST, incubated with the appropriate HRP-conjugated secondary antibody at 1:4500, and detected by enhanced chemiluminescence (Pierce, Thermo Scientific) and autoradiography. Protein bands were quantitated by densitometry using NIH Image J software and normalized to actin.
Reverse phase protein array (RPPA)
RPPA was performed as described by Mannsperger et al. [30]. 2.7 ng cytoplasm or 2.8 ng nuclear protein extract per spot was printed with a non-contact spotter onto nitrocellulose slides (Oncyte Avid, Grace Bio-labs, Bend OR) using an Aushon 2470 Microarrayer (Billerica, MA). Slides were mounted in a customized incubation chamber (Metecon, Mannheim Germany), blocked for 1 hr at room temperature with 50% (v/v) Odyssey blocking buffer in PBS and individually stained with 37 validated primary antibodies at 1:300 in blocking buffer at 4°C overnight and Alexa 680-labeled secondary antibodies (Invitrogen) at 1:8000 in PBS with 0.05% Tween for 1 hr at room temperature. Slides were scanned with the Licor Odyssey system and spot intensities were calculated with GenePix Pro 5.0 microarray analysis software (Molecular Devices). To estimate the total protein concentration per spot, a slide from each run was stained with Fast Green FCF (Sigma-Aldrich) as described by Loebke et al. [31]. Data analysis was done using R with the RPPanalyzer package from CRAN ( http://cran.r-project.org, [32]). For each antibody the logged mean of the raw foreground pixel intensities of a single spot was subtracted by the corresponding logged Fast Green FCF signal to normalize for the total protein per spot.
Results
Colorectal tumors have higher triplex DNA-binding activity than corresponding normal tissue
A summary of clinical characteristics of the 63 study patients are shown in Table 1. To examine purine-motif triplex DNA-binding proteins, cytoplasmic and nuclear extracts from 63 colorectal cancer patients’ tumor and corresponding normal tissues were isolated and examined by gel shifts (EMSA). Figure 1 presents examples of EMSAs from eight patients representing all four tumor stages, where in most samples one major band (H3) is present in varying amounts. In some patients, tumor cytoplasmic extracts contained a higher amount of the major H3 complex than normal or tumor nuclear extracts (patients 1 and 5), while in other patients, tumor nuclear extracts contained a higher amount of the major H3 complex (patients 6 and 8). Cytoplasmic and nuclear extracts from HeLa cells were included as positive controls. Normalized EMSA H3 values are listed below each sample. To verify that the major EMSA H3 band is specific for the triplex DNA probe, the 33P-labeled parent duplex DNA probe lacking G*G base pairs did not produce the major H3 complex in patient tissue or HeLa nuclear extracts (Additional file 1: Figure S1). EMSA H3 binding values were generally higher in tumor than normal tissue, whether evaluating cytoplasmic extracts (mean = 0.512, median = 0.509 for tumor tissue; mean = 0.386, median = 0.384 for normal tissue) or nuclear extracts (mean = 0.361, median = 0.368 for tumor tissue; mean = 0.264, median = 0.228 for normal tissue) as shown in Figure 2. Wilcoxon sign rank test results showed significantly higher triplex DNA EMSA binding activity in tumor than normal extracts when examining total measures (p = 0.001), cytoplasmic extracts only (p = 0.001) and nuclear extracts only (p = 0.012)(Additional file 2). We also performed EMSA analysis of cytoplasmic and nuclear extracts of eight colorectal cancer cell lines (GEO, SW480, HT29, HCT116, Colo206F, wiDR, Colo320, and RKO) and found that all eight cell lines had a triplex DNA-binding protein pattern that was very similar to HeLa extracts, with a moderate amount of the major H3 band produced by cytoplasmic extracts and an abundant amount of the H3 band produced by nuclear extracts (Additional file 1: Figure S2a).
Table 1.
Patient clinical characteristics
| Tumor characteristics | Absolute (n = 63) | Relative (%) | |
|---|---|---|---|
| Sex |
Male |
46 |
73% |
| |
Female |
17 |
27% |
| Localization |
Colon |
36 |
57.1% |
| |
Rectum |
27 |
42.9% |
| TNM Staging |
pT1 |
10 |
15.9% |
| |
pT2 |
5 |
7.9% |
| |
pT3 |
34 |
54% |
| |
pT4 |
16 |
25.4% |
| Lymph Node Status |
pN0 |
37 |
58.7% |
| |
pN1 |
16 |
25.4% |
| |
pN2 |
10 |
15.9% |
| Metastasis Staging |
M0 |
42 |
66.7% |
| |
M1 |
21 |
33.3% |
| Status |
alive |
43 |
68.1% |
| dead | 20 | 31.7% |
Figure 1.
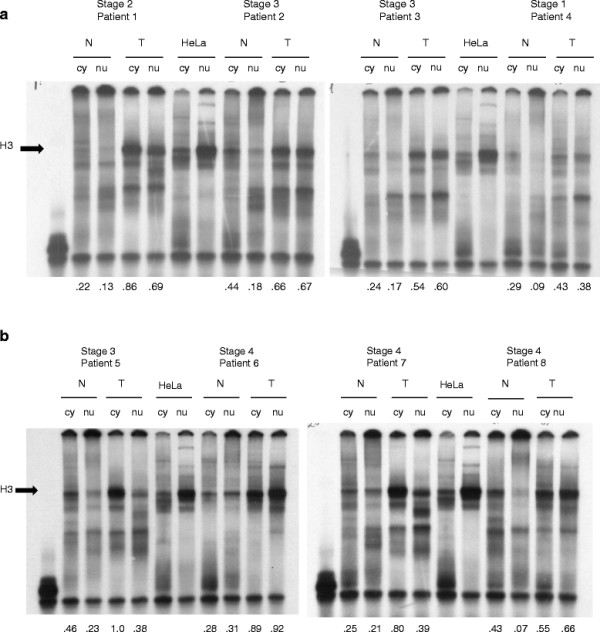
Electrophoretic Mobility Shift Assay (EMSA) of Resected Tissue Extracts with Purine Triplex DNA. 33P-labeled purine-motif triplex DNA (1 nM) was complexed with 5 μg total protein from normal cytoplasmic (N cy), normal nuclear (N nu), tumor cytoplasmic (T cy) or tumor nuclear (T nu) extracts of tissues obtained from eight selected colorectal cancer patients. 1.25 μg HeLa cytoplasmic and nuclear extracts were used as positive (+) controls. The major EMSA band was H3, indicated with an arrow. Normalized EMSA H3 values are listed below the corresponding samples. The purine-motif triplex probe alone is shown in lane 1.
Figure 2.
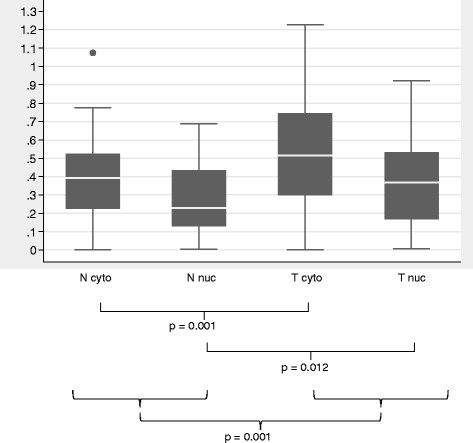
Normalized EMSA H3 values for all 63 colorectal cancer patient extracts. Box plots indicating the median normalized EMSA H3 values (white lines in the boxes), upper and lower quartiles (25th through 75th percentiles defined by the shaded box), and ranges of data values for each extract type. N cyto, cytoplasmic normal tissue extracts; N nuc, nuclear normal tissue extracts; T cyto, cytoplasmic tumor tissue extracts; Tnuc, nuclear tumor tissue extracts.
Increased triplex DNA-binding activity in colorectal tumors correlates with lymph node disease, metastasis, and overall survival
We wanted to investigate whether the amount of the EMSA H3 complex correlated with patient clinicopathological data and overall survival. Median follow-up time for patient clinical data was 28.9 months. Normalized EMSA data of patient samples were correlated with clinical risk factors and computed for univariate prognostic impact. We observed that lymph node disease (N-Stage) was significantly associated with the ratio of tumor/normal (T/N) triplex-binding activity for cytoplasmic and nuclear extracts and total values (p = 0.026; 0.019; 0.017, respectively, Table 2a). This meant that all patients without lymph node disease at diagnosis had significantly decreased binding ratios (T/N) in both cytoplasmic and nuclear extracts. Also, the triplex DNA-binding activity in tumor nuclear extracts and total tumor extracts correlated significantly with metastasis (p = 0.031, p = 0.046, respectively, Table 2b). Kaplan-Meier survival analysis using a median cut-off of 1.5 (rounded-up) for the nuclear binding activity ratio (T/N) showed significantly lower overall survival in patients whose T/N nuclear binding activity ratio was greater than 1.5 (n = 30; p = 0.026) than in patients whose ratio was less than 1.5 (n = 33, Figure 3, Additional file 2). This suggested that although triplex DNA-binding protein(s) were present in normal colorectal tissue extracts, they were more abundant in tumor extracts. It also suggested that an abundance of the major triplex-binding EMSA complex (H3) in the nuclei of tumor cells was associated with metastasis and reduced overall survival (Additional file 3).
