

Abstract
Purpose
Endoglin (ENG, CD105) is a membranous protein overexpressed in tumor-associated endothelial cells, chemoresistant populations of ovarian cancer cells, and potentially stem cells. Our objective was to evaluate the effects and mechanisms of targeting endoglin in ovarian cancer.
Experimental Design
Global and membranous endoglin expression was evaluated in multiple ovarian cancer lines. In vitro, the effects of siRNA-mediated endoglin knockdown with and without chemotherapy were evaluated by MTT assay, cell-cycle analysis, alkaline comet assay, γ-H2AX foci formation, and qPCR. In an orthotopic mouse model, endoglin was targeted with chitosan-encapsulated siRNA with and without carboplatin.
Results
Endoglin expression was surprisingly predominantly cytoplasmic, with a small population of surface-positive cells. Endoglin inhibition decreased cell viability, increased apoptosis, induced double-stranded DNA damage, and increased cisplatin sensitivity. Targeting endoglin downregulates expression of numerous DNA repair genes, including BARD1, H2AFX, NBN, NTHL1, and SIRT1. BARD1 was also associated with platinum resistance, and was induced by platinum exposure. In vivo, anti-endoglin treatment decreased tumor weight in both ES2 and HeyA8MDR models when compared to control (35-41% reduction, p<0.05). Endoglin inhibition with carboplatin was associated with even greater inhibitory effect when compared to control (58-62% reduction, p<0.001).
Conclusions
Endoglin downregulation promotes apoptosis, induces significant DNA damage through modulation of numerous DNA repair genes, and improves platinum sensitivity both in vivo and in vitro. Anti-endoglin therapy would allow dual treatment of both tumor angiogenesis and a subset of aggressive tumor cells expressing endoglin and is being actively pursued as therapy in ovarian cancer.
Keywords: Endoglin, CD105, platinum resistance, ovarian cancer, siRNA, cancer stem cells
Introduction
Epithelial ovarian carcinoma (EOC) remains the most lethal gynecologic malignancy.1 While initial response to first-line therapy (consisting of surgical cytoreduction and combination platinum/taxane therapy) is usually effective, the majority of patients will ultimately recur with chemotherapy-resistant cancer and succumb to disease. This emphasizes the need for novel therapies aimed at targeting the population of cancer cells most resistant to initial therapy.
Endoglin (ENG) is a 180kDa disulfide-linked homodimer transmembrane protein most prominently expressed on proliferating endothelial cells. It is a well-characterized angiogenic marker that is upregulated during angiogenesis, and is overexpressed in vascular endothelium in malignancies including ovarian, leukemia, gastrointestinal stromal tumors (GIST), melanoma, and laryngeal cancers, but is rarely expressed in non-endothelial cells.2-3 It is a co-receptor of TGFBR2 that binds TGF-B and is an important mediator of fetal vascular/endothelial development.4 Recently, anti-angiogenic agents have received extensive attention as new therapeutic modalities, and CD105 has become an additional target by which intratumoral angiogenesis may be targeted.5-6 However, endoglin may serve in a capacity beyond angiogenesis alone. Studies in GIST7 and breast cancer8 suggest that endoglin is upregulated not only in tumor endothelial cells, but also in actual tumor cells, and is associated with poor prognosis. Soluble endoglin has also been noted in ovarian cancer ascites,9 and increased endoglin expression in ovarian cancer endothelial cells is associated with poor prognosis.10 Additionally, we have recently shown that while endoglin is rarely expressed in primary ovarian cancer cells, it is frequently expressed in recurrent platinum-resistant tumor cells, as compared to the primary untreated tumor.11 These findings suggest a broader role of endoglin in tumor cell biology beyond that of endothelial expression alone. The goal of our current study is to evaluate the effects of targeting tumor-specific endoglin in ovarian cancer both in vitro and in vivo and explore the mechanisms by which endoglin may contribute to chemoresistance.
Methods and Materials
Evaluation of endoglin expression in ovarian cancer cell lines
Multiple ovarian cancer cell lines were evaluated for the presence of endoglin, including HeyA8, HeyA8MDR, ES2, A2780ip2, A2780cp20, A2780cp55, SKOV3ip1, SKOV3TRp2, IGROV-AF1, and HIO-180. Cells were maintained in RPMI-1640 medium with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT). The taxane-resistant cell line HeyA8MDR was maintained in the same media with the addition of 150 ng/ml of paclitaxel. Cell lines were routinely screened for Mycoplasma (GenProbe detection kit; Fisher, Itasca, IL) and all experiments performed on 70-80% confluent cultures. Cells less than 20 passages from confirmation of genotype by STR analysis were used.
Cell lysates were collected in modified radioimmunoprecipitation assay lysis buffer with protease inhibitor cocktail (Roche, Manheim, Germany). Immunoblot analysis was performed using rabbit anti-endoglin antibody (Sigma, St. Louis, MO) at 1:500 dilution overnight at 4°C. A loading control was performed with mouse anti-β-actin antibody (Clone AC-15, Sigma) at 1:20,000 dilution for 1 hour at RT. After washing, membranes were incubated in HRP-conjugated goat anti-rabbit (for Endoglin) or goat anti-mouse (for β-actin) secondary antibodies (Bio-Rad, Hercules, CA). Visualization was performed by enhanced chemiluminescence (Pierce Thermo Scientific, Rockford, IL).
