

Abstract
Transforming growth factor-β (TGF-β) and its signaling pathways are important mediators in the suppression of cancers of the gastrointestinal (GI) tract. TGF-β is released from cells in a latent complex consisting of TGF-β, the TGF-β propeptide (LAP) and a latent TGF-β binding protein (LTBP). We previously generated mice in which the LTBP-binding cysteine residues in LAP TGF-β1 were mutated to serine precluding covalent interactions with LTBP. These Tgfb1C33S/C33S mice develop multiorgan inflammation and tumors consistent with reduced TGF-β1 activity. To test whether further reduction in active TGF-β levels would yield additional tumors and a phenotype more similar to Tgfb1-/- mice, we generated mice that express TGF-β1C33S and are deficient in either integrin β8 or TSP-1, known activators of latent TGF-β1. In addition we generated mice that have one mutant allele and one null allele at the Tgfb1 locus, reasoning that these mice should synthesize half the total amount of TGF-β1 as Tgfb1C33S/C33S mice and the amount of active TGF-β1 would be correspondingly decreased compared to Tgfb1C33S/C33S mice. These compound mutant mice displayed more severe inflammation and higher tumor numbers than the parental Tgfb1C33S/C33S animals. The level of active TGF-β1 in compound mutant mice appeared to be decreased compared to Tgfb1C33S/C33S mice as determined from analyses of surrogate markers of active TGF-β, such as P-Smad2, C-Myc, KI-67, and markers of cell cycle traverse. We conclude that these mutant mice provide a useful system for modulating TGF-β levels in a manner that determines tumor number and inflammation within the GI tract.
Keywords: TGF-β1, Integrin β8, GI Tumors, Inflammation
Introduction
Transforming growth factor β (TGF-β) has divergent effects on tumor generation and progression (1, 2). During the early stages of tumorigenesis, TGF-β acts as a tumor suppressor presumably because TGF-β is a powerful inhibitor of the growth of epithelial cells (3, 4). As tumors progress in their evolution, the growth suppressive property of TGF-β can be lost allowing a growth enhancing effect of TGF-β to predominate (5-8). Thus, the participation of TGF-β in tumorigenesis is complicated and dependent upon multiple factors including cell type, location, and the phase of tumor development.
After synthesis and secretion, TGF-β undergoes a series of modifications in order to produce the active growth factor (9). Mature TGF-β is a 25 kDa homodimer derived from a larger precursor by intracellular proteolytic processing (9). The cleaved propeptide, the latency associated protein (LAP), remains non-covalently bound to the mature growth factor, rendering the TGF-β latent, even after secretion. Dissociation from LAP is required for TGF-β receptor binding. LAP is usually covalently bound to a second protein, a latent TGF-β binding protein (LTBP), through cysteine residues at the amino terminal region of the LAP monomers and a pair of cysteines in the LTBP. There are four Ltbp genes (Ltbp1, 2, 3, and 4), but only LTBP-1, -3 and -4 bind to LAP (10, 11). The complex of TGF-β, LAP, and LTBP is called the large latent complex (LLC), whereas the complex of TGF-β and LAP is called the small latent complex (SLC). There are three TGF-β genes – Tgfb1, b2 and b3, and all three proteins are secreted as latent complexes (12). The conversion of LLC to active TGF-β is mediated by several mechanisms, including proteolytic processing of LTBP and/or LAP to release active cytokine, the interaction of competitive LAP-binding sequences of thrombospondin-1 (TSP-1) or F-spondin with the latent complex, and the binding of integrins to RGD sequences in TGF-β1 and TGF-β3 LAPs (9, 13). Several different β integrins including β3, β5, β6 and β8 bind LAP and may activate the latent complex, but two different mechanisms of activation appear to be employed. β5 and β6 integrin-mediated activation utilizes force supplied by the cell cytoskeleton acting through the integrin upon the LLC tethered to the matrix via the LTBP (14, 15). The application of force distorts the LLC thereby exposing or liberating the TGF-β. SLC binding to LTBP is required for latent TGF-β activation by the integrins αvβ5 and αvβ6 (15, 16). The integrin β8 activates SLC, but activation requires a metalloprotease in addition to the integrin (17). In this activation process, the integrin serves to co-localize the latent complex and the protease, thereby enhancing the rate of protease-mediated activation. TSP-1 also activates both LLC and SLC (18).
Elucidating the role of LTBPs in TGF-β biology has been the focus of a number of investigations (10, 19). Mice and/or people missing LTBP-1, -3 or -4 have phenotypes consistent with alterations in TGF-β levels indicating the importance of LTBPs in TGF-β biology (20-24). However, because binding to LTBP facilitates SLC secretion, the cause of LTBP-related phenotypes is unclear; i.e., are they related to decreased SLC secretion or impaired latent TGF-β activation. To address this question, we generated mice in which the cysteines at position 33 in the TGF-β1 LAP, which normally bind to LTBP, were changed to serines (25). Animals (Tgfb1C33S/C33S) with this mutation should produce all of their TGF-β1 as SLC. If LTBP is required for proper TGF-β1 sequestration and activation, these mice should display a TGF-β1-null-like phenotype. If LTBP is not required for TGF-β1 generation, the mice should display a normal phenotype. The mutation of the cysteines to serines also allows the SLC to be readily secreted; therefore mutant phenotypes would not be caused by decreased extracellular latent TGF-β1 (14, 26).
Tgfb1C33S/C33S mice displayed phenotypes, such as shortened life span, lack of epidermal Langerhans cells, and multiorgan inflammation, consistent with decreased active TGF-β1 (25). The mutant mice also developed tumors of the stomach, colon, cecum, rectum and anus. Measurement of active TGF-β1 in serum revealed a decrease in the amount of active TGF-β1, but little change in the amount of total (latent plus active) secreted TGF-β1. These results indicated that LTBP, as part of the LLC, is required for proper latent TGF-β1 activation. We suggested that these animals were hypomorphs rather than nulls for TGF-β1 because the inflammation and shortened life span were not as severe as observed in TGF-β1 null mice. We hypothesized that the Tgfb1C33S/C33S animals activated some secreted TGF-β1 SLC, albeit inefficiently.
Here, we report a series of experiments designed to test this contention. We reasoned that if we further lowered active TGF-β1 levels in Tgfb1C33S/C33S mice by eliminating activators of the SLC or by deleting one Tgfb1 allele, the abnormal phenotype would be enhanced to resemble more closely the Tgfb1-/- phenotype.
Materials and Methods
Mouse Lines and Reagents
Tgfb1+IC33S, Tgfb1+/-, Tsp1+/-, and Itgb8+/- mice have been described previously (25, 27-30). All mice were housed in a Specific Pathogen Free facility and routinely checked for infections and parasites. All procedures were conducted according to the regulations of the NYU Langone Medical Center IACUC.