Table 2.
Correlation of the ratio of tumor (T) to normal (N) (T/N) EMSA H3 values for each patient with clinical features: test statistics (a) by presence of disease in lymph nodes (N-Stage) and (b) by presence of metastasis in distant organs (distant metastasis)
| | |||
|---|---|---|---|
|
(a) Grouping Variable: presence of disease in lymph nodes (N Stage) dichotomized | |||
| |
Ratio T/N |
Ratio T/N |
Ratio T/N |
| cytoplasm | nucleus | total | |
|
Mann-Whitney U |
322.000 |
313.000 |
310.500 |
|
Wilcoxon W |
1025.000 |
1016.000 |
1013.500 |
|
Z |
-2.220 |
-2.345 |
-2.380 |
|
Asymp. Sig (2-tailed) |
0.026 |
0.019 |
0.017 |
|
(b) Grouping Variable: presence of metastasis in distant organs (distant metastasis) | |||
| |
Cytoplasm tumor |
Nucleus tumor |
Total tumor |
|
Mann-Whitney U |
342.500 |
309.500 |
321.000 |
|
Wilcoxon W |
1245.500 |
1212.500 |
1224.000 |
|
Z |
-1.689 |
-2.156 |
-1.993 |
| Asymp. Sig (2-tailed) | 0.091 | 0.031 | 0.046 |
Figure 3.
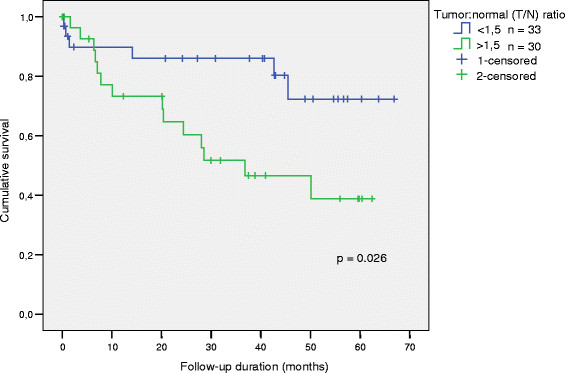
Overall Survival according to the tumor:normal (T/N) colorectal tissue nuclear triplex DNA-binding activity ratio. Cut-off = 1.5 (rounded-up median).
Identification of U2AF65 as the protein present in the EMSA H3 complex
We wished to identify the protein(s) responsible for binding the triplex DNA probe in the major EMSA H3 complex. We isolated biotinylated purine-motif triplex DNA-protein complexes from RKO cells with streptavidin-conjugated agarose, separated the complexes by SDS-PAGE, and stained with Coomassie Blue. Protein bands were analyzed by nano-HPLC ESI-MS-MS and identified using MASCOT database searches. We identified (1) 100-kDa and (2) 60-kDa proteins from nuclear extracts and a (3) 65-kDa protein from cytoplasmic extracts. These corresponded to the following proteins:
(1) PSF (polypyrimidine tract binding-associated splicing factor, or SFPQ) [NCBI Protein AAH04534]
(2) P54nrb (nuclear RNA-binding protein) or NonO [NCBI Protein NP_031389]
(3) U2AF65 (U2 small nuclear RNA auxiliary factor 2 isoform b) [NCBI Protein NP_001012496]
PSF and p54nrb are known to function as RNA polymerase II-associated splicing factors, bind as heterodimers, and are implicated in the regulation of expression of the Myc family of oncoproteins, COX2, etc. They also bind to and stimulate topoisomerase I and promote homologous DNA pairing and the incorporation of a single-stranded oligonucleotide into homologous superhelical double-stranded DNA D-loop formation [33,34]. U2AF65, identified from cytoplasmic extracts, is also an RNA polymerase II-associated splicing factor that can associate with mRNAs that include a predominance of transcription factors and cell cycle regulators, and shuttle continuously between the nucleus and cytoplasm [35,36].
Super-shift EMSA with a well-characterized monoclonal antibody against U2AF65 [37] consistently produced a super-shifted H3 band in all human extracts tested that were known to express U2AF65 by Western blot analysis (RKO and tumor tissue cytoplasmic and nuclear extracts are shown in Figure 4). This confirmed that U2AF65 is present in the H3 triplex DNA-protein complex observed by EMSA (Figure 4). Available antibodies against PSF or p54nrb did not produce any super-shifted bands in our EMSA analysis (Additional file 1: Figure S3).
Figure 4.
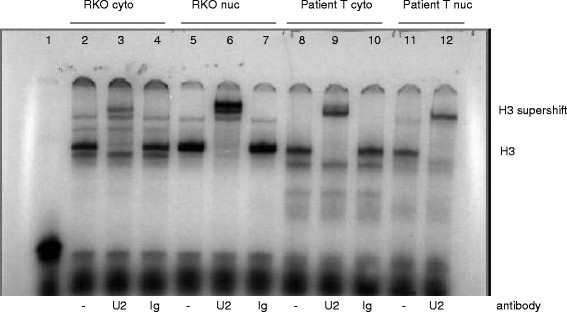
Production of a super-shifted H3 band in RKO and patient tissue extracts by super-shift EMSA with a monoclonal antibody against U2AF65. 33P-labeled triplex DNA (1 nM) was complexed with 1.5 μg total protein from RKO cytoplasmic (lanes 2-4), RKO nuclear (lanes 5-7), 5 μg tumor cytoplasm (T cyto lanes 8-10) or tumor nuclear (T nuc lanes 11-12) extracts. Lanes 2, 5, 8, and 11, no antibody; lanes 3, 6, 9, and 12, 400 ng anti-U2AF65 antibody MC3; lanes 4, 7, and 10, mouse IgG antibody (negative control). Each reaction also contained 2 μg poly (dI-dC) carrier DNA. Lane 1, triplex DNA probe alone.
U2AF65 expression correlates with EMSA H3 values and p54nrb and PSF expression in tumor tissues and with a higher tumor stage
We measured expression of the three splicing factors in normal and tumor colorectal tissue extracts obtained from 51 of the 63 patients using Western blotting to determine if triplex DNA-binding activity in EMSA correlates directly with U2AF65, PSF, and/or p54nrb total protein expression. Spearman correlations indicated that U2AF65 expression correlated significantly with EMSA H3 values, and that the correlation was highly significant in tumor extracts (cytoplasmic p = 1.8e-8; nuclear p = 5.9e-5; total p = 1.8e-8; Table 3a, Additional file 4). In comparison, PSF and p54nrb were highly expressed in nuclear extracts but seldom detected in cytoplasmic extracts, and their expression correlated with EMSA H3 values only in tumor nuclear extracts (p = 0.036 and 0.0071, respectively) (Table 3a). When correlating the expressions of the three splicing factors with each other, PSF and p54nrb were highly significantly associated in nuclear extracts of both normal and tumor tissue (p = 1e-6 in both) as expected, as they are known to bind and function as heterodimers. Also, U2AF65 expression was highly significantly correlated with p54nrb expression in both normal and tumor nuclear extracts (p = 0.00037 and 1e-6, respectively)(Table 3b), but with PSF expression only in tumor nuclear extracts (p = 0.0005), suggesting a unique functional aspect of U2AF65 and PSF in tumor cell nuclei. We also examined expression of the three splicing factors identified by biotin triplex DNA affinity in the eight colorectal cancer cell lines using Western blotting. Consistent with patient tissue data, U2AF65 expression from all cell line extracts most closely matched the abundance of the EMSA H3 band, with moderate expression in all cytoplasmic extracts and abundant expression in all nuclear extracts (Additional file 1: Figure S2b).
Table 3.