Immunohistochemistry
Cell lines in culture were washed with ice cold PBS twice, then fixed by applying 100% ice cold methanol for 10 min. Cells were rehydrated with PBS. Endogenous peroxidase was blocked with 3% H2O2 in methanol for 15min at RT. The slides were incubated in 10% normal goat serum for 1 hr at RT. The primary anti-endoglin antibody (Sigma HPA011862) was diluted in 10% normal goat serum at 1:50. The slides were kept at 4°C overnight. Biotin-labeled secondary antibody was applied on cells at the concentration of 1:2000 for 1hr at RT, followed by avidin-biotin peroxidase buffer. DAB (3,3’-diaminobenzidine) was used as chromophore to detect the staining. To visualize endoglin expression in tumor sections, formalin-fixed paraffin-embedded tissue was cut in sections of 5uM thickness. Slides were warmed for 15 minutes and sequentially deparaffinized. Antigen retrieval was carried out in Citrate buffer (pH6.0) in a pressure cooker at high pressure for 5 min. Endogenous peroxidase was quenched by 3% H2O2 in methanol for 15 min. Slides were incubated in 10% normal goat serum for 1hr at RT. Slides were then incubated (4°C, Overnight) in antibody against endoglin (Sigma HPA011862) in 10% normal goat serum at 1:200 dilution. Detection was carried out using biotin labeled secondary antibody against rabbit at dilution of 1:2000 incubated at RT for 1 hr, followed by avidin-biotin peroxidase buffer. DAB (3,3’-diaminobenzidine) was used as chromophore.
Flow cytometry
After trypsinisation and centrifugation, the cell pellet was washed and resuspended in washing buffer (PBS containing 2% FBS and 0.1% sodium azide).1×107 cells were resuspended in 50μls of 10% goat buffer for 1hr kept on ice. Cells were incubated in antibody against endoglin 1:100 (Sigma HPA011862) in 10% goat serum for 1hr on ice. Alexa-488-conjugated anti rabbit antibody was applied on cells for 30 minutes and incubated on ice. The cells were washed twice in PBS and analyzed by FACS.
Endoglin Downregulation by siRNA transfection
In order to determine the effects of endoglin downregulation in ovarian cancer cells, transient knockdown was accomplished with anti-endoglin siRNA. Lipofectamine 2000 (Invitrogen) transfection was performed on Hey8MDR and ES2 cell lines using control siRNA (target sequence: 5’-UUCUCCGAACGUGUCACGU-3’, Sigma) lacking known human or mouse targets, or one of two different Endoglin-targeting constructs (5’-CAAUGAGGCGGUGGCAAU-3’ [“ENG_A”} or 5’-CAGAAACAGUCCAUUGUGA-3’ [“ENG_B”], Sigma). These anti-human sequences have no more than 8 consecutive bp homology with murine CD105 (by BLASTN) and therefore should not affect murine endoglin expression. Lipofectamine was added to 5μg siRNA at a 3:1 v/v ratio (or as otherwise specified, as in Figure 1E) were incubated for 20 min at RT, added to cells in serum-free RPMI to incubate for 12 hours in 6- well plates, then maintained in 10% FBS/RPMI for an additional 12 hours, trypsinized and re-plated on a 96-well plate at a concentration of 2,000 cells per well. Cells were treated with vehicle or increasing doses of carboplatin or paclitaxel to generate an IC 50 curve. After 5 days, cells were washed and incubated with MTT reagent (Sigma) for 2 hours at 37°C. Media was then removed, cells dissolved in DMSO, and optical density measurements at 570 nm read with a spectrophotometer. The IC50 was the chemotherapy concentration giving the ODIC50 reading, calculated by the formula ODIC50 = [(ODMAX − ODMIN)/2 + ODMIN]. Assays were repeated in triplicate.
FIGURE 1.