TGF-β Assays
The preparation of sera and measurement of TGF-β1 with the Quantine (R & D Systems) kit have been described previously (25). Tissue extracts were prepared from wet tissue samples. The samples were homogenized at 4°C in Tissue Protein Extraction Reagent and Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientifc, Rockford, USA). The extracts were centrifuged at 8000 × g for 10 min at 4°C. After the protein concentrations in the supernatants were determined, the levels of total and active TGF-β1 were measured using a mouse TGF-β1 duo-set (DY1679) according to the manufacturer’s instructions (R&D Systems, Minneapolis, USA). Total TGF-β1 levels were determined by acid activation (final concentration 0.2 M hydrochloric acid, 10 min, room temperature) of the latent TGF-β1 in the homogenate. Untreated and activated samples from the same homogenate were assayed in parallel.
Immunoblotting
Western blotting was performed as described by Yoshinga et al. (25).
Immunohistochemistry
Preparation of tissues, sectioning, and antibody staining were performed as described in Yoshinaga et al. (25).
Quantitative Real-Time PCR (qPCR) Analysis
RNA was extracted from 5 pairs of mice from each genotype using TRIzol (Invitrogen). Reverse transcription was performed using 1 μg of RNA and the SuperScript III Reverse Transcriptase (Invitrogen) (50 °C, 60 min). The resulting cDNA was used for qPCR analysis. qPCR was performed using specific primers and QuantiFast SYBR Green PCR Kit (Qiagen) on an iCycler Thermal Cycler (Bio-Rad). Each target transcript expression was quantified by comparing the threshold cycle (Cq) with that of β-actin by using the comparative Cq method (31). The primers used are described in Supplemental Table 1.
Statistical Analysis
Descriptive statistics were performed with StatView J-4.5 program (SAS Institute). The Kaplan–Meier method was used to estimate all survival curves from mouse studies. The log-rank statistic was used to compare the overall survival distributions.
Pathological Scoring of Inflammation
A scoring system in which three parameters were monitored in individual histological sections was used for quantification of inflammation (25). The three parameters were; Inflammation, which was measured as cell infiltration of the mucosa by mixed populations of inflammatory cells and edema, hyperplasia, which was monitored by the hyperplasia of the mucosal epithelium including lengthening of crypts, increased density of epithelial cells and crypts and thickening of mucosa, and necrosis/ulceration, which was monitored my examining for necrosis of mucosal epithelial cells with attenuation, erosion, or ulceration of the epithelial barrier. A scoring system for each parameter was used in which 0 = within normal limits, 1 = minimal to mild, 2 = moderate and 3 = severe. Two individuals monitored slides in a blinded fashion. Within the stomach tissue, we observed no necrosis and both the wild type and mutants showed hyperplasia. Therefore, only the inflammation parameter was used.
Results
Tumor production in Tgfb1C33S/C33S;thrombospondin1-/- mice
To test our hypothesis that Tgfb1C33S/C33S activate TGF-β1 SLC, we first generated Tgfb1C33S/C33S mice deficient in either of two known activators of SLC, the matricellular protein thrombospondin-1 (TSP-1) or the integrin β8 (17, 18), and examined these animals for changes in inflammation, tumor number, and markers for TGF-β activity.
Tgfb1C33S/C33S;Tsp1-/- mice displayed a phenotype that was essentially unchanged compared to Tgfb1C33S/C33S mice (Supplemental Table 2). (Tsp1+/- mice, used as controls, show no phenotype.) There was a slight increase in the degree of inflammation in the colon and rectum when Tgfb1C33S/C33S;Tsp1+/- and Tgfb1C33S/C33S;Tsp1-/- mice were compared, but inflammation in all other organs was essentially equivalent amongst mice with the two genotypes (Supplemental Table 2A (12 weeks) and Supplemental Fig. 1). There was no change in survival of Tgfb1C33S/C33S;Tsp1-/- mice compared to Tgfb1C33S/C33S mice (data not shown). When these mice were examined for presence of tumors, a slight but statistically insignificant increase in tumor incidence was observed upon elimination of TSP-1 (Supplemental Table 2B). The distribution of tumor types was approximately equivalent in Tgfb1C33S/C33S;Tsp1+/- and Tgfb1C33S/C33S;Tsp1-/- mice and similar to results in our previous studies (25). Therefore, Tsp1 appears to make only a minor, if any, contribution to the level of active TGF-β in tumor production in Tgfb1C33S/C33S mice.
Tumor production in Tgfb1C33S/C33S;Itgb8-/- mice
We next examined the degree of inflammation and tumor production in Tgfb1C33S/C33S versus Tgfb1C33S/C33S;Itgb8-/- mice. The integrin αvβ8 is an important modulator of TGF-β1 levels within the intestine (32, 33). Unlike Tgfb1C33S/C33S;Tsp1-/- mice, Tgfb1C33S/C33S;Itgb8-/- mice presented with significant changes compared to Tgfb1C33S/C33S animals. The compound mutant mice had a shortened survival compared to Tgfb1C33S/C33S mice but similar to Itgb8-/- mice (Fig. 1A). The level of inflammation was increased in the stomach, cecum, colon and rectum (Table 1A). The increase in inflammation was clearly apparent when tissues from the different groups were compared by a scoring method that quantified inflammation, hyperplasia and necrosis/ulceration (Supplemental Fig. 2A). Tumor incidence also was substantially increased from 19% to 53% when Tgfb1C33S/C33S animals were compared to Tgfb1C33S/C33S;Itgb8-/- animals (Table 1B). Although only rectal tumors were observed in Tgfb1C33S/C33S mice, more than half of the tumor-bearing Tgfb1C33S/C33S;Itgb8-/- mice had multiple tumors and these occurred in several different organs. In Figure 2, images of a rectal adenosquamous cell carcinoma (Fig. 2Aa) and rectal adenocarcinoma (Fig. 2Ac) are illustrated. All of the tumors were invasive, as tumor cells were observed in the submucosal muscle layer.
Figure 1.