(a) Spearman correlation p values of EMSA H3 values with expression of triplex DNA-binding proteins (3BP) and (b) correlations of U2AF65 expression to PSF and p54nrb expression in normal and tumor tissue extracts
|
3BP expression correlated |
U2AF65 |
p54nrb |
PSF |
|---|---|---|---|
| With EMSA H3 | |||
|
Normal cytoplasm |
0.0028 |
0.91 |
0.66 |
|
Normal nucleus |
0.015 |
0.11 |
0.075 |
|
Normal total |
0.016 |
0.26 |
0.095 |
|
Tumor cytoplasm |
1.8e-08 |
0.53 |
0.019 |
|
Tumor nucleus |
5.9e-05 |
0.0071 |
0.036 |
|
Tumor total |
1.8e-08 |
0.0048 |
7.5e-05 |
|
Correlation to U2AF65 |
p54nrb |
PSF |
|
|
Normal cytoplasm |
0.00066 |
0.085 |
|
|
Normal nucleus |
0.00037 |
0.073 |
|
|
Normal total |
0.00040 |
0.094 |
|
|
Tumor cytoplasm |
0.0041 |
0.0002 |
|
|
Tumor nucleus |
1e-06 |
0.0005 |
|
| Tumor total | 1e-06 | 1e-06 | |
Having shown that the EMSA H3 complex was increased in tumor compared to adjacent normal tissue, we wished to determine if U2AF65, p54nrb and PSF expression was associated with tumor stage. U2AF65 protein expression according to extract type and tumor stage in all colon tumors is shown in Figure 5. Colon tumors in Figure 5 in advanced clinical stages, UICC Stage III and IV (Dukes C and D) express significantly higher U2AF65 in the cytoplasm and overall than did tumors at early stages (mean value of U2AF65 tumor cytoplasm UICC Stage I and II expression = 0.349 vs. UICC Stage III and IV = 0.491; p = 0.024 [Mann-Whitney U-Test, Additional file 5]). PSF and p54nrb expression were not significantly correlated with tumor stage. While both p54nrb and PSF expression were significantly correlated with EMSA H3 values in tumor but not normal tissue extracts, the antibodies against these proteins that we tested were unable to produce a super-shifted EMSA band. Thus the relevance of p54nrb and PSF as triplex DNA-binding proteins remains to be determined.
Figure 5.

U2AF65 protein expression by colon tumor stage. Total protein (25 μg) from cytoplasmic (cyto) and nuclear (nuc) colon tumor and normal tissue extracts were separated using 10% SDS-PAGE and electro-transferred to nitrocellulose membranes. Blots were incubated with anti-U2AF65 antibody MC-3 and detected using chemiluminescence and autoradiography. Blots were reprobed with an anti-actin antibody, and densitometry was performed using NIH Image J software. U2AF65 expression values were normalized by dividing the actin expression values in each extract, and plotted according to colon tumor stage using the R program (Additional file 6).
Expression of the WRN helicase correlates with EMSA H3 binding activity
We wanted to test the hypothesis that proteins that bind to or stabilize triplexes and G-quadruplexes can act in a yin-yang fashion (in complementary opposition) with proteins such as helicases that unwind or destabilize these structures, and that expression and/or function of these binding and unwinding proteins may be imbalanced in tumors that could contribute to genomic instability. We tested 51 patient colorectal tumor and normal tissue extracts for expression of the RecQ-family helicase WRN because it is known to act preferentially on aberrant structures such as triplexes and G-quadruplexes and to promote genomic integrity [19]. We used the Wilcoxon sign rank test to determine if WRN is differentially expressed in normal and tumor tissue extracts and Spearman’s rho to correlate WRN helicase expression in normal and tumor tissue extracts with EMSA H3 data. We detected no significant differences in normalized WRN expression between normal and tumor extracts or according to tumor stage (mean cytoplasmic expression in tumor tissue = 0.424, in normal tissue = 0.283; mean nuclear expression in tumor tissue = 0.275, in normal tissue = 0.196; total expression mean in tumor tissue = 0.679, in normal tissue = 0.465). However, we did observe that total WRN expression correlated significantly with total EMSA H3 binding values in both normal tissue (rho 0.296, p = 0.03) and tumor extracts (rho 0.460, p < 0.001).
Reverse-phase protein array and western blot analysis of tissue extracts show a correlation of U2AF65 expression with total and truncated beta-catenin expression
Another goal of our study was to measure the expression of numerous cancer-relevant proteins in patient tissue extracts and correlate it with EMSA H3 values and expression of the three splicing factors identified using biotin triplex DNA affinity as a screen to identify potentially relevant functional relationships among these splicing factors and other well-characterized proteins. Using reverse-phase protein array (RPPA) analysis, we examined extracts from 51 patients (because not all extracts met the minimum concentration needed for accurate measurement) for expression of cancer-related proteins with 37 previously validated antibodies. Spearman correlation of the expression of multiple signaling proteins was calculated. Significant correlations after Bonferroni correction for multiple testing were found with both EMSA H3 values and U2AF65 expression, including NF- B p65, GSK3 beta, beta-catenin, Src, and PI3K p110 alpha (Table 4; exact p values are shown in Additional file 7: Table S1). The expression levels of a distinct set of proteins were found to correlate significantly with both p54nrb and PSF expression, such as cyclin D1, c-Myc, JNK1, CDK4, Akt1, and Stat3. Expression of all three splicing factors and EMSA H3 values also significantly correlated with another set of proteins including p38 alpha, ErbB1 (EGFR), mTOR, PTEN, and Stat5.
Table 4.
Spearman correlations of EMSA H3 values and triplex DNA-binding protein expression to other proteins by reverse phase protein array (RPPA)
| EMSAH3 | U2AF65 | p54nrb | PSF | |
|---|---|---|---|---|
|
Ph-Erk |
|
** |
|
|
|
NF-κB p65 |
*** |
*** |
|
|
|
Cyclin D1 |
|
|
*** |
*** |
|
GSK3β |
*** |
* |
|
|
|
c-myc |
|
|
** |
*** |
|
JNK1 |
|
|
*** |
*** |
|
PCNA |
*** |
*** |
|
** |
|
β-catenin |
*** |
*** |
|
|
|
Ph-Raf |
|
|
|
** |
|
Src |
** |
** |
|
|
|
p 38 α |
** |
** |
*** |
*** |
|
Cdk4 |
|
|
** |
*** |
|
Akt1 |
|
|
** |
*** |
|
ErbB1 |
*** |
*** |
** |
*** |
|
Bcl-2 |
|
** |
|
|
|
mTor |
*** |
*** |
*** |
*** |
|
P13K p110 α |
** |
*** |
|
|
|
PLC γ |
|
|
|
*** |
|
PTEN |
*** |
*** |
*** |
*** |
|
Stat3 |
* |
|
** |
*** |
| Stat5 | *** | *** | *** | *** |
*p < 0.05; **p < 0.01; ***p < 0.001.
The most highly significant correlation in our RPPA analysis was that between U2AF65 expression and beta-catenin (p = 9e-10), known to be deregulated and a major player in the etiology of colorectal cancer. To confirm our RPPA results, we compared Western blots of beta-catenin and U2AF65 expression in tissue extracts from 50 patients. Representative Western blots for six patients are shown in Figure 6, which includes some patient samples also shown in Figure 1 EMSAs. These data were quantitated by densitometry and graphed in Additional file 1: Figure S4. According to Spearman’s rho, we observed that total beta-catenin and U2AF65 expression are highly significantly correlated in cytoplasmic and nuclear tumor extracts (p = 5.7e-6 and p = 3.1e-6, respectively), while their expression correlated significantly in normal nuclear extracts (p = 0.0018), and showed no significant correlation in normal cytoplasmic extracts (p = 0.15). In addition, beta-catenin expression was higher in cytoplasmic and nuclear extracts of stage III and IV colon tumors than in those of stage I and II colon tumors (Additional file 1: Figure S5). Western blots of beta-catenin expression showed truncated bands (65-80- kDa) for some extracts but not for others, which was consistent with previous reports of truncated or novel spliceforms of beta-catenin mRNA [38,39] and an 80-kDa truncated beta-catenin protein [40] in colorectal cancer. In addition to a significant correlation between full-length beta-catenin (92-kDa) expression and U2AF65 expression, we found a significant correlation between truncated beta-catenin and U2AF65 expression, particularly in the cytoplasm (p = 0.0047) and nuclei (p = 0.022) of tumor cells.
Figure 6.