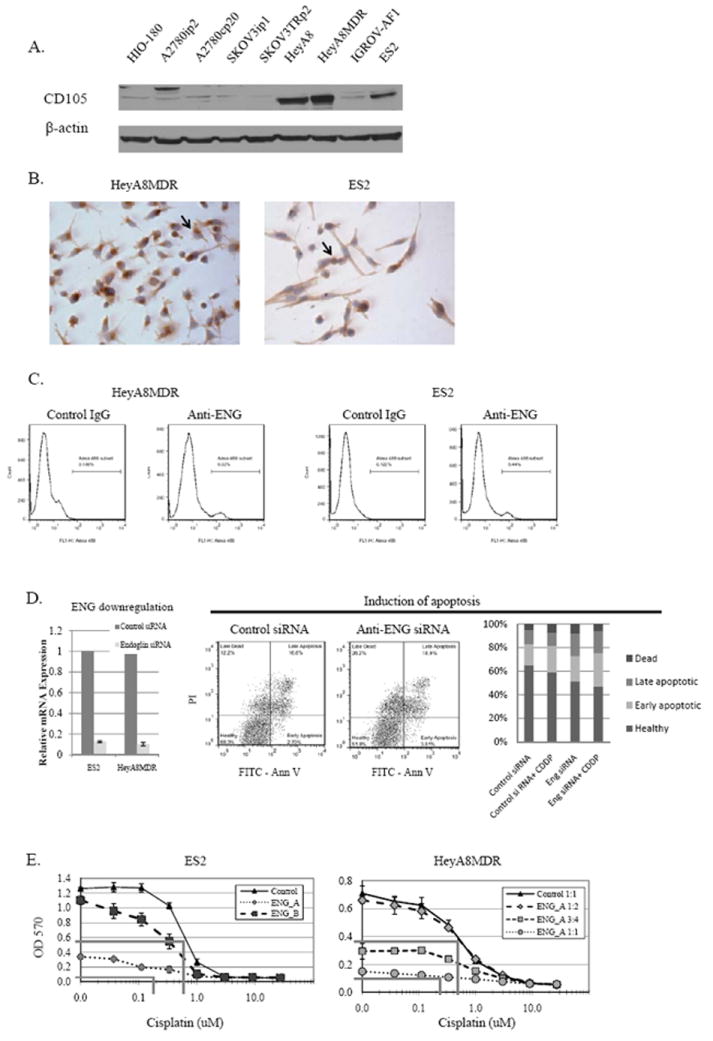
A) Endoglin expression in multiple ovarian cancer cell lines, as measured by Western blot. B) As assessed by IHC, endoglin expression is predominantly cytoplasmic, though some cells with strong membranous staining are noted (arrows). C) A small but distinct endoglin-positive population is seen by flow cytometry. D) Endoglin was effectively downregulated with siRNA. By TUNEL assay, Annexin V/PI co-fluorescence demonstrate a decrease in viable cells, and an increase in both early and late apoptosis, both alone and in combination with cisplatin. E) Cells treated with increasing doses of cisplatin after endoglin downregulation were also assessed by MTT, with the OD570 reflecting the absorbance produced by viable cells. Endoglin downregulation resulted in a significant reduction in cell viability, and increased cisplatin chemosensitivity about 4-fold in ES2 model and 2-fold in HeyA8MDR. Lines denoting the calculated IC50 for control and endoglin-siRNA treatment are shown (grey lines).
Apoptosis analysis
Analysis of apoptosis was performed with the Annexin V assay combined with propidium iodide (PI, eBiosciences #88-8005-74). ES2 and HeyA8MDR cells were transfected with either control siRNA or anti-endoglin siRNA in serum-free RPMI growth media for 12 hours, followed by maintenance in 10% FBS/RMPI. Cells were trypsinized 96 hours following transfection, washed twice in PBS, and then resuspended in 200μL 1× binding buffer containing 5μL of Annexin V. 10μL of PI was added, cells were incubated for 10 minutes at RT in the dark. Fluorescent signal (FITC and PI) in cells were analyzed by FACS and data were analyzed with FlowJo v.7.6.1 (Ashland, OR).
Alkaline comet assay
ES2 cells (n=400,000 in 6-well plate) were transfected with endoglin and control siRNA. Twenty-four hours following transfection, cells were exposed to cisplatin without supplemental SVF at a concentration of 1μM (the approximate IC80 level for this line) for either 1 or 4 hours, carefully rinsed to remove the drug, and cultured in regular media. Vehicle or control siRNA were included in all experiments. At the indicated time points, cells were collected and subjected to alkaline comet assay according to the manufacturer’s instructions (catalog # 4250-050-K; Trevigen). Briefly, cells were combined with low melting agarose onto CometSlides (Trevigen). After lysis, cells were subjected to electrophoresis and stained with SYBR green. Subsequently, cells were visualized using fluorescent microscopy (Carl Zeiss, Thornwood, NY). At least 200 comet images were analyzed for each time point using Comet Score software (version 1.5; TriTek Corp.). The number of tail-positive cells with small and large nuclei was manually counted by an examiner blinded to treatment group, and expressed as a percentage of all cells evaluated. Experiments were repeated in triplicate.
γ-H2AX foci formation
ES2 cell lines were cultured and seeded on sterile cover slips. Twenty-four hours following transfection with control or anti-endoglin siRNA, cells were exposed to 1μM cisplatin for either 1 or 4 hours, carefully rinsed to remove the drug, and cultured in regular media. Following the treatment period, IHC was performed as previously described12-13 with slight modification for foci staining. Briefly, cells were rinsed in phosphate buffered saline (PBS) and incubated for 5 minutes at 4°C in ice-cold cytoskeleton buffer (10mM Hepes/KOH, pH 7.4, 300mM sucrose, 100mM NaCl, 3mM MgCl2) supplemented with 1mM PMSF, 0.5mM sodium vanadate and proteasome inhibitor (Sigma, 1:100 dilution) followed by fixation in 70% ethanol for 15 minutes. The cells were blocked and incubated with primary antibody (1:500 dilution, anti-phosphoH2AX Ser139, Millipore, catalog # MI-07-164). The secondary antibody was anti-rabbit Alexa Fluor 488 conjugated antibody (1:2000 dilution; Invitrogen). DAPI (Invitrogen, catalog # D21490) was used for nuclear staining. The cover slips were subsequently mounted onto slides with mounting media (Aqua poly mount, Polysciences, Inc. catalog # 18606) and analyzed via fluorescence microscopy (Carl Zeiss, Thornwood, NY). Positive and negative controls were included on all experiments. A total of 500 cells were assessed. For foci quantification, cells with greater than 10 foci were counted as positive according to the standard procedure. Experiments were repeated in triplicate.