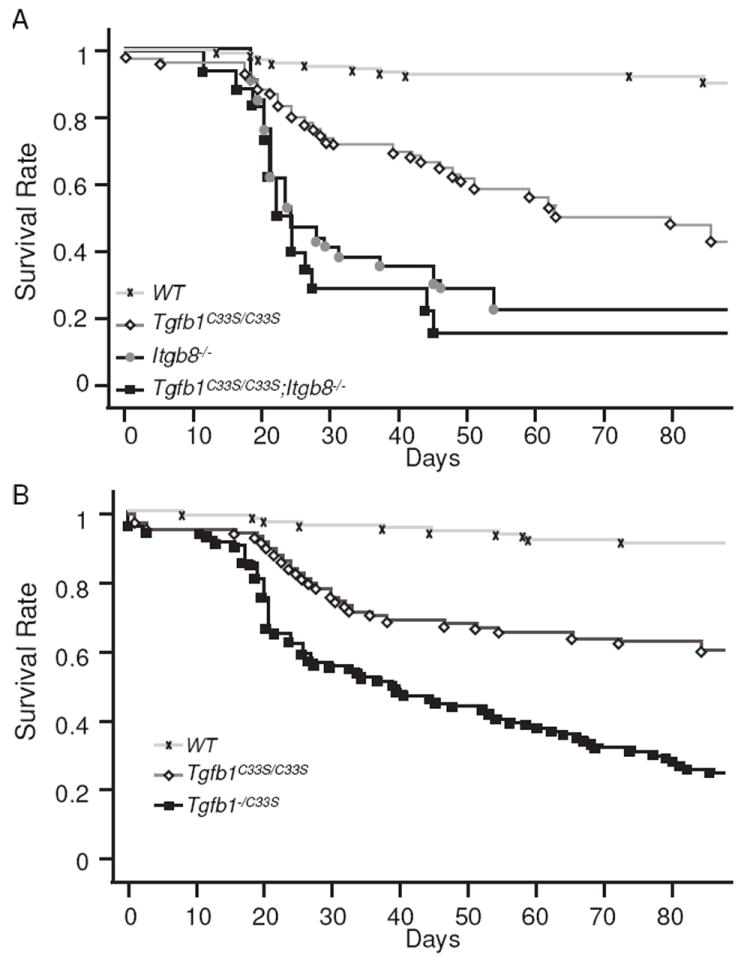
Survival of Tgfb1 Mutant Mice. Survival curves were constructed according to the Kaplan-Meier method. A. WT, 129 mice; Tgfb1C33S/C33S, 60 mice; Itgb8-/-, 33 mice; Tgfb1C33S/C33S; Itgb8-/-, 18 mice. B. WT, 168 mice; Tgfb1C33S/C33S, 112 mice, Tgfb1-/C33S, 138 mice.
Table 1.
Inflammation and Tumor Production in Tgfb1 Mutant Mice
| A. Inflammation in Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- micea | |||||||
| Genotype | N | Lung | Heart | Stomach | Cecum | Colon | Rectum |
| WT | 16 | - | - | - | - | - | - |
| Itgb8-/- | 6 | - | - | - | - | - | - |
| Tgfb1C33S/C33S | 16 | -~++ | -~++ | +~++ | +~++ | -~++ | +~++ |
| Tgfb1C33S/C33S; Itgb8-/- | 15 | +~++ | -~++ | ++ | ++ | ++ | ++ |
| B. Tumors in Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- micea | |||||||
| Genotype | N | Stomach | Cecum | Colon | Rectum | Total | (%) |
| WT | 16 | 0 | 0 | 0 | 0 | 0 | (0) |
| Itgb8-/- | 6 | 0 | 0 | 0 | 0 | 0 | (0) |
| Tgfb1C33S/C33S | 16 | 0 | 0 | 0 | 3 | 3 | (19) |
| Tgfb1C33S/C33S; Itgb8-/- | 15 | 3 | 2 | 2 | 7 | 8 | (53) b, c |
| C. Inflammation in Tgfb1C33S/C33S and Tgfb1-/C33S micea | |||||||
| Genotype | Lung | Heart | Stomach | Cecum | Colon | Rectum | Liver |
| WT | - | - | - | - | - | - | - |
| Tgfb1C33S/C33S | -~+ | -~+ | -~+ | ++ | -~+ | ++ | - |
| Tgfb1-/C33S | +~++ | +~++ | ++ | ++ | +~++ | ++ | +>++ |
| D. Tumors in Tgfb1C33S/C33S and Tgfb1-/C33S micea | |||||||
| Genotype | N | Stomach | Cecum | Colon | Rectum | Total | (%) |
| WT | 22 | 0 | 0 | 0 | 0 | 0 | (0) |
| Tgfb1C33S/C33S | 56 | 5 | 1 | 0 | 7 | 11 | (20) b |
| Tgfb1-/C33S | 15 | 8 | 1 | 0 | 4 | 11 | (73) c, d |
8-12 weeks of age
= no inflammation,
= mild inflammation,
= moderate inflammation,
WT = Control Tgfb1+/+ or Tgfb1+/- animals
indicates an intermediate value,
12 weeks of age
Total refers to total number of animals with tumors and includes animals with multiple tumors.
P<0.05 vs. Tgfb1C33S/C33S.
5 mice had multiple tumors.
12 weeks of age
indicates greater prevalence
Total refers to total number of mice with tumors and includes mice with multiple tumors.
2 mice had multiple tumors.
P = 0.0004 vs. Tgfb1C33S/C33S
2 mice had multiple tumors.
Figure 2.
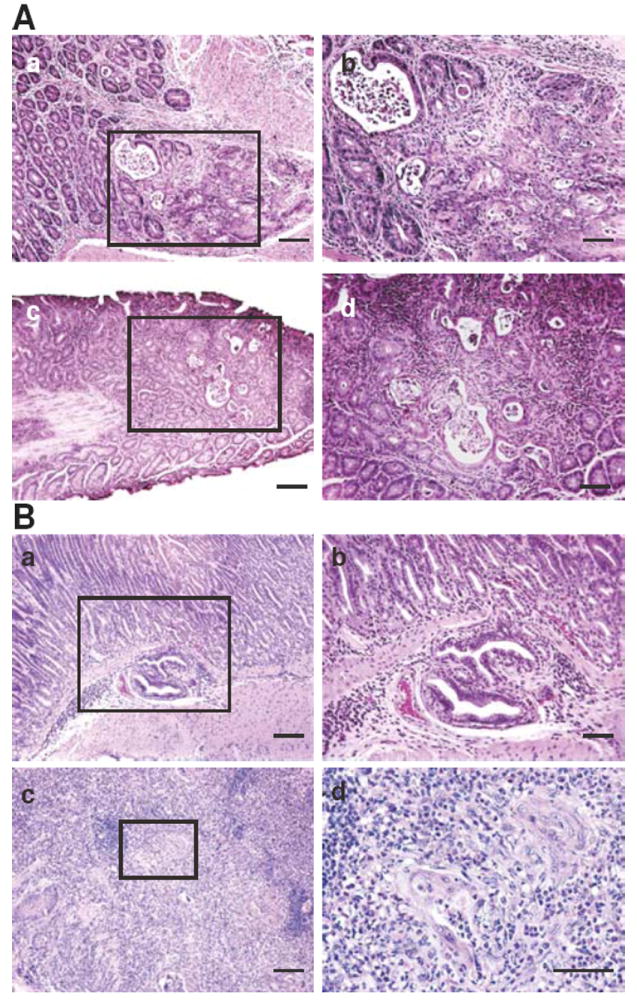
Tumor Histology in Tgfb1C33S/C33S;Itgb8-/- and Tgfb1-/C33S mice. A. Tumors in Tgfb1C33S/C33S;Itgb8-/- animals. Illustrated are an H&E stained rectal adenosquamous cell carcinoma (a and b) and a rectal adenocarcinoma (c and d).