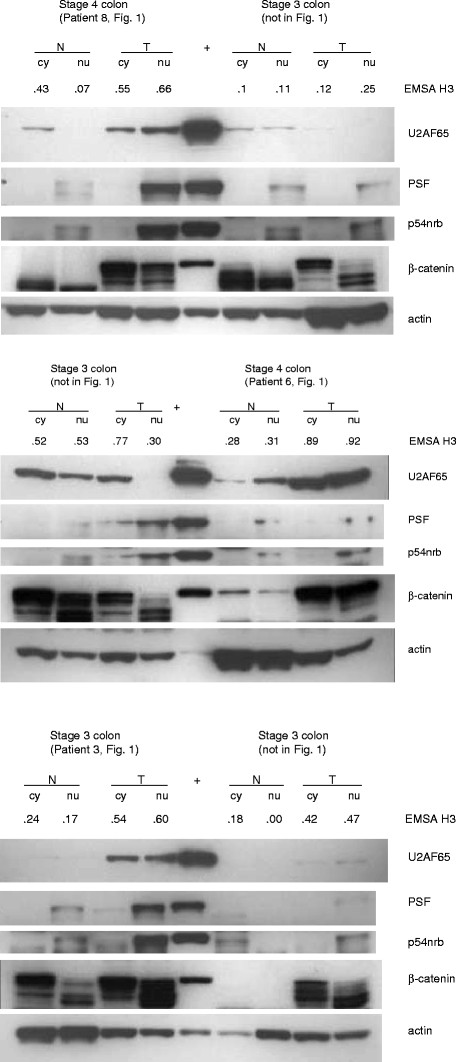
Western blots of U2AF65, PSF, p54nrb, and beta-catenin expression in normal and tumor colorectal tissue extracts. Total protein (25 μg) from cytoplasmic (cy) and nuclear (nu) tissue extracts obtained from six selected patients were separated using 10% SDS-PAGE and electro-transferred to nitrocellulose membranes. Blots were incubated with the antibodies against U2AF65, PSF, p54nrb, beta-catenin, and actin, then the appropriate secondary antibody and detected using chemiluminescence and autoradiography. Each patient’s tumor stage and number which are also included in Figure 1, and corresponding EMSA H3 values are shown above the samples.
Discussion
The data provides support to the hypothesis that the major triplex DNA-binding protein in human cells is more abundant and has higher binding activity in vitro in extracts from colorectal cancer tissues compared to adjacent normal tissues. This increased binding activity correlated significantly with the expression of triplex/G-quadruplex DNA-unwinding helicase WRN, and with the spread of cancer to the lymph nodes, metastasis, and reduced overall survival. The major triplex DNA-binding protein in gel shifts was identified as the U2AF65 splicing factor. U2AF65 expression was higher in more advanced colon tumor stages and correlated significantly with total and truncated beta-catenin expression.
U2AF is a non-small nuclear ribonucleoprotein (snRNP) splicing factor required for the binding of U2 snRNP to the pre-mRNA branch site [41,42]. Purified U2AF is comprised of two polypeptides of 65- (U2AF65) and 35-kDa (U2AF35), respectively. U2AF65 binds to the polypyrimidine (Py) tract adjacent to the 3’ splice site using RNA-recognition motifs and cross-links to the branch point in an ATP-independent manner at the earliest stage of spliceosome formation [43]. Both subunits of U2AF are essential for the viability of many model organisms, such as zebra fish, Drosophila, C. elegans, and S. pombe[44]. Both U2AF65 and U2AF35 shuttle continuously between the nucleus and cytoplasm by a mechanism that involves carrier receptors and is independent from binding to mRNA. It has also been suggested that U2AF participates in the nuclear export of mRNA [45].
U2AF65 binds to single-stranded RNA and recognizes a wide variety of pyrimidine (Py)-tracts. The Py-tracts of higher eukaryotic pre-mRNAs are often interrupted with purines, yet U2AF65 must identify these degenerate Py-tracts for accurate pre-mRNA splicing. Based on in vitro studies, investigators have proposed that U2AF35 assists U2AF65 recruitment to nonconsensus polypyrimidine tracts. Pacheco et al. analyzed the roles of the two U2AF subunits in vivo in the selection of alternative 3' splice sites associated with polypyrimidine tracts of different strengths. Their results revealed a feedback mechanism by which RNA interference-mediated depletion of U2AF65 triggers down regulation of U2AF35 expression. They also showed that knockdown of each U2AF subunit inhibits weak 3' splice site recognition, while over-expression of U2AF65 alone is sufficient to activate selection of this splice site [46,47]. It would be interesting to examine if over-expression of U2AF65 alone in the context of cancer activates splicing of weak or nonconsensus polypyrimidine tracts that could tip the balance of splicing regulation in a subset of cellular transcripts which could promote tumorigenesis.
The proteins we identified in RKO nuclear extracts using biotin triplex DNA affinity were PSF, a 100-kDa protein that also binds to the polypyrimidine tract, and its heterodimeric binding partner p54nrb. We speculate that the 100- and 60-kDa proteins identified in previous studies using Southwestern blotting with HeLa nuclear extracts [16] probed with the same purine triplex DNA probe used in this study are indeed PSF and p54nrb, but this has yet to be tested. Both PSF and p54nrb bind to double-stranded (ds)DNA, single-stranded (ss)DNA, and RNA, and contain DNA- and RNA-binding domains. PSF participates in constitutive pre-mRNA splicing and is a component of later spliceosomal B and C complexes (when U2AF65 is no longer present). PSF and p54nrb also bind and function in nuclear retention of defective RNAs and are involved in transcriptional regulation and the DNA damage response [48-51]. Interestingly, PSF also functions in DNA annealing, where PSF requires ssDNA and dsDNA with sequence homology for their in vitro pairing activity as well as divalent cations. PSF can promote the incorporation of ssDNA within the two separated strands of a homologous superhelical DNA duplex and produce a three-stranded D-loop structure, which is required for homologous recombination. Other splicing factors SF2/ASF and U2AF65 also caused DNA annealing but could not form D loops [52]. PSF and p54nrb, as well as GRSF-1, YB-1, and polypyrimidine tract-binding protein (PTB) also bind to the MYC family of internal ribosome entry sites (IRES) and positively regulate translation of the Myc family of oncoproteins in vitro and in vivo[53]. Protein array data in this study showed that expression of both PSF and p54nrb in colorectal tissue extracts correlated significantly with c-Myc expression levels, which is consistent with a role for PSF and p54nrb in the regulation of c-Myc protein expression.
Researchers identified both U2AF and PSF, as well as hnRNP C and PTB, as RNA-binding proteins that bind to two regions 3’ of the (CUG)n repeat expansion in the 3’-UTR of the DMPK gene, where expansion of this trinucleotide repeat causes the neuromuscular disorder myotonic dystrophy [54]. Their study explored RNA-binding proteins interacting with non-CUG regions or higher order structures in the DMPK 3’-UTR that may be involved in RNA-mediated pathogenesis. Their finding that both U2AF and PSF can bind near this triplet repeat sequence with the potential to form higher order structures such as triplexes is consistent with our data on biotin triplex DNA affinity identification of both U2AF65 and PSF. Another group identified an RNA/protein complex in both Drosophila and 293 cells that consisted of expanded CAG RNA, U2AF65, and the NXF1 nuclear export receptor, providing further evidence that in other models, U2AF65 interacts with these triplet repeat sequences [55]. We believe that the purine triplex DNA EMSA probe can be a surrogate multiplex nucleic acid structure that acts as a “bait and hook” to capture proteins that may be binding D-loops, R-loops, triplexes, G-quadruplexes, or other multi-stranded structures containing Hoogsteen or reverse Hoogsteen base pairs in vivo.
PTB also binds to polypyrimidine tracts in pre-mRNAs, and numerous studies have shown that PTB competes with U2AF65 for binding to these sequences [56-61]. Since PSF is a PTB-associated protein, binding competition between PSF and U2AF65 may be possible as well, which may explain why we identified both PSF with the biotinylated triplex DNA in RKO nuclear extracts and U2AF65 in RKO cytoplasmic extracts. Gama-Carvalho and colleagues performed immunoprecipitation of U2AF65- and PTB-associated RNAs from HeLa cells followed by microarray analysis to determine which mRNAs are associated with these two splicing factors that can compete for binding to polypyrimidine tracts [36]. Among U2AF65-associated mRNAs was a predominance of transcription factors and cell cycle regulators, whereas PTB-associated transcripts were enriched in mRNAs that encode proteins implicated in intracellular transport, vesicle trafficking, and apoptosis.
Related to cancer, researchers found that 2 of 14 patients with malignant mesothelioma, a pulmonary malignancy, had antibodies against U2AF65 using the SEREX technique (serologic identification by recombinant expression cloning) [62]. Additionally, a patient with liver cirrhosis that progressed to hepatocellular carcinoma had antinuclear antibodies that recognized a nuclear protein putatively identified as U2AF65 [63]. Other splicing factors, most notably SFRS1 (ASF/SF2), are reported to be over-expressed in colon, thyroid, kidney, lung and breast cancer cells [64]. Other splicing factors shown to be over-expressed in colorectal cancer cells are hnRNP-F and –K, SPF45, and SRPK1 [64]. However, the present report is the first to describe correlation of increased expression or binding activity of U2AF65 in primary colorectal tumors with tumor stage, lymph node disease, metastasis and reduced overall survival.