RNA extraction from cell lines
Total RNA was isolated from ovarian cancer cell lines using Trizol reagent (Invitrogen, Carlsbad, CA) per manufacturer’s instructions. RNA was then DNase treated and purified using the RNeasy Mini Kit (QIAGEN, Hilden, Germany). RNA was eluted in 50 μL of RNase-free water and stored at -80°C. The concentration of all RNA samples was quantified by spectrophotometric absorbance at 260/280 nm using an Epoch Microplate Spectrophotometer (BioTek Instruments, Winooski, VT).
DNA repair qPCR array
ES2 and HeyA8 cells in culture were exposed to siRNA against endoglin in Lipofectamine 2000 as described above. After 48 hours, cells were collected and mRNA extracted. Two replicates per cell line were performed. These four samples were then subjected to a quantitative PCR array consisting of 84 genes from DNA damage/repair pathways (plus additional housekeeping genes; the RT2 Profiler PCR Array Human DNA Damage Signaling Pathway, SA Biosciences Cat# PAHS-209Z, performed per manufacturer’s instructions). Briefly, extracted RNA was converted to cDNA and amplified using the RT2 FFPE PreAMP cDNA Synthesis Kit (SABiosciences, Frederick, MD). Quality of cDNA was confirmed with the Human RT2 RNA QC PCR Array (SABiosciences). Gene expression was analyzed using the Human DNA Damage Signaling Pathway RT2 Profiler PCR Array (SABiosciences), which profiles the expression of 84 genes involved in pluripotent cell maintenance and differentiation14. Functional gene groupings consist of the ATM/ATR signaling, nucleotide excision repair, base-excision repair, mismatch repair, double strand break repair, apoptosis, and cell cycle checkpoint regulators. PCR amplification was performed on an ABI Prism 7900HT sequence detection system and gene expression was calculated using the comparative CT method15.
Reverse transcription and quantitative PCR
Extracted RNA samples were diluted to 20 ng/μL using RNase-free water. cDNA was prepared using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The resulting cDNA samples were analyzed using quantitative PCR. Primer and probe sets for ENG (PPH01140F) ATM (PPH00325C), BARD1 (PPH09451A), DDIT3 (PPH00310A), H2AFX (PPH12636B), NBN (PPH00946C), NTHL1 (PPH02720A), PPP1R15A (PPH02081E), SIRT1 (PPH02188A), ATP7B (PPH06148A), and RPLP0 (Hs99999902_m1, housekeeping gene) were obtained from SABiosciences and used according to manufacturer’s instructions. PCR amplification was performed on an ABI Prism 7900HT sequence detection system and gene expression was calculated using the comparative CT method.
Orthotopic Mouse Model
Female athymic nude mice (nu-nu) were obtained from the National Cancer Institute Frederick Cancer Research and Development Center (Frederick, MD). Mice were cared for in accordance with American Association for Accreditation of Laboratory Animal Care guidelines, the United States Health Services Commissioned Corps “Policy on Human Care and Use of Laboratory Animals,” and University of Alabama at Birmingham Institutional Animal Care and Use Committee policies. ES2 tumors were established by intraperitoneal (IP) injection of 1×106 cells suspended in 200μL of serum free RPMI media. Hey8MDR tumors were established in a similar way, using 5×105 cells. To evaluate the effectiveness of endoglin-targeted therapy in vivo, siRNA was incorporated into chitosan nanoparticles as previously described.16-17 Therapy was initiated 1 week after tumor cell injection. Mice were randomized to one of four treatments (n=10 per group): a) control siRNA alone (150 ug/kg twice weekly injected IV), b) control siRNA with IP carboplatin (160 mg), c) anti-endoglin siRNA (150 ug/kg twice weekly) alone, or d) anti-endoglin siRNA with carboplatin. All treatments were suspended in 100 μL 0.9% normal saline (NS). Mice were monitored for adverse effects, and all treatment groups sacrificed when control mice became uncomfortable with tumor burden. ES2 tumors behaved aggressively, and were harvested following 2 weeks of treatment. Hey8MDR tumors were harvested after 3 weeks of therapy. Mouse weight, ascites volume, tumor weight and distribution of tumor were recorded. Representative tumor samples were obtained from 5 mice in each treatment group, formalin-fixed, paraffin-embedded, and cut into 5 micron sections for evaluation of Proliferating Cell Nuclear Antigen (PCNA), Terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling assay (TUNEL), γ H2AX (phosphorylation of Histone 2A protein) and 53BP1 (a mediator of the DNA damage checkpoint).