B. Tumors in Tgfb1-/C33S mice. Illustrated are an H&E stained gastric adenocarcinoma (a and b), as well as a squamous cell carcinoma (c and d). Higher magnifications of the boxed area in each tumor are illustrated in b and d. Bars; a and c, 100 μm, b and d, 50 μm.
To determine if the increased tumor incidence in Tgfb1C33S/C33S;Itgb8-/- mice correlated with changes in TGF-β levels and/or TGF-β signaling, we analyzed stomach and rectal tissue from wild type, Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- animals for markers of TGF-β1 activity. Because TGF-β1 is an inhibitor of epithelial cell growth, decreases in TGF-β1 signaling should result in enhanced epithelial proliferation. Consistent with potentially diminished levels of active TGF-β1, Tgfb1C33S/C33S;Itgb8-/- mice displayed increased (more than twice that observed in wild type (WT) tissues) cell proliferation of the epithelial cells of the mucosal layer in the rectal epithelium, as monitored by immunostaining for the proliferation marker KI-67 (Fig. 3A). We observed a progressive increase in the number of proliferating cells when WT samples were compared to Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- samples. (Fig. 3A). Similar results were found in stomach tissues from the three genotypes (data not shown). TGF-β signaling is usually marked by increases in the level of P-Smad2, an intracellular mediator of the canonical TGF-β signaling pathway. P-Smad2 immunostaining in the rectum was heterogeneous in Tgfb1C33S/C33S;Itgb8-/- mice with some areas showing decreased intensity, whereas other areas appeared to have normal levels (Fig. 4Aa-d). In general there appeared to be less P-Smad2 in the rectal tissue of each of the two mutant genotypes and significantly less staining in the double mutant. However, in order to survey the entire rectum for P-Smad levels, we performed immunoblotting and scanning after SDS-PAGE on the soluble proteins from rectal tissue (Fig. 4Ae). The results show a clear loss of P-Smad2 reactivity in the double mutant tissue, whereas tissue from animals with only the Tgfb1C33S/C33S or Itgb8-/- mutations appeared to contain amounts of P-Smad2 close to or slightly more than that of the wild type sample. The level of C-Myc expression, which normally is suppressed by TGF-β, was enhanced in the Tgfb1C33S/C33S;Itgb8-/- rectal tissue compared to wild type tissue (Fig. 5A). We also measured the transcript levels for the cell cycle regulators p15, p21, and p27, which are known to be regulated by TGF-β, in the three genotypes (Fig. 5A). There was only a slight decrease, which was not statistically significant, in p21 expression in Tgfb1C33S/C33S;Itgb8-/- mice compared to either wild type or Tgfb1C33S/C33S mice. However, there were pronounced decreases in the expression of the negative regulators p15 and p27 compared to wild type in both Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- rectal tissues consistent with a decrease in active TGF-β1.
Figure 3.
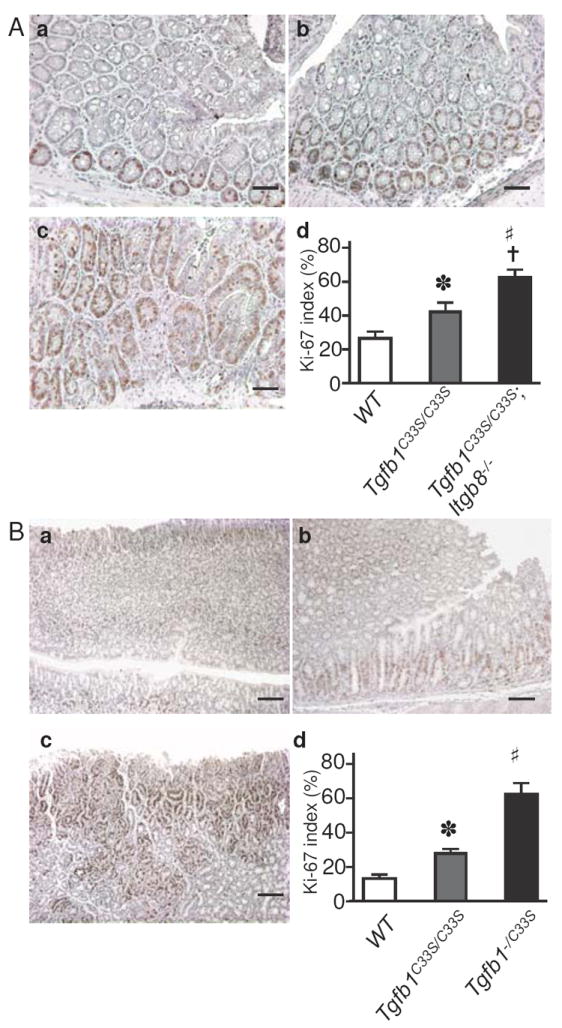
KI-67 staining increases in mutant tumor tissue compared to WT. A. KI-67 staining of Tgfb1C33S/C33S;Itgb8-/- tissues. Sections from WT (a), Tgfb1C33S/C33S (b), and Tgfb1C33S/C33S;Itgb8-/- (c) rectal tissues are illustrated after staining for KI-67 to detect proliferating cells. Bars, 100 μm. d. The percentage of positive cells in each of the three genotypes was computed by counting the number of positive cells versus total cells in three random fields of sections from the appropriate tissues. The tissue from Tgfb1C33S/C33S had significantly more positive cells than did WT tissue (*; P<0.05) and tissue from Tgfb1C33S/C33S;Itgb8-/- had more than 2 times the number of positive cells than did WT tissue (#; P<0.0001) or Tgfb1C33S/C33S (†; P<0.001). N = 3 animals per group.
B. KI-67 staining of Tgfb1-/C33S tissues. Sections from WT (a), Tgfb1C33S/C33S (b), and Tgfb1-/C33S (c) stomach tissues are illustrated after staining for KI-67 to detect proliferating cells. Bars, 100 μm. d. The percentage of positive cells was computed as described above. The Tgfb1C33S/C33S tissue had significantly more positive cells than did WT (*; P<0.0001) and the Tgfb1-/C33S tissue displayed more positive cells than Tgfb1C33S/C33S tissue (#; P<0.0001). N = 3 animals per group.
Figure 4.