Why U2AF65 is over-expressed in colorectal tumor cells, and whether this over-expression is important to the development and/or progression of colorectal cancer or a passive effect of general gene deregulation are unknown. About 75% of sporadic colorectal cancers are characterized by a chromosomal instability (CIN) phenotype. The most common reported chromosomal losses involve 5q (APC), 18q (DCC), and 17p (p53), while the most common gains involve 8q and 20q. The gene encoding U2AF65 (U2AF2) is located at c19q13.42. Chromosomal amplifications at c19q13.42 have been found in a rare embryonal tumor using array CGH and FISH [65,66]. Other groups have reported amplifications or aberrations at c19q13 in colorectal tumors, particularly in liver metastases compared to primary tumors [67], and in other solid tumors including pancreatic [68] and ovarian [69].
Regarding genomic instability, Vasquez and colleagues recently showed that both non-B DNA sequences and WRN helicase deficiency induce mutations characterized by single base changes, mostly at C-G base pairs, in an additive but not synergistic manner [70]. Because no synergy was observed, the authors concluded that a role for WRN in reducing mutation frequencies via a mechanism dependent on its cellular helicase activity (for example, of non-B DNA sequences) is unlikely. Their data do not directly support our present hypothesis, which is similar to their hypothesis that if one function of the WRN helicase were to resolve non-B (triplex and Z-DNA) structures, as observed in vitro, then mutation frequencies may be higher in WRN-deficient cells than in WRN-wild type cells because both the number and stability of such structures would be greater in WRN-deficient cells. However, they did verify that purified WRN protein was able to unwind the third purine-rich strand of a synthetic triplex in vitro. Although our data suggest a correlation between expression of the WRN helicase with triplex DNA-binding activity in both normal and tumor tissue extracts, defining a functional role and mechanism of non-B DNA unwinding activity by WRN helicase and G*G multiplex binding (for example, by U2AF65) will require further study.
Beta-catenin, as a transcription factor complexed with TCF4, is known to upregulate expression of many relevant proteins in colorectal cancer, such as c-myc, cyclin D1, LEF-1, CD44, and c-jun. Whether beta-catenin influences the expression of U2AF65 is unknown, but a search of transcription factor binding sites in the U2AF65 (U2AF2) gene promoter did not indicate any beta-catenin or TCF family transcription factor sites among the 55 high-scoring (>85%) sites we identified (Cold Spring Harbor Laboratory Mammalian Promoter Database http://rulai.cshl.edu/CSHLmpd2/; Transcription Factor Search http://www.cbrc.jp/research/db/TFSEARCH.html). Similarly, mining through microarray expression studies revealed no reports describing U2AF65 (U2AF2) as a beta-catenin, TCF4, or Wnt target gene (NCBI GEO; R Nusse Wnt/Beta catenin targets list: http://www.stanford.edu/~rnusse/pathways/targets.html). The biological significance of the correlation of U2AF65 and beta-catenin expression in colorectal tumor tissues, such as if beta-catenin as a transcription factor affects U2AF65 expression, or if U2AF65 as a splicing factor affects the splicing or expression of beta-catenin, remains to be determined.
Several studies have examined the interaction of beta-catenin with splicing factors and the role of beta-catenin in mRNA splicing. Researchers identified alternative splicing of SLC39A14, a divalent cation transporter, in colorectal tumors and found it to be regulated by the Wnt pathway, probably through regulation of splicing factor SRSF1 [71]. The beta-catenin/TCF4 pathway also modifies alternative splicing through modulation of expression of splicing factors SRp20 [72] and SF1 [73] and direct interaction with FUS/TLS (translocated in liposarcoma) and various other RNA-binding proteins, including p54nrb [74]. Others have shown that beta-catenin regulates multiple steps of RNA metabolism in colon cancer cells and may coordinate RNA metabolism [75].
Authors have also reported identification of truncated beta-catenin isoforms, mostly in colorectal cancer cells. In primary colorectal tumors, a relatively small percent (7 of 58 examined) contained somatic interstitial deletions that included all or part of exon 3 of the beta-catenin gene, and RT-PCR analysis from 3 of the 7 tumors detected transcripts that lacked exon 3 and the presence of the normal transcript [39]. Researchers also detected two novel beta-catenin mRNA splice variants in the SW480 colon cancer cell line and in primary colorectal tumors [38]. A truncated beta-catenin protein of 80-kDa was also detected in three colorectal metastases to the liver [40]. Several of these isoforms have truncations in the NH2-terminus of the protein that produce deletions of key serine and threonines that are phosphorylated by GSK-3 beta, which is important for proteosomal degradation, which was hypothesized to stabilize the protein and have a dominant oncogenic effect [76]. Data from this and other studies lead us to speculate that U2AF65 could be binding to a multi-stranded nucleic acid structure such as R-loops, D-loops, or G-quartet mRNA in vivo that is mimicked by the purine triplex DNA probe in our study, and that overexpression or increased EMSA binding activity of U2AF65 in tumor tissues could cause deregulation of mRNA splicing and protein isoform expression, such as beta-catenin, that could contribute to colorectal cancer initiation and/or progression.
Conclusions
We found that increased triplex DNA-binding activity in colorectal tumor extracts in vitro is associated with WRN helicase expression, increased total beta-catenin expression, lymph node disease, metastasis, and reduced overall survival in patients with colorectal cancer. Multifunctional splicing factor U2AF65 was identified as the major triplex-binding protein in human tissues and cell lines. Increased expression of U2AF65 is also associated with expression of splicing factors PSF and p54nrb, a higher tumor stage, and increased truncation of beta-catenin in colorectal tumors. We believe that our results contribute to and generate interest in the growing fields of alternative non-B DNA structures and genomic instability, aberrantly regulated splicing factors, mRNA splicing and protein isoforms related to cancer both as basic research objectives regarding the etiology of cancer and cancer diversity and as novel translational research in the search for promising prognostic, diagnostic and targeting tools.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LDN, HM, MWVD and HA designed the study; CB, DB, PK, and GM performed all statistical analysis and collected patient clinical data; LDN and HM performed all experiments; UK supervised reverse phase protein array experiments; LDN, DPH, MWVD and HA wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Figure S1. Electrophoretic Mobility Shift Assay (EMSA) of patient tissue lysates and HeLa nuclear extract with triplex and parent duplex DNA probes. 33P‐labeled purine‐motif duplex or triplex DNA (1 nM) was complexed with 5 μg protein from normal tissue cytoplasmic (N cy), normal nuclear (N nu), tumor tissue cytoplasmic (T cy) or tumor nuclear (T nu) extracts of colorectal cancer patients. 1.25 μg HeLa nuclear extract (H) was used as a control in lanes 6 and 12. Purine triplex probe alone is in lane 1 and duplex probe alone is in lane 7. Figure S2a. Electrophoretic Mobility Shift Assay (EMSA) of Cytoplasmic and Nuclear Extracts from Eight Colorectal Cancer Cell Lines with Purine triplex DNA. 33P‐labeled purine‐motif triplex DNA (1 nM) was complexed with 1.25 μg total protein from cytoplasmic (cy) or nuclear (nuc) extracts from eight colorectal cancer cell lines. 1.25 μg HeLa cytoplasmic and nuclear extracts were used as positive (+) controls. Each reaction also contained 2 μg poly (dI‐dC) carrier DNA. The purine triplex DNA probe alone is shown in lane 1. Figure S2b. Western blots showing expression of three candidate triplex DNA‐binding proteins in eight colorectal cancer cell lines. Total protein (25 μg) from cytoplasmic (cy) and nuclear (nu) extracts from eight colorectal cancer cell lines were separated using 10% SDS‐PAGE and electro‐transferred to nitrocellulose membranes. Blots were incubated with the antibodies against PSF, U2AF65, p54nrb, beta‐catenin, and actin, then the appropriate secondary antibody and detected using chemiluminescence and autoradiography. Figure S3. Lack of a super‐shifted H3 band in RKO nuclear extract by super‐shift EMSA with antibodies against PSF and p54nrb. 33P‐labeled triplex DNA (1 nM) was complexed with 1.5 μg total protein from RKO nuclear extracts (lanes 2‐9). Lane 1, triplex DNA probe alone; Lane 2, no antibody; lane 3, 400 ng anti‐U2AF65 antibody MC3; lane 4, 1000 ng anti‐U2AF65 antibody MC3; lane 5, 400 ng anti‐PSF antibody; lane 6 1000 ng anti‐PSF antibody; lane 7, 400 ng anti‐p54nrb antibody; lane 8, 1000 ng anti‐p54nrb antibody; lane 9, mouse IgG antibody (negative control). Each reaction also contained 2 μg poly (dI‐dC) carrier DNA. Figure S4. Quantitation of Protein Expression of PSF, U2AF65, p54nrb, and beta‐catenin obtained from six colorectal cancer patients’ tissue extracts. Autoradiographs from Western blots in Figure 6 were scanned, and protein expression bands were quantitated using NIH Image J. Protein expression was normalized by dividing by the samples’ corresponding actin value and graphed using Graph Pad. Figure S5. Beta‐catenin Expression by Tumor type and Stage. Western blots using an anti‐beta‐catenin antibody to examine expression in patient extracts were described for Figure 6. Beta‐catenin expression values were normalized by dividing the actin expression value in each extract, and plotted according to colon or rectum tumor stage using the R program. N cyto, cytoplasmic normal tissue extracts; N nuc, nuclear normal tissue extracts; T cyto, cytoplasmic tumor tissue extracts; T nuc, nuclear tumor tissue extracts.