Tumor PCNA Immunohistochemistry and TUNEL
Sections were deparaffinized and re-hydrated, and antigen retrieval was performed with citrate buffer (pH 6.0) in pressure cooker for 5 minutes. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide solution in methanol for 15 minutes. Sections were blocked with CytoQ immune diluent and block and probed with PCNA primary antibody (PCNA-PC10, Cell signaling Technology, 1:5000 dilution) at 4°C overnight. Sections were washed and incubated with the Mach 3 mouse HRP polymer system. After rinsing, the sections were incubated with DAB chromophoric solution (Scytek Labs, Utah, USA) for 5 min at room temperature, then counterstained with Gill’s hematoxylin (Ricca chemicals). Four 40× microscopic fields were counted from each section, averaged over 5 mice in each treatment group, and expressed as a percentage of the total number of tumor cells. Apoptosis was determined by TUNEL assay with a colorimetric apoptotic cell detection kit (Promega), per manufacturer’s instruction. As with PCNA IHC, 4 microscopic fields at 40x magnification were evaluated from each section. Stained cells were recorded as a percentage of the total number of tumor cells.
Tumor γH2AX and 53BP1 IHC
Formalin fixed tissues were heated at 60°C for 1hr and rehydrated according to standard protocol. Subsequently, the tissues were permeabilized in 0.5% Triton X-PBS for 10 min, blocked in 2% BSA-0.1% Triton-X-PBS for 1 hr, and incubated with primary antibodies (1:500 dilution, anti phospho H2AX Ser139, Millipore, catalog # MI-07-164; 1:500 dilution, anti-53BP1, Novus Biologicals, catalog # NB100-304). The secondary antibody was anti-rabbit Alexa Fluor 488 conjugated antibody (1:2000 dilution; Invitrogen). DAPI (Invitrogen, catalog # D21490) was used for nuclear staining. The slides were subsequently mounted using mounting media (Aqua poly mount, Polysciences, Inc. catalog # 18606) and analyzed via fluorescence microscopy (Carl Zeiss, Thornwood, NY). Positive and negative controls were included on all experiments. A total of 500 cells were assessed. For foci quantification, cells with greater than 10 foci were counted as positive according to the standard procedure. Experiments were repeated in triplicate. Data show the mean and SEM.
Statistics
Analysis of normally distributed continuous variable was performed using a two-tailed Student’s t-test. Those data with alternate distribution were examined using a nonparametric Mann-Whitney U test. A p<0.05 was considered statistically significant.
Results
Effects of endoglin downregulation on cell viability and platinum sensitivity
Endoglin is expressed by multiple ovarian cancer cell lines (Figure 1A), most prominently in HeyA8, HeyA8MDR, and ES2 cells. Weak expression was detected in the HIO-180, A2780ip2, A2780cp20, SKOV3ip1, SKOV3TRp2, and IGROV-AF1 cell lines. This was previously demonstrated at the mRNA level by quantitative PCR11. To confirm that expression was predominantly at the cell surface, consistent with its function as a co-receptor for TGFβ, we performed immunohistochemistry on the ES2 and HeyA8MDR cell lines. Surprisingly, the predominant staining was noted n the perinuclear cytoplasm (Figure 1B). This was confirmed by flow cytometry, where interestingly not only was membranous staining rare, but there was a very distinct separate population with 100-fold fluorescent intensity (rather than a global shift among all cells), consistent with a separate small population of cells with strong endoglin surface expression (Figure 1C). This population represented 6.0% of HeyA8MDR and 5.4% of ES2 cells. On close examination of IHC on cultured cells, a minority of the cells could be seen to have strong membranous expression of CD105 (arrows, Figure 1B). A separate endoglin-positive population has previously been noted in renal cell carcinoma cells, which did exhibit stem-cell properties.18 However, these data are conclusive that the majority of endoglin expression in ovarian cancer is cytoplasmic, suggesting a role other than just as a co-receptor for TGF-beta.
To determine whether siRNA-mediated downregulation of endoglin had significant effects on viability and chemosensitivity, two different siRNA constructs (ENG_A siRNA and ENG_B siRNA) were examined. Both effectively reduced endoglin expression at 48 hours at the mRNA (Figure 1D) and protein level11). Both were previously shown to reduce cell viability11. To determine the mechanism by which endoglin knockdown reduced viability, evaluation of apoptosis was performed by the TUNEL assay. Annexin V/PI co-fluorescent staining performed 48 hours following transfection indicated significantly fewer viable cells in those treated with anti-endoglin siRNA than those treated with control siRNA (47.2% vs. 65.1%, p<0.05). A sample flow cytometry plot and a graph of average over three experiments are shown in Figure 1D. Those treated with anti-endoglin siRNA had increased percentages of cells in both early apoptosis (21.5% vs. 17.9%, p<0.05) and late apoptosis (18.9% vs. 12.0%, p<0.05). Effects were more pronounced when combined with cisplatin. In order to determine whether Endoglin knockdown had an effect on viability in combination with chemotherapy, cells were exposed to siRNA, then re-plated after 24 hours, and incubated with increasing concentrations of cisplatin or paclitaxel. Because endoglin downregulation alone was associated with substantial cell death in the HeyA8MDR model, knockdown was performed with several dilutions of siRNA in an effort to more clearly delineate effects on platinum sensitivity. In both ES2 (normal IC50 for cisplatin = 0.7μM) and HeyA8MDR (normal IC50 for cisplatin = 0.65μM) models, increased cisplatin chemosensitivity was noted (up to 4-fold and 2-fold reduction in IC50, respectively, Figure 1E). Similar experiments were performed with paclitaxel, which did not show an increased sensitization with endoglin downregulation (data not shown).