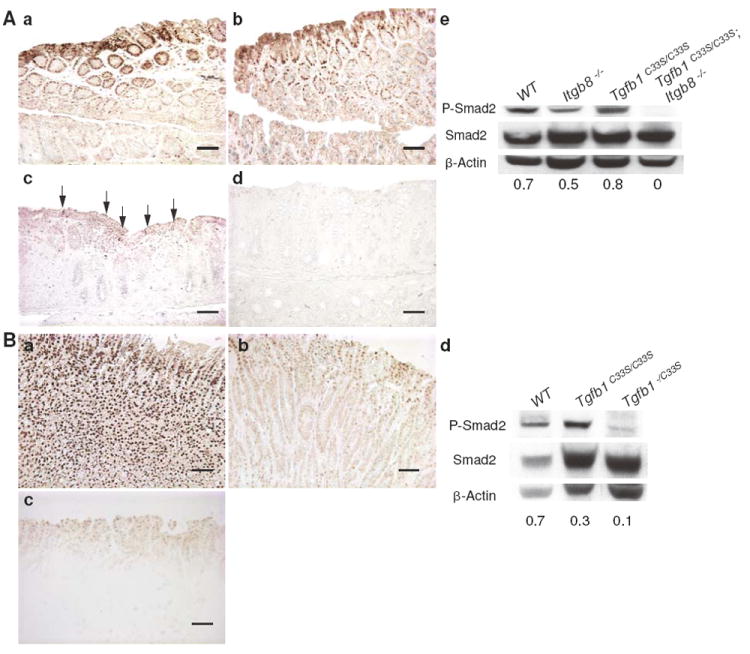
P-Smad levels are decreased in tumor tissue compared to WT. A. P-Smad2 staining in rectal tissue. Staining of WT and Itgb8-/- tissues (a and b) indicated higher levels of P-Smad2 in the epithelium than observed in tissues from Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- mice (c and d). e. Data from scans of immunoblots are presented under each lane as the ratio of P-Smad2 and Smad2. The results indicate no difference between WT and Tgfb1C33S/C33S samples, but there is almost a complete loss of signaling in the Tgfb1C33S/C33S;Itgb8-/- tissue. Bars, 100 μm. Arrows indicate P-Smad2-positive areas in panel c. B. P-Smad staining in stomach tissue. a. WT, b. Tgfb1C33S/C33S, and c. Tgfb1-/C33S. d. To measure TGF-β signaling, the amount of P-Smad2 was quantified in gastric tissue after extraction, immunoblotting after SDS-PAGE, and densitometry. The ratio of P-Smad2 to Smad2 is indicated at the bottom of each gel lane. The amount of P-Smad2 was less in Tgfb1-/C33S tissue than in Tgfb1C33S/C33S. Bars, 100 μm.
Figure 5.
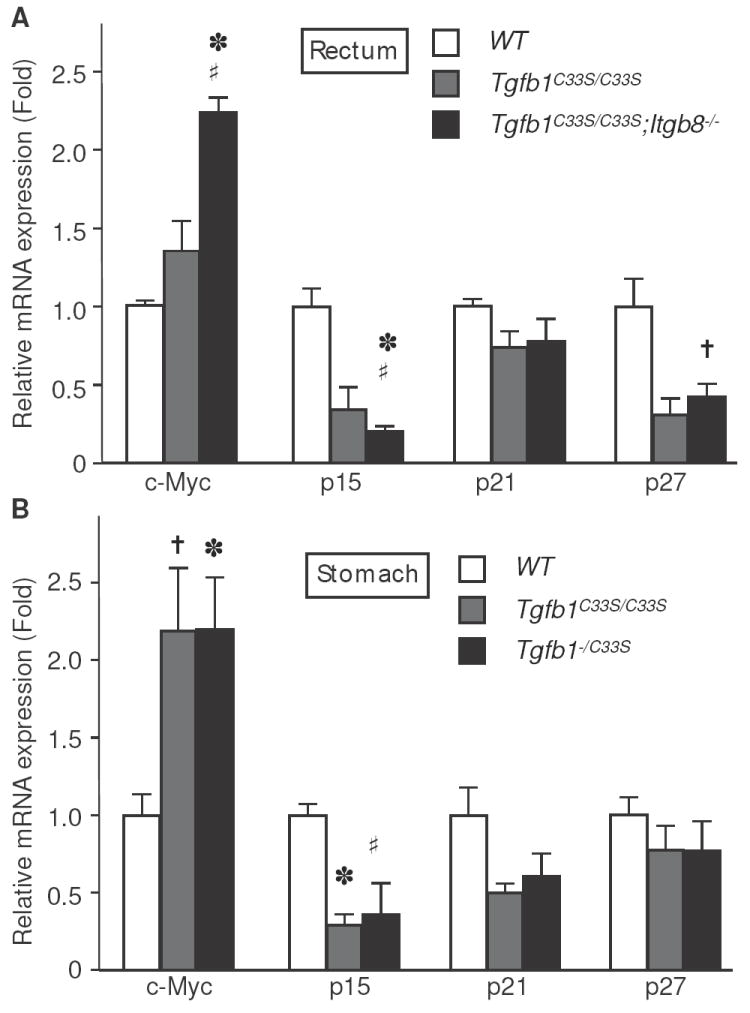
Transcript levels of cell growth regulators are altered in tumor tissue compared to WT. Rectal (A) or gastric (B) tissue from WT and mutant animals was extracted and expression levels of the indicated markers were measured by qPCR as described in Methods. A. WT, Tgfb1C33S/C33S, and Tgfb1C33S/C33S;Itgb8-/-. *; P<0.01 vs. WT; †; P<0.05 vs. WT; #; P<0.05 vs. Tgfb1C33S/C33S. B. WT, Tgfb1C33S/C33S, and Tgfb1-/C33S. *; P<0.01 vs. WT, †; P<0.05 vs. WT, #; P=0.07 vs. WT.
Next, we measured TGF-β1 in sera from WT, Itgb8-/-, Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- mice (Fig. 6A). When total TGF-β1 was quantified after acid activation of mouse sera, the Tgfb1C33S/C33S, Itgb8+/+ and Tgfb1C33S/C33S;Itgb8-/- samples were equivalent and slightly lower than the control WT animal samples (Fig. 6A). When active TGF-β1 in serum was quantified, the samples from Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- mice were approximately half of the control values (Fig. 6B). However, we did not detect a difference between those two samples. This may indicate that the contribution of integrin αvβ8 to the activation of latent TGF-β1 in serum is negligible. We also evaluated the amount of total and active TGF-β1 in rectal tissue extracts (Supplemental Fig. 3A and B). Although there were small differences with respect to total TGF-β1 amongst the four experimental groups, there was no statistical difference in the amount of active TGF-β1 amongst the samples. The reasons for this are not known.
Figure 6.