DB-Triplexdata.
PK Statistical analysis Triplex.
DBuergy Correlations(1).
Daniel Apr 5(1).
histograms_proteins_groups.
Table S1. RPPA antibodies and Spearman correlation p values.
Contributor Information
Laura D Nelson, Email: lnelson@mdanderson.org.
Christian Bender, Email: christianbender81@googlemail.com.
Heiko Mannsperger, Email: hmannsperger@metasystems.de.
Daniel Buergy, Email: Daniel.Buergy@umm.de.
Patryk Kambakamba, Email: patryk.kambakamba@yahoo.de.
Giridhar Mudduluru, Email: Mudduluru@umm.de.
Ulrike Korf, Email: u.korf@dkfz-heidelberg.de.
Dennis Hughes, Email: dphughes@mdanderson.org.
Michael W Van Dyke, Email: mvandyke@email.wcu.edu.
Heike Allgayer, Email: Heike.Allgayer@umm.de.
Acknowledgments
We thank Mohammed Abba, Irfan Asangani, Nitin Patil, Christian Schmidt, and Frederick Wenz for insightful discussions and assistance. We also thank Donald Norwood for critical reading of the manuscript.
LDN is supported by a Guest Scientist Scholarship from the German Cancer Research Center (DKFZ) and the National Cancer Institute (NCI; 1RO1CA149501-01A1). CB is supported by the network SB-Cancer in the Helmholtz Alliance on Systems Biology, Heidelberg Germany. HM is supported by the German Federal Ministry of Education and Science in the framework of the program for medical genome research (01GS0890 and 01GS0864). DB is supported by the Department of Anesthesiology and Intensive Care Medicine, Medical Faculty Mannheim, University of Heidelberg, Mannheim Germany.
PK is supported by Experimental Surgery, Medical Faculty Mannheim, University of Heidelberg, Mannheim Germany. GM is supported by the Stiftung fur Krebs- und Scharlachforschung Mannheim, University of Heidelberg, Mannheim Germany and Wilhelm Sander Foundation, Munich, Germany. UK is supported by the German Federal Ministry of Education and Science in the framework of the program for medical genome research (01GS0890 and 01GS0864), Heidelberg. DPH is supported by the National Cancer Institute (NCI; 1RO1CA149501-01A1).
MWVD is supported by the North Carolina Biotechnology Center. HA is supported by Alfried Krupp von Bohlen and Halbach Foundation (Award for Young Full Professors), Essen, Hella-Bühler-Foundation, Heidelberg, Dr. Ingrid zu Solms Foundation, Frankfurt/Main, the Hector Foundation, Weinheim, Germany, the FRONTIER Excellence Initiative of the University of Heidelberg, the BMBF, Bonn, Germany, the Walter Schulz Foundation, Munich, Germany, the German-Israeli Project Cooperation, DKFZ Heidelberg, and the Deutsche Krebshilfe, Germany. Wilhelm Sander Foundation, Munich, Germany, and the DKFZ-MOST German Israel Program.
References
- Frank-Kamenetskii MD, Mirkin SM. Triplex DNA structures. Annu Rev Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- Patel DJ, Phan AT, Kuryavyi V. Human telomere, oncogenic promoter and 5'-UTR G-quadruplexes: diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J Biol Chem. 2007;282:32433–32441. doi: 10.1074/jbc.M704618200. [DOI] [PubMed] [Google Scholar]
- Nikolova EN, Kim E, Wise AA, O'Brien PJ, Andricioaei I, Al-Hashimi HM. Transient Hoogsteen base pairs in canonical duplex DNA. Nature. 2011;470:498–502. doi: 10.1038/nature09775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Bacolla A, Wells RD. Non-B DNA conformations as determinants of mutagenesis and human disease. Mol Carcinog. 2009;48:273–285. doi: 10.1002/mc.20507. [DOI] [PubMed] [Google Scholar]
- Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- Napierala M, Bacolla A, Wells RD. Increased negative superhelical density in vivo enhances the genetic instability of triplet repeat sequences. J Biol Chem. 2005;280:37366–37376. doi: 10.1074/jbc.M508065200. [DOI] [PubMed] [Google Scholar]
- Zhao J, Bacolla A, Wang G, Vasquez KM. Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci. 2010;67:43–62. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Wang G, Vasquez KM. DNA triple helices: biological consequences and therapeutic potential. Biochimie. 2008;90:1117–1130. doi: 10.1016/j.biochi.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Collins JR, Gold B, Chuzhanova N, Yi M, Stephens RM, Stefanov S, Olsh A, Jakupciak JP, Dean M. et al. Long homopurine*homopyrimidine sequences are characteristic of genes expressed in brain and the pseudoautosomal region. Nucleic Acids Res. 2006;34:2663–2675. doi: 10.1093/nar/gkl354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joos S, Haluska FG, Falk MH, Henglein B, Hameister H, Croce CM, Bornkamm GW. Mapping chromosomal breakpoints of Burkitt's t(8;14) translocations far upstream of c-myc. Cancer Res. 1992;52:6547–6552. [PubMed] [Google Scholar]
- Wang G, Vasquez KM. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cer RZ, Bruce KH, Mudunuri US, Yi M, Volfovsky N, Luke BT, Bacolla A, Collins JR, Stephens RM. Non-B DB: a database of predicted non-B DNA-forming motifs in mammalian genomes. Nucleic Acids Res. 2011;39:D383–D391. doi: 10.1093/nar/gkq1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/S0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- Musso M, Nelson LD, Van Dyke MW. Characterization of purine-motif triplex DNA-binding proteins in HeLa extracts. Biochemistry. 1998;37:3086–3095. doi: 10.1021/bi9717486. [DOI] [PubMed] [Google Scholar]
- Nelson LD, Musso M, Van Dyke MW. The Yeast STM1 Gene Encodes a Purine Motif Triple Helical DNA-binding Protein. J Biol Chem. 2000;275:5573–5581. doi: 10.1074/jbc.275.8.5573. [DOI] [PubMed] [Google Scholar]
- Van Dyke MW, Nelson LD, Weilbaecher RG, Mehta DV. Stm1p, a G4 Quadruplex and Purine Motif Triplex Nucleic Acid-binding Protein, Interacts with Ribosomes and Subtelomeric Y' DNA in Saccharomyces cerevisiae. J Biol Chem. 2004;279:24323–24333. doi: 10.1074/jbc.M401981200. [DOI] [PubMed] [Google Scholar]
- Frantz JD, Gilbert W. A yeast gene product, G4p2, with a specific affinity for quadruplex nucleic acids. J Biol Chem. 1995;270:9413–9419. doi: 10.1074/jbc.270.16.9413. [DOI] [PubMed] [Google Scholar]
- Ozgenc A, Loeb LA. Current advances in unraveling the function of the Werner syndrome protein. Mutat Res. 2005;577:237–251. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Frank B, Hoffmeister M, Klopp N, Illig T, Chang-Claude J, Brenner H. Colorectal cancer and polymorphisms in DNA repair genes WRN, RMI1 and BLM. Carcinogenesis. 2010;31:442–445. doi: 10.1093/carcin/bgp293. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, Liu Z, Yamamoto H, Loda M, Fuchs CS, Ogino S. WRN promoter methylation possibly connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod Pathol. 2008;21:150–158. doi: 10.1038/modpathol.3800996. [DOI] [PubMed] [Google Scholar]
- Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–499. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- Martin ES, Tonon G, Sinha R, Xiao Y, Feng B, Kimmelman AC, Protopopov A, Ivanova E, Brennan C, Montgomery K. et al. Common and distinct genomic events in sporadic colorectal cancer and diverse cancer types. Cancer Res. 2007;67:10736–10743. doi: 10.1158/0008-5472.CAN-07-2742. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J. et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N. et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with 'mini-extracts', prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM. Current protocols in molecular biology. Pa: Greene Pub. Associates. J. Wiley, order fulfillment, Brooklyn, N.Y. Media; 1987. [Google Scholar]