Downregulation of endoglin induces DNA damage in vitro
Because platinum toxicity is mediated primarily through induction of DNA damage, we evaluated whether the enhanced cisplatin sensitivity from endoglin knockdown was a result of increased DNA damage. DNA damaging agents can induce both single-stranded breaks (SSBs) and double stranded breaks (DSBs) which can lead to initiation of apoptotic pathways. DNA damage in the ES2 line was first assessed via an alkaline comet assay, which detects both SSB and DSB. As quantified in Figure 2A, increased DNA damage over 24 hours was observed with cisplatin, endoglin downregulation with siRNA, and the combination (although combination therapy was not significantly increased compared to either single-agent treatment). A representative section demonstrating common effects on nearly all cells is shown (Figure 2B). Because a long comet tail can be the result of either DNA damage without death or apoptosis-associated DNA release, the nucleus size was also quantified. A small nucleus would be associated with apoptosis, whereas a long comet tail associated with a normal (larger) nucleus would indicate just DNA damage. As shown in Figure 2C for cells treated for 24 hours, those cells with a long tail present predominantly still had a large nucleus. Because most toxic effects on viability noted previously were assessed at 48 hours or longer, this DNA damage may be a precursor to apoptosis induction. But it does demonstrate that DNA damage is the inciting event, rather than a result of apoptosis triggered by other mechanisms.
FIGURE 2.
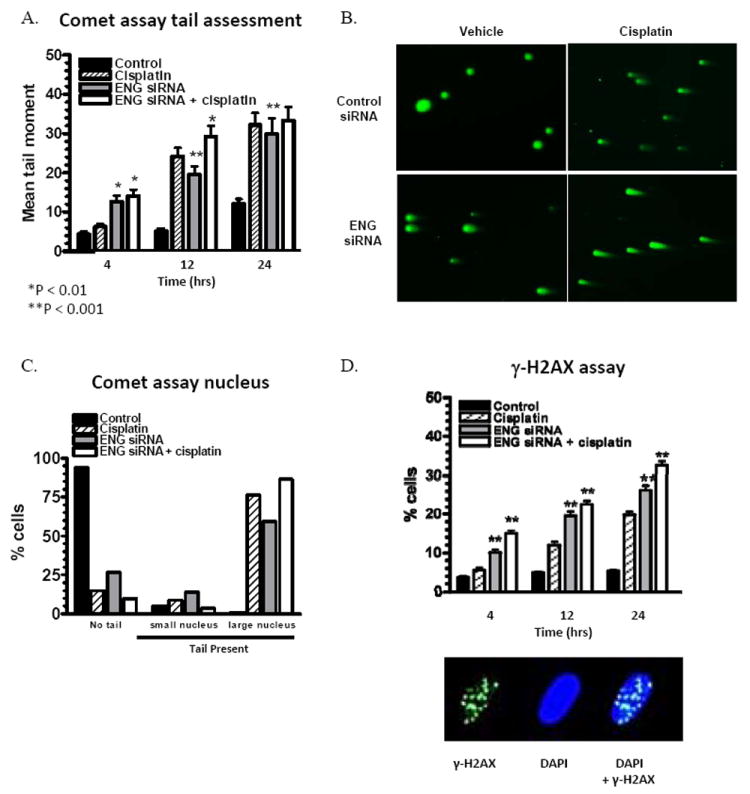
ES2 cells were evaluated for DNA damage after endoglin targeting. SiRNA-mediated endoglin downregulation induces significant persistent DNA damage, as indicated by alkaline comet assay mean tail moment (A), and visually at 24 hours (B, Original magnification, ×100). This is not a result of immediate apoptosis, as demonstrated by a predominance of large nuclei despite a prominent comet tail (C). Downregulation also induces activation of γ-H2AX foci, a specific measure of double-stranded DNA damage (D). The combination of endoglin downregulation and cisplatin on induction of γ-H2AX foci was greater than either agent alone. Error bars represent SEM.
To further characterize the specific nature of DNA damage, development of foci of activated γ-H2AX was performed (Figure 2D). ES2 cells were employed, due to the rapid toxicity and cell death noted with endoglin downregulation with HeyA8. Phosphorylation of the histone protein H2AX on serine 139 (γ-H2AX) occurs at sites flanking DNA DSBs. The phosphorylation of thousands of H2AX molecules forms a focus in the chromatin flanking the DSB site that can be detected in situ. A higher proportion of cells with persistent γ-H2AX foci was noted with endoglin downregulation, to an even greater extent than cisplatin alone. The combination of cisplatin and endoglin downregulation induced more DSB repair than either agent alone. Collectively, these data suggest that a primary mechanism of DNA damage after endoglin downregulation is through induction of double-strand breaks in DNA.