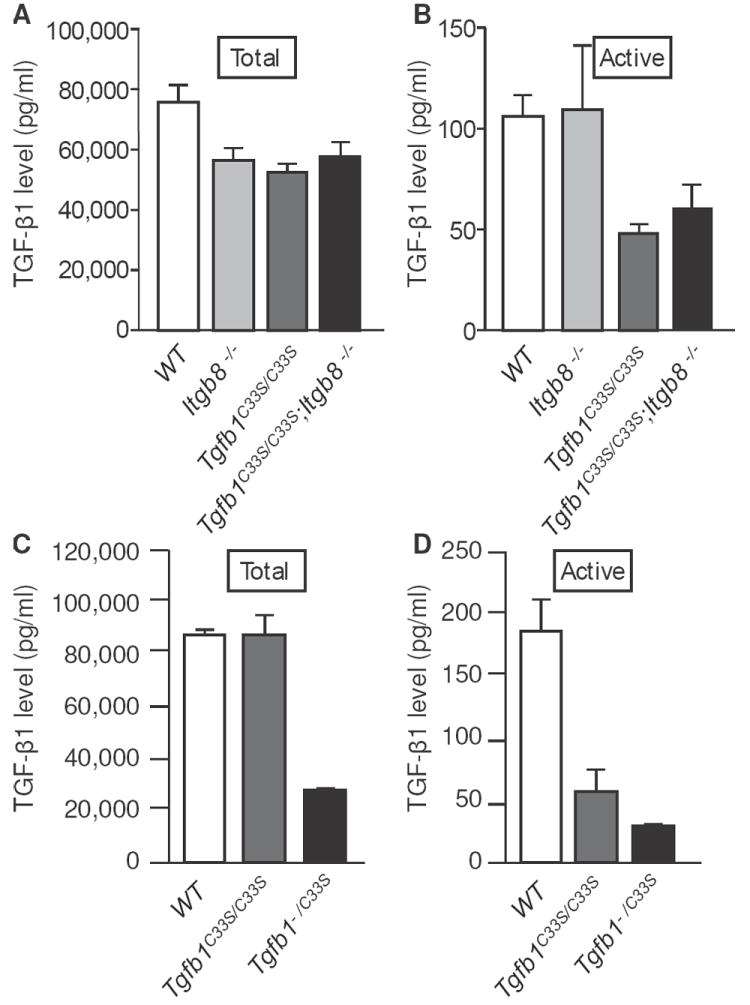
Active TGF-β1 levels are decreased in sera from mutant versus WT mice. TGF-β1 levels from WT, Itgb8-/-, Tgfb1C33S/C33S, and Tgfb1C33S/C33S;Itgb8-/- mouse sera measured by ELISA (A and B). Total TGF-β1 was measured after acid treatment of serum samples to activate all of the latent TGF-β1 present (A), whereas active TGF-β1 was measured in sera without acid treatment (B). There was little difference amongst the four genotypes with respect to total TGF-β1. However, Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- sera had significantly less active TGF-β1 than did WT or Itgb8-/- sera. There was no statistical difference in levels of TGF-β1 in the Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- sera. TGF-β1 levels in WT, Tgfb1C33S/C33S, and Tgfb1-/C33S mice were also measured by ELISA (C and D). WT and Tgfb1C33S/C33S sera had approximately equivalent amounts of total TGF-β1, while Tgfb1-/C33S sera had less than half the amount of total TGF-β1 as did WT sera. When serum active TGF-β1 levels were quantified, Tgfb1C33S/C33S sera had slightly less than a third the amount found in WT, whereas Tgfb1-/C33S sera had approximately 20% of WT.
Tumor production in Tgfb1-/C33S mice
We reasoned that if enhanced tumorigenesis in Tgfb1C33S/C33S;Itgb8-/- versus Tgfb1C33S/C33S mice was the result of lower TGF-β1 levels, a second approach to decrease active TGF-β1 levels would be to generate Tgfb1-/C33S mice. As these animals would have only one functional TGF-β1 allele and that would be the C33S allele, they should produce approximately half as much TGF-β1 as Tgfb1C33S/C33S mice. Therefore, we generated Tgfb1-/C33S mice by crossing Tgfb1+/C33S and Tgfb1-/+ animals. Tgfb1-/C33S mice were produced at the expected Mendelian frequency (Data not shown) and had a shortened life span compared to wild type or Tgfb1C33S/C33S mice (Fig. 1B). Tgfb1-/C33S mice displayed more severe inflammation in their lungs, heart, stomach, liver and colon than did Tgfb1C33S/C33S mice (Table 1C; Supplemental Fig. 2B). Tumor incidence in Tgfb1-/C33S mice was also significantly enhanced from 20% to 73% when Tgfb1C33S/C33S mice were compared with Tgfb1-/C33S mice (Table 1D). Interestingly, there were only 2 Tgfb1-/C33S mice with multiple tumors. The reason for this is unclear. The distribution of tumor types in Tgfb1-/C33S mice was also different compared to Tgfb1C33S/C33S mice. By 12 weeks of age 53% of the Tgfb1-/C33S animals had gastric adenocarcinomas compared to 9% in Tgfb1C33S/C33S mice. (The incidence of gastric adenocarcinomas in Tgfb1C33S/C33S;Itgb8-/- mice was 20% (Table 1B)). Two gastric tumors are shown in figure 2B. One tumor is an adenocarcinoma (Fig. 2Ba and b) and one is a squamous cell carcinoma (Fig. 2Bc and d). Both tumors were invasive, as tumor cells were found in the submucosal muscular layer. The rectal tumors in Tgfb1-/C33S mice were similar to those observed in animals with the Tgfb1C33S/C33S;Itgb8-/- genotype (Data not shown).
We next measured a number of parameters to characterize signaling and TGF-β1 levels in Tgfb1-/C33S mice, as we did with the Tgfb1C33S/C33S;Itgb8-/- mice. We focused on the gastric tumors because of their high incidence and relative rarity in other mouse models. As we observed with Tgfb1C33S/C33S;Itgb8-/- mice, stomach tissue from Tgfb1-/C33S mice had higher levels of KI-67 positive cells than did wild type or Tgfb1C33S/C33S tissues (Fig. 3B). Quantification of the number of KI-67 positive cells revealed that there was a 4-fold increase in Tgfb1-/C33S tissue compared to controls and a 2-fold increase compared to Tgfb1C33S/C33S tissue (Fig. 3B). P-Smad2 staining in the stomachs of wild type, Tgfb1C33S/C33S, and Tgfb1-/C33S mice revealed that there was progressively less staining, as the expected level of total TGF-β1 decreased (Fig. 4B). We observed that C-Myc expression in the stomach tissue of Tgfb1-/C33S mice was elevated compared to wild type mice, but was not increased compared to Tgfb1C33S/C33S animals (Fig. 5B). We also measured tumor suppressor gene expression. As observed with the Tgfb1C33S/C33S and Tgfb1C33S/C33S;Itgb8-/- tissues, there was little difference in expression of p21 when WT, Tgfb1C33S/C33S, and Tgfb1-/C33S stomach tissues were compared by qPCR (Fig. 5B). Similar to the samples in figure 5A, there was a decrease in the expression levels of p15 amongst WT, Tgfb1C33S/C33S, and Tgfb1-/C33S tissue (Fig. 5B).