- Mannsperger HA, Uhlmann S, Schmidt C, Wiemann S, Sahin O, Korf U. RNAi-based validation of antibodies for reverse phase protein arrays. Proteome Sci. 2010;8:69. doi: 10.1186/1477-5956-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loebke C, Sueltmann H, Schmidt C, Henjes F, Wiemann S, Poustka A, Korf U. Infrared-based protein detection arrays for quantitative proteomics. Proteomics. 2007;7:558–564. doi: 10.1002/pmic.200600757. [DOI] [PubMed] [Google Scholar]
- Mannsperger HA, Gade S, Henjes F, Beissbarth T, Korf U. RPPanalyzer: Analysis of reverse-phase protein array data. Bioinformatics. 2010;26:2202–2203. doi: 10.1093/bioinformatics/btq347. [DOI] [PubMed] [Google Scholar]
- Straub T, Grue P, Uhse A, Lisby M, Knudsen BR, Tange TO, Westergaard O, Boege F. The RNA-splicing factor PSF/p54 controls DNA-topoisomerase I activity by a direct interaction. J Biol Chem. 1998;273:26261–26264. doi: 10.1074/jbc.273.41.26261. [DOI] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR, Frank J, Manley JL. Molecular architecture of the human pre-mRNA 3' processing complex. Mol Cell. 2009;33:365–376. doi: 10.1016/j.molcel.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Carvalho M, Carvalho MP, Kehlenbach A, Valcarcel J, Carmo-Fonseca M. Nucleocytoplasmic Shuttling of Heterodimeric Splicing Factor U2AF. J Biol Chem. 2001;276:13104–13112. doi: 10.1074/jbc.M008759200. [DOI] [PubMed] [Google Scholar]
- Gama-Carvalho M, Barbosa-Morais NL, Brodsky AS, Silver PA, Carmo-Fonseca M. Genome-wide identification of functionally distinct subsets of cellular mRNAs associated with two nucleocytoplasmic-shuttling mammalian splicing factors. Genome Biol. 2006;7:R113. doi: 10.1186/gb-2006-7-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Carvalho M, Krauss RD, Chiang L, Valcarcel J, Green MR, Carmo-Fonseca M. Targeting of U2AF65 to Sites of Active Splicing in the Nucleus. J Cell Biol. 1997;137:975–987. doi: 10.1083/jcb.137.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisil H, Herrmann A, Butherus K, Pirson S, Reich JG, Kemmner W. Verification of predicted alternatively spliced Wnt genes reveals two new splice variants (CTNNB1 and LRP5) and altered Axin-1 expression during tumour progression. BMC Genomics. 2006;7:148. doi: 10.1186/1471-2164-7-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwao K, Nakamori S, Kameyama M, Imaoka S, Kinoshita M, Fukui T, Ishiguro S, Nakamura Y, Miyoshi Y. Activation of the beta-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res. 1998;58:1021–1026. [PubMed] [Google Scholar]
- Hugh TJ, Dillon SA, O'Dowd G, Getty B, Pignatelli M, Poston GJ, Kinsella AR. beta-catenin expression in primary and metastatic colorectal carcinoma. Int J Cancer. 1999;82:504–511. doi: 10.1002/(SICI)1097-0215(19990812)82:4<504::AID-IJC6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Green MR. Biochemical characterization of U2 snRNP auxiliary factor: an essential pre-mRNA splicing factor with a novel intranuclear distribution. EMBO J. 1991;10:207–214. doi: 10.1002/j.1460-2075.1991.tb07937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin B, Zamore PD, Green MR. A factor, U2AF, is required for U2 snRNP binding and splicing complex assembly. Cell. 1988;52:207–219. doi: 10.1016/0092-8674(88)90509-0. [DOI] [PubMed] [Google Scholar]
- Gaur RK, Valcarcel J, Green MR. Sequential recognition of the pre-mRNA branch point by U2AF65 and a novel spliceosome-associated 28-kDa protein. RNA. 1995;1:407–417. [PMC free article] [PubMed] [Google Scholar]
- Sridharan V, Heimiller J, Singh R. Genomic mRNA profiling reveals compensatory mechanisms for the requirement of the essential splicing factor U2AF. Mol Cell Biol. 2011;31:652–661. doi: 10.1128/MCB.01000-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin AS, Tan W, Bear J, Smulevitch S, Felber BK. U2AF participates in the binding of TAP (NXF1) to mRNA. J Biol Chem. 2002;277:3935–3942. doi: 10.1074/jbc.M107598200. [DOI] [PubMed] [Google Scholar]
- Pacheco TR, Moita LF, Gomes AQ, Hacohen N, Carmo-Fonseca M. RNA interference knockdown of hU2AF35 impairs cell cycle progression and modulates alternative splicing of Cdc25 transcripts. Mol Biol Cell. 2006;17:4187–4199. doi: 10.1091/mbc.E06-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco TR, Coelho MB, Desterro JMP, Mollet I, Carmo-Fonseca M. In Vivo Requirement of the Small Subunit of U2AF for Recognition of a Weak 3' Splice Site. Mol Cell Biol. 2006;26:8183–8190. doi: 10.1128/MCB.00350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shav-Tal Y, Zipori D. PSF and p54(nrb)/NonO–multi-functional nuclear proteins. FEBS Lett. 2002;531:109–114. doi: 10.1016/S0014-5793(02)03447-6. [DOI] [PubMed] [Google Scholar]
- Salton M, Lerenthal Y, Wang SY, Chen DJ, Shiloh Y. Involvement of matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle. 2010;9:1568–1576. doi: 10.4161/cc.9.8.11298. [DOI] [PubMed] [Google Scholar]
- Li S, Kuhne WW, Kulharya A, Hudson FZ, Ha K, Cao Z, Dynan WS. Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 2009;37:6746–6753. doi: 10.1093/nar/gkp741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladen CL, Udayakumar D, Takeda Y, Dynan WS. Identification of the polypyrimidine tract binding protein-associated splicing factor.p54(nrb) complex as a candidate DNA double-strand break rejoining factor. J Biol Chem. 2005;280:5205–5210. doi: 10.1074/jbc.M412758200. [DOI] [PubMed] [Google Scholar]
- Akhmedov AT, Lopez BS. Human 100-kDa homologous DNA-pairing protein is the splicing factor PSF and promotes DNA strand invasion. Nucleic Acids Res. 2000;28:3022–3030. doi: 10.1093/nar/28.16.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold LC, Spriggs KA, Haines SJ, Dobbyn HC, Hayes C, de Moor CH, Lilley KS, Bushell M, Willis AE. Identification of internal ribosome entry segment (IRES)-trans-acting factors for the Myc family of IRESs. Mol Cell Biol. 2008;28:40–49. doi: 10.1128/MCB.01298-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Mahadevan MS. Myotonic dystrophy: the role of the CUG triplet repeats in splicing of a novel DMPK exon and altered cytoplasmic DMPK mRNA isoform ratios. Mol Cell. 2000;5:959–967. doi: 10.1016/S1097-2765(00)80261-0. [DOI] [PubMed] [Google Scholar]
- Tsoi H, Lau CK, Lau KF, Chan HY. Perturbation of U2AF65/NXF1-mediated RNA nuclear export enhances RNA toxicity in polyQ diseases. Hum Mol Genet. 2011;20:3787–3797. doi: 10.1093/hmg/ddr297. [DOI] [PubMed] [Google Scholar]
- Castelo-Branco P, Furger A, Wollerton M, Smith C, Moreira A, Proudfoot N. Polypyrimidine tract binding protein modulates efficiency of polyadenylation. Mol Cell Biol. 2004;24:4174–4183. doi: 10.1128/MCB.24.10.4174-4183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo JM, Majos N, Bonnal S, Martinez C, Castelo R, Guigo R, Bilbao D, Valcarcel J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19:475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Sharma S, Falick AM, Black DL. Polypyrimidine tract binding protein blocks the 5' splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol Cell. 2005;19:485–496. doi: 10.1016/j.molcel.2005.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauliere J, Sureau A, Expert-Bezancon A, Marie J. The polypyrimidine tract binding protein (PTB) represses splicing of exon 6B from the beta-tropomyosin pre-mRNA by directly interfering with the binding of the U2AF65 subunit. Mol Cell Biol. 2006;26:8755–8769. doi: 10.1128/MCB.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinescu V, Loomis PA, Ehmann S, Beales M, Potashkin JA. Regulation of retention of FosB intron 4 by PTB. PLoS One. 2007;2:e828. doi: 10.1371/journal.pone.0000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosme RS, Yamamura Y, Tang Q. Roles of polypyrimidine tract binding proteins in major immediate-early gene expression and viral replication of human cytomegalovirus. J Virol. 2009;83:2839–2850. doi: 10.1128/JVI.02407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson C, Callow M, Stevenson S, Scott B, Robinson BW, Lake RA. Serologic responses in patients with malignant mesothelioma: evidence for both public and private specificities. Am J Respir Cell Mol Biol. 2000;22:550–556. doi: 10.1165/ajrcmb.22.5.3930. [DOI] [PubMed] [Google Scholar]