Endoglin-targeting DNA damage is through effects on multiple mediators of DNA repair
In order to determine the mechanism by which downregulation of endoglin induces DNA damage, we first subjected both ES2 and HeyA8MDR cells treated with control siRNA or endoglin-siRNA for 48 hours to a qPCR-based array of 84 genes participating in DNA damage and repair pathways. This exploratory analysis found multiple genes that were either downregulated or upregulated in response to decreased endoglin, some of which were only associated with changes in one cell line (Supplemental Table 1). Select genes were then chosen for confirmatory assessment with qPCR (Figure 3). Genes for these analyses were selected based on the degree to which they were altered, the associated p-value, and whether the change was noted in both cell lines. With endoglin downregulation, significant concurrent downregulation was noted by qPCR in H2AFX (36-43%), BARD1 (47-71%), NBN (38-41%), NTHL1 (39-53%), and SIRT1 (34-49%). A significant induction of mRNA was noted in DDIT3 (1.9-2.6-fold) and PPP1R15A (1.27-1.74-fold). There was no single DNA repair pathway subclass that comprised all affected genes, but consistent with data from the γ-H2AX assay, most were participants in either the double stranded break repair (BARD1, H2AFX, NBN) or nucleotide excision repair (SIRT1, NTHL1).
FIGURE 3.
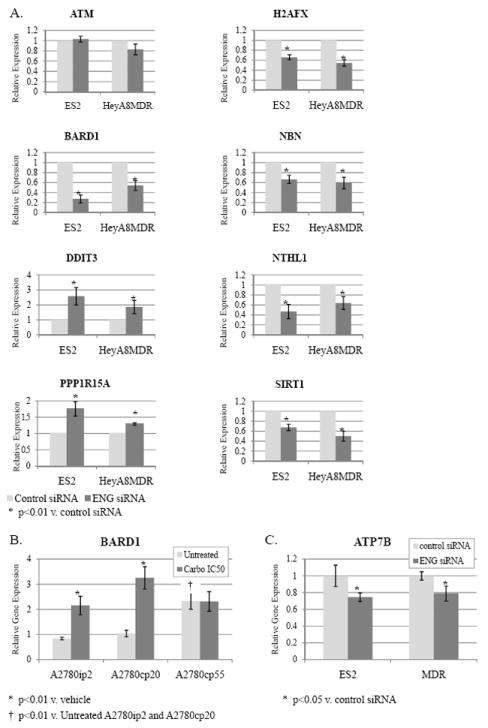
A) ES2 and HeyA8MDR cells were exposed to endoglin-targeting siRNA or control siRNA, mRNA extracted 48 hours later, and subjected to quantitative PCR for selected genes. Each collection was performed in triplicate, and the mean change over housekeeping gene presented. Significant decreases were noted in H2AFX, BARD1, NBN, NTHL1, and SIRT1. Induction of DDIT3 and PPP1R15A was also significant. B) BARD1 mRNA was assessed by qPCR in a triad of progressively platinum-resistant A780 cell lines, and noted to be significantly increased in A2780cp55 at baseline, and in A2780ip2 and A2780cp20 with exposure to carboplatin. C) The copper transporter ATP7B was also modestly, but significantly, reduced with endoglin downregulation.
The downregulation of BARD1 was particularly interesting. BARD1 is an oncogenic regulator of BRCA1, and downregulation would be expected to result in export of BRCA1 from the nucleus and impairment of DNA repair. Furthermore, BARD1 was noted to be significantly upregulated in chemoresistant tumor samples from patients, compared to their primary tumors.11 BARD1 expression is prominent in ES2 and HeyA8MDR, which follows if it is under transcriptional regulation by endoglin. Therefore, we examined BARD1 induction in response to platinum treatment in a progressively platinum-resistant triad of cell lines derived from A2780: A2780ip2 (which generates IP tumors more consistently than the parental line but is chemosensitive), A2780cp20 (having a platinum IC50 of 20μM), and A2780cp55 (with an IC50 of 55μM). The A2780cp20 and cp55 lines are stably platinum-resistant, and not chronically maintained in platinum. BARD1 expression is minimal in the parental A2780ip2 line, but increases at baseline (“Untreated”) with each degree of platinum resistance (Figure 3B).
Additionally, when exposed to an IC50 concentration of carboplatin, BARD1 mRNA production is significant increased in both A2780ip2 and A2780cp20. Levels were unchanged with carboplatin exposure in A2780cp55, likely due to its high baseline expression. A significant reduction in BARD1 with endoglin downregulation and an induction of BARD1 in response to platinum exposure strongly implicate this gene and its control on BRCA1 as a major mechanism through which endoglin downregulation may lead to DNA damage, apoptosis, and sensitivity to platinum.
In addition to enhanced DNA repair mechanisms, a major mechanism of platinum resistance is through increased export of platinum agents through copper transporters such as ATP7B.19 Therefore we also examined the effects of endoglin downregulation on ATP7B by qPCR. SiRNA-mediated targeting of endoglin resulted in a significant downregulation of ATP7B (by 20-24%, p<0.05, Figure 3C). While significant, this was not to the same extent many DNA repair genes were induced or activated.