Finally, we measured the level of total and active TGF-β1 in sera from wild type, Tgfb1C33S/C33S, and Tgfb1-/C33S mice (Fig. 6C and D). The total TGF-β1 in sera from Tgfb1C33S/C33S mice, as measured after acid activation, was equivalent to wild type as described earlier (25). The Tgfb1-/C33S mouse sera had less than half of the total amount of TGF-β1 consistent with the loss of one allele. Both the mutant sera (Tgfb1C33S/C33S and Tgfb1-/C33S) had less than 30% of the amount of active TGF-β1 than the wild type sample (Fig. 6D). The amount of active TGF-β1 in the Tgfb1-/C33S mouse sera was slightly more than half of that found in the Tgfb1C33S/C33S mouse sera. When the amount of total and active TGF-β1 was measured in tissue, there were small differences in the total and active concentrations of cytokine between the genotypes and the amount of active was approximately 10% of total (Supplemental Fig. 3C and D). There were no statistically significant differences between the Tgfb1C33S/C33S and Tgfb1-/C33S samples, although both of these samples were lower than the wild type control sample.
Discussion
The association of TGF-β with cancers of the GI tract is well established. Extensive work has shown that elimination of the growth factor or interference with its signaling either by TGF-β receptor mutations or mutations in the intracellular signaling pathway yield carcinomas (34). Our previous work showing that blocking the formation of the disulfide bond between the TGF-β1 propeptide and its matrix localizing proteins, the LTBPs, resulted in the production of GI cancers, was consistent with the hypothesis that binding to LTBP was required for proper TGF-β1 generation from its latent complex (25). Interference with active TGF-β1 production yields results similar to ablation of TGF-β1 signaling. Because the inflammatory response observed in Tgfb1C33S/C33S mice was not as severe as that observed in Tgfb1-/- mice (28, 29), we presumed that some latent TGF-β1 was activated in our mutant animals. Indeed, by further impairing active TGF-β1 production either by eliminating an activator of latent TGF-β or by simply decreasing the amount of total TGF-β1 produced, we enhanced both the tumor and inflammatory phenotypes. Therefore, these mutant animals provide a hypomorphic series in which the relationship of different TGF-β1 levels, inflammation, and relatively rapid tumor production can be explored. In addition, the results re-enforce our earlier conclusions of the importance of TGF-β LLC in TGF-β biology as well as highlighting the role of the integrin αvβ8 in activation of TGF-β1 SLC.
Both integrins αvβ6 and αvβ8 are important for active TGF-β1 formation (27, 33). Because Tgfb1C33S/C33S animals produce only the SLC of TGF-β1 and this complex is not activated by αvβ6 (14), we focused on the effects of αvβ8 loss. Mice that were deficient for integrin β8 and that produced TGF-β1C33S displayed a higher degree of inflammation and considerably more tumors than Tgfb1C33S/C33S mice. The enhanced inflammation is consistent with published reports that αvβ8 is crucial for latent TGF-β1 activation by dendritic cells (33) and supports the observed association of inflammation and tumorigenesis (35, 36). Similar results were observed on the degree of inflammation with Tgfb1C33S/C33S mice that were housed in helicobacter-free conditions (Data not shown). Yet, Itgb8-/- mice do not get tumors, indicating that αvβ8 is not the exclusive latent TGF-β1 activator in the GI tract. The other potential activator of TGF-β1 SLC, TSP-1, appeared to make no contribution to the level of TGF-β in Tgfb1C33S/C33S mice as determined by our assays. Perhaps analysis of a larger cadre of animals might reveal a minor contribution. Our second approach to diminish TGF-β1 levels, using mice with one null allele and one Tgfb1C33S allele, also enhanced both inflammation and tumor frequency. Together, our results with the different mutant animals indicate that tumor production is dependent upon the level of TGF-β1 signaling.
A limitation of our study is that we do not know the precise level of active TGF-β1 in the specific tissues examined. The TGF-β1 levels in serum reflect the expected changes, the secondary markers for TGF-β1 signaling in the tissues reflect decreased TGF-β1, and the level of active TGF-β1 from stomach is decreased when Tgfb1C33S/C33S mice are compared to wild type mice. However, we were unable to detect statistically significant differences in active TGF-β1 when tissue extracts from Tgfb1C33S/C33S animals were compared to either Tgfb1-/C33S or Tgfb1C33S/C33S;Itgb8-/- tissue extracts. The reason for this is not apparent but might reflect the fact that the amount of active TGF-β1 in the tissue is close to the limit of sensitivity of the assay. The availability of an assay that directly monitored TGF-β1 signaling within the tissue would resolve the question of tissue levels of active cytokine.
An interesting question is why the tumor types and distribution appeared to be different between Tgfb1C33S/C33S;Itgb8-/- and Tgfb1-/C33S mice, as each mutation should lower the amount of active TGF-β1. This may reflect the fact that in one case, Tgfb1-/C33S mice, the total amount of TGF-β1 was diminished, whereas in the second case, Tgfb1C33S/C33S;Itgb8-/-, the total TGF-β was not decreased; only one of the potential activators was decreased compared to Tgfb1C33S/C33S animals. The contribution of integrin β8 to active TGF-β1 levels also may vary throughout the GI tract, thereby yielding a unique pattern of tumorigenesis. Alternatively, the differences in tumor incidence may reflect the different mouse strain backgrounds of the two sets of mutant and control animals generated for our studies. The Itgb8 null mutation is an embryonic lethal when placed in the C57BL genetic background (27). Only when the mutation is placed into mixed background of C57BL plus ICR, do pups with the null mutation survive for up to 8 months; this precludes using inbred animals. Therefore, differences in genetic backgrounds might account for differences in results observed between the two sets of animals. Finally, the fact that the microenvironment in the stomach and rectum may be different and may differentially affect tumorogenesis.
It is interesting to speculate on the actual cause of tumors in these mutant mice. Tumor onset is relatively rapid but the frequency is not particularly high. In this respect, our model differs from other models where tumor frequency and rate are quite robust. Usually these other models involve the alteration of tumor suppressor genes and/or oncogenes. TGF-β1-/C33S-mediated gastric tumor production, therefore, is in some ways closer to that seen in humans. Since all of the epithelial cells have the same genotype, why are there not more tumors or what is unique about the cells comprising the tumors that occur? Thus far, we have not explored what additional genetic changes may have occurred within these tumors because at the time of sacrifice (12 weeks), the tumors are all microscopic. Additional experiments need to be done using macroscopic tumors from older animals and analyzing the involved tissue for genetic alterations.
Tumorigenesis within the GI tract due to loss of TGF-β1 is interesting to consider with respect to the cell type responsible for initiation of the lesion. The loss of TGF-β1 signaling within the epithelium removes a potent inhibitor of cell growth and potentially allows for the early growth of initiated cells. Additionally, the presence of inflammatory cells of several types in Tgfb1-/- mice due to increased inflammation may supply mediators that promote tumorogenesis. However, removal of T and B cells by crossing with Rag2-/- mice appears not to delay colon tumor appearance in Tgfb1-/- mice by a significant degree (37). Rather, the absence of TGF-β1 yields disorganized crypt architecture that may predispose the tissue for malignant transformation. Alternatively, two other reports describing tumor production using cells deficient in TGF-β signaling describe contributions of either mutant stroma or T cells to GI tumor production (38, 39). It is unclear in the mutant mice we have examined if the decreases in TGF-β have a direct effect on the epithelium thereby promoting carcinoma formation or whether the lack of TGF-β produced by or signaling through other cell types promotes tumor formation. This question can be further explored with respect to hematopoietic cell contribution to tumor development using bone marrow from Tgfb1-/C33S mice transferred to WT animals. Such experiments are planned.