- Imai H, Chan EK, Kiyosawa K, Fu XD, Tan EM. Novel nuclear autoantigen with splicing factor motifs identified with antibody from hepatocellular carcinoma. J Clin Invest. 1993;92:2419–2426. doi: 10.1172/JCI116848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosso AR, Martins S, Carmo-Fonseca M. The emerging role of splicing factors in cancer. EMBO Rep. 2008;9:1087–1093. doi: 10.1038/embor.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov A, Remke M, Gessi M, Ryzhova M, Hielscher T, Witt H, Tobias V, Buccoliero AM, Sardi I, Gardiman MP. et al. Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic rosettes. Acta Neuropathol. 2010;120:253–260. doi: 10.1007/s00401-010-0688-8. [DOI] [PubMed] [Google Scholar]
- Pfister S, Remke M, Castoldi M, Bai AH, Muckenthaler MU, Kulozik A, von Deimling A, Pscherer A, Lichter P, Korshunov A. Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol. 2009;117:457–464. doi: 10.1007/s00401-008-0467-y. [DOI] [PubMed] [Google Scholar]
- Sayagues JM, Abad MM, Melchor HB, Gutierrez ML, Gonzalez-Gonzalez M, Jensen E, Bengoechea O, Fonseca E, Orfao A, Munoz-Bellvis L. Intratumoural cytogenetic heterogeneity of sporadic colorectal carcinomas suggests several pathways to liver metastasis. J Pathol. 2010;221:308–319. doi: 10.1002/path.2712. [DOI] [PubMed] [Google Scholar]
- Kuuselo R, Simon R, Karhu R, Tennstedt P, Marx AH, Izbicki JR, Yekebas E, Sauter G, Kallioniemi A. 19q13 amplification is associated with high grade and stage in pancreatic cancer. Genes Chromosomes Cancer. 2010;49:569–575. doi: 10.1002/gcc.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayani J, Marrano P, Graham C, Zheng Y, Li L, Katsaros D, Lassus H, Butzow R, Squire JA, Diamandis EP. Genomic instability and copy-number heterogeneity of chromosome 19q, including the kallikrein locus, in ovarian carcinomas. Mol Oncol. 2011;5:48–60. doi: 10.1016/j.molonc.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Wang G, Jain A, Chuzhanova NA, Cer RZ, Collins JR, Cooper DN, Bohr VA, Vasquez KM. Non-B DNA-forming sequences and WRN deficiency independently increase the frequency of base substitution in human cells. J Biol Chem. 2011;286:10017–10026. doi: 10.1074/jbc.M110.176636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsen K, Mansilla F, Schepeler T, Oster B, Rasmussen MH, Dyrskjot L, Karni R, Akerman M, Krainer AR, Laurberg S. et al. Alternative splicing of SLC39A14 in colorectal cancer is regulated by the Wnt pathway. Mol Cell Proteomics. 2011;10:M110 002998. doi: 10.1074/mcp.M110.002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves V, Matos P, Jordan P. The beta-catenin/TCF4 pathway modifies alternative splicing through modulation of SRp20 expression. RNA. 2008;14:2538–2549. doi: 10.1261/rna.1253408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitashige M, Naishiro Y, Idogawa M, Honda K, Ono M, Hirohashi S, Yamada T. Involvement of splicing factor-1 in beta-catenin/T-cell factor-4-mediated gene transactivation and pre-mRNA splicing. Gastroenterology. 2007;132:1039–1054. doi: 10.1053/j.gastro.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Sato S, Idogawa M, Honda K, Fujii G, Kawashima H, Takekuma K, Hoshika A, Hirohashi S, Yamada T. beta-catenin interacts with the FUS proto-oncogene product and regulates pre-mRNA splicing. Gastroenterology. 2005;129:1225–1236. doi: 10.1053/j.gastro.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kwak HY, Hur J, Kim IA, Yang JS, Park MW, Yu J, Jeong S. beta-catenin regulates multiple steps of RNA metabolism as revealed by the RNA aptamer in colon cancer cells. Cancer Res. 2007;67:9315–9321. doi: 10.1158/0008-5472.CAN-07-1128. [DOI] [PubMed] [Google Scholar]
- Wagenaar RA, Crawford HC, Matrisian LM. Stabilized beta-catenin immortalizes colonic epithelial cells. Cancer Res. 2001;61:2097–2104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Electrophoretic Mobility Shift Assay (EMSA) of patient tissue lysates and HeLa nuclear extract with triplex and parent duplex DNA probes. 33P‐labeled purine‐motif duplex or triplex DNA (1 nM) was complexed with 5 μg protein from normal tissue cytoplasmic (N cy), normal nuclear (N nu), tumor tissue cytoplasmic (T cy) or tumor nuclear (T nu) extracts of colorectal cancer patients. 1.25 μg HeLa nuclear extract (H) was used as a control in lanes 6 and 12. Purine triplex probe alone is in lane 1 and duplex probe alone is in lane 7. Figure S2a. Electrophoretic Mobility Shift Assay (EMSA) of Cytoplasmic and Nuclear Extracts from Eight Colorectal Cancer Cell Lines with Purine triplex DNA. 33P‐labeled purine‐motif triplex DNA (1 nM) was complexed with 1.25 μg total protein from cytoplasmic (cy) or nuclear (nuc) extracts from eight colorectal cancer cell lines. 1.25 μg HeLa cytoplasmic and nuclear extracts were used as positive (+) controls. Each reaction also contained 2 μg poly (dI‐dC) carrier DNA. The purine triplex DNA probe alone is shown in lane 1. Figure S2b. Western blots showing expression of three candidate triplex DNA‐binding proteins in eight colorectal cancer cell lines. Total protein (25 μg) from cytoplasmic (cy) and nuclear (nu) extracts from eight colorectal cancer cell lines were separated using 10% SDS‐PAGE and electro‐transferred to nitrocellulose membranes. Blots were incubated with the antibodies against PSF, U2AF65, p54nrb, beta‐catenin, and actin, then the appropriate secondary antibody and detected using chemiluminescence and autoradiography. Figure S3. Lack of a super‐shifted H3 band in RKO nuclear extract by super‐shift EMSA with antibodies against PSF and p54nrb. 33P‐labeled triplex DNA (1 nM) was complexed with 1.5 μg total protein from RKO nuclear extracts (lanes 2‐9). Lane 1, triplex DNA probe alone; Lane 2, no antibody; lane 3, 400 ng anti‐U2AF65 antibody MC3; lane 4, 1000 ng anti‐U2AF65 antibody MC3; lane 5, 400 ng anti‐PSF antibody; lane 6 1000 ng anti‐PSF antibody; lane 7, 400 ng anti‐p54nrb antibody; lane 8, 1000 ng anti‐p54nrb antibody; lane 9, mouse IgG antibody (negative control). Each reaction also contained 2 μg poly (dI‐dC) carrier DNA. Figure S4. Quantitation of Protein Expression of PSF, U2AF65, p54nrb, and beta‐catenin obtained from six colorectal cancer patients’ tissue extracts. Autoradiographs from Western blots in Figure 6 were scanned, and protein expression bands were quantitated using NIH Image J. Protein expression was normalized by dividing by the samples’ corresponding actin value and graphed using Graph Pad. Figure S5. Beta‐catenin Expression by Tumor type and Stage. Western blots using an anti‐beta‐catenin antibody to examine expression in patient extracts were described for Figure 6. Beta‐catenin expression values were normalized by dividing the actin expression value in each extract, and plotted according to colon or rectum tumor stage using the R program. N cyto, cytoplasmic normal tissue extracts; N nuc, nuclear normal tissue extracts; T cyto, cytoplasmic tumor tissue extracts; T nuc, nuclear tumor tissue extracts.
DB-Triplexdata.
PK Statistical analysis Triplex.
DBuergy Correlations(1).
Daniel Apr 5(1).
histograms_proteins_groups.
Table S1. RPPA antibodies and Spearman correlation p values.