Evaluation of tumor growth with anti-endoglin treatment in an orthotopic murine model
In order to determine if endoglin downregulation was an effective therapy in vivo, an orthotopic murine model was utilized using human specific anti-endoglin siRNA delivered within a chitosan nanoparticle. Chitosan (CH) is a natural nanoparticle that has been previously demonstrated to result in efficient delivery of siRNA to tumor after IV administration, with subsequent protein downregulation and gene-specific modulation.16, 20-22 Because the siRNA delivered is specific to the human endoglin mRNA, any observed effect would be expected to be due to targeting the tumor cells, rather than the vasculature, which would require murine-specific siRNA. ES2 and HeyA8MDR cells were injected IP, and treatment was started 1 week later with a) control siRNA-CH alone, b) control siRNA-CH plus carboplatin, c) anti-endoglin siRNA-CH alone, or d) anti-endoglin siRNA-CH plus carboplatin. Carboplatin was used instead of cisplatin because of its preferable side-effect profile in vivo, which has led to its choice as standard of care in ovarian cancer patients. Tumors demonstrated reduced growth both with endoglin downregulation alone and in combination with platinum. In the ES2 model (Figure 4A), mice treated with carboplatin had similar tumor burden to control (p=0.555), an expected result due to the highly platinum-resistant nature of the ES2 cell line, which is derived from a patient with clear cell carcinoma. Mice treated with anti-endoglin siRNA alone had a significantly reduced tumor weight, by 35.6% (p=0.014). Combined END-siRNA-CH with carboplatin was more effective that either agent alone, with a 57.7% reduction in tumor weight compared to control (p<0.001). Furthermore, combination therapy was more effective than siRNA-endoglin-CH alone, with an additional 34.3% reduction (p=0.033) In the HeyA8MDR model (Figure 4B), mice treated with carboplatin, endoglin-siRNA-CH, or combination therapy had significantly less tumor weight when compared to control (34% reduction p=0.027, 41.2% reduction p=0.002, and 61.2% reduction p<0.01, respectively). Those treated with carboplatin and control siRNA-CH had similar tumor burden reduction as those treated with endoglin-siRNA-CH (p=0.628). Combination therapy was again more effective than either single-agent carboplatin (additional 40.6% reduction, p=0.069), or endoglin-siRNA alone (34%, p=0.048). In the resected tumors, reduced expression of endoglin was confirmed with immunohistochemistry, in both groups of tumors treated with endoglin-siRNA-CH. Representative sections are pictured (Figure 4C). With both models, there was not a significant difference in mouse weight in any group. The distribution of tumor was also similar in all groups, suggesting there was not a significant effect on particular site of growth, adhesion, or migration.
FIGURE 4.
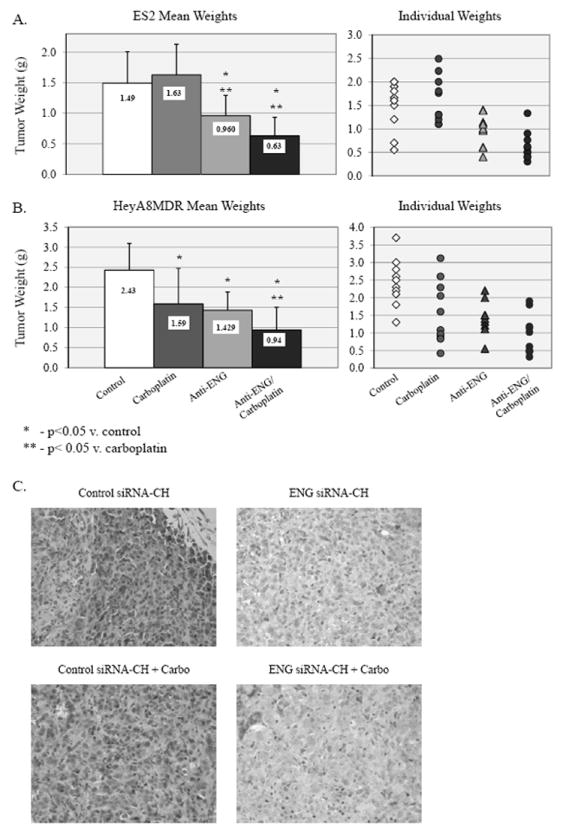
An orthotopic murine model using ES2 and HeyA8MDR cell lines was employed to evaluate treatment with control siRNA-CH alone, control siRNA-CH with carboplatin, anti-endoglin siRNA-CH alone, or anti-endoglin siRNA-CH plus carboplatin. A) In the ES2 model, carboplatin was ineffective, as expected given the platinum-resistant nature of the ES2 cell line. Mice treated with anti-endoglin siRNA-CH alone and combined with carboplatin demonstrated less tumor burden when compared to control or carboplatin alone. Those treated with both anti-endoglin siRNA-CH and carboplatin also demonstrated reduced tumor burden when compared to those endoglin-siRNA-CH alone (p=0.03). B) In the HeyA8MDR model, tumors were smaller in mice treated with carboplatin or anti-endoglin siRNA-CH alone, and again combination therapy was more effective than either agent alone (p<0.05). C) By qualitative assessment with IHC, endoglin expression was reduced in the tumors treated with endoglin-siRNA-CH therapy.
Endoglin downregulation induces DNA damage and apoptosis in vivo
Our in vitro findings suggest a role of DNA damage and apoptosis following endoglin downregulation. To validate these findings in