Supplementary Material
Acknowledgments
The authors thank Melinda Vassallo and Joseph Ambrogio for their technical assistance.
Grant Support. This work was supported by National Institutes of Health grants CA034282, GM083220, and CA139238 to DBR. The authors wish to acknowledge the use of the NYUMC Histopathology and Transgenic Mouse Cores supported by the NYUMC Cancer Center grant CA016087. K.S. and K.Y. were supported by a fellowship from the Uehara Foundation.
Footnotes
Author contributions. K.S., M.O., K.Y. and D.B.R. conceived and designed the experiments. K.S., M.O., M.H., and K.Y. developed the methodology and acquired data. K.S., M.O., M.H., K.Y., J.M., and D.B.R. analyzed the data. K.S., M.O., M.H., and D.B.R. wrote and reviewed the manuscript.
Conflict of Interest Statement: Disclosure of potential conflicts of interest. No conflicts of interest were disclosed.
References
- 1.Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006 Feb-Apr;17(1-2):29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007 Oct;12(4):313–27. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietenpol JA, Stein RW, Moran E, Yaciuk P, Schlegel R, Lyons RM, et al. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell. 1990 Jun 1;61(5):777–85. doi: 10.1016/0092-8674(90)90188-k. [DOI] [PubMed] [Google Scholar]
- 4.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003 Nov;3(11):807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 5.Bachman KE, Park BH. Duel nature of TGF-beta signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol. 2005 Jan;17(1):49–54. doi: 10.1097/01.cco.0000143682.45316.ae. [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001 Oct;29(2):117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 7.Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, et al. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003 Oct;112(7):1116–24. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002 Feb;12(1):22–9. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 9.Dabovic B, Rifkin DB. TGF-ß bioavailability: latency, targeting, and activation. In: Derynck R, Miyazono K, editors. The TGF-ß Family. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
- 10.Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005 Mar 4;280(9):7409–12. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- 11.Saharinen J, Taipale J, Keski-Oja J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 1996;15(2):245–53. [PMC free article] [PubMed] [Google Scholar]
- 12.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003 Jan 15;116(Pt 2):217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 13.Attur MG, Palmer GD, Al-Mussawir HE, Dave M, Teixeira CC, Rifkin DB, et al. F-spondin, a neuroregulatory protein, is up-regulated in osteoarthritis and regulates cartilage metabolism via TGF-beta activation. FASEB J. 2009 Jan;23(1):79–89. doi: 10.1096/fj.08-114363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004 Jun 7;165(5):723–34. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF- 1 from the extracellular matrix. J Cell Biol. 2007 Dec 17;179(6):1311–23. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munger JS, Huang XZ, Kawakatsu H, Griffiths MJD, Dalton SL, Wu JF, et al. The integrin avb6 binds and activates latent TGFb1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1998;96:319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 17.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157(3):493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schultz-Cherry S, Ribeiro S, Gentry L, Murphy-Ullrich E. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-ß in a chemically defined system. J Biol Chem. 1994;269(43):26775–82. [PubMed] [Google Scholar]
- 19.Chandramouli A, Simundza J, Pinderhughes A, Cowin P. Choreographing Metastasis to the Tune of LTBP. J Mammary Gland Biol Neoplasia. 2011 Jun;16(2):67–80. doi: 10.1007/s10911-011-9215-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dabovic B, Chen Y, Colarossi C, Obata H, Zambuto L, Perle MA, et al. Bone abnormalities in latent TGF-[beta] binding protein (Ltbp)-3-null mice indicate a role for Ltbp-3 in modulating TGF-[beta] bioavailability. J Cell Biol. 2002 Jan 21;156(2):227–32. doi: 10.1083/jcb.200111080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noor A, Windpassinger C, Vitcu I, Orlic M, Rafiq MA, Khalid M, et al. Oligodontia is caused by mutation in LTBP3, the gene encoding latent TGF-beta binding protein 3. Am J Hum Genet. 2009 Apr;84(4):519–23. doi: 10.1016/j.ajhg.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sterner-Kock A, Thorey IS, Koli K, Wempe F, Otte J, Bangsow T, et al. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002 Sep 1;16(17):2264–73. doi: 10.1101/gad.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todorovic V, Frendewey D, Gutstein DE, Chen Y, Freyer L, Finnegan E, et al. Long form of latent TGF-{beta} binding protein 1 (Ltbp1L) is essential for cardiac outflow tract septation and remodeling. Development. 2007 Oct;134(20):3723–32. doi: 10.1242/dev.008599. [DOI] [PubMed] [Google Scholar]
- 24.Urban Z, Hucthagowder V, Schurmann N, Todorovic V, Zilberberg L, Choi J, et al. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am J Hum Genet. 2009 Nov;85(5):593–605. doi: 10.1016/j.ajhg.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshinaga K, Obata H, Jurukovski V, Mazzieri R, Chen Y, Zilberberg L, et al. Perturbation of transforming growth factor (TGF)-beta1 association with latent TGF-beta binding protein yields inflammation and tumors. Proc Natl Acad Sci U S A. 2008 Dec 2;105(48):18758–63. doi: 10.1073/pnas.0805411105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000 Mar 17;86(5):571–9. doi: 10.1161/01.res.86.5.571. [DOI] [PubMed] [Google Scholar]
- 27.Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, et al. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci. 2009 Jan 15;122(Pt 2):227–32. doi: 10.1242/jcs.035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993 Jan 15;90(2):770–4. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-ß1 gene results in mulitfocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, et al. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest. 1998 Mar 1;101(5):982–92. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009 Apr;55(4):611–22. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 32.Melton AC, Bailey-Bucktrout SL, Travis MA, Fife BT, Bluestone JA, Sheppard D. Expression of alphavbeta8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J Clin Invest. 2010 Dec;120(12):4436–44. doi: 10.1172/JCI43786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007 Sep 20;449(7160):361–5. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grady WM, Markowitz S. TGF-ß Signaling Pathway and Tumor Suppression. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
- 35.Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link? Biochem Pharmacol. 2006 Nov 30;72(11):1605–21. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 36.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010 Mar 19;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999 Jul;59(15)(14):3379–86. [PubMed] [Google Scholar]
- 38.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004 Feb 6;303(5659):848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 39.Kim BG, Li C, Qiao W, Mamura M, Kasperczak B, Anver M, et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006 Jun 22;441(7096):1015–9. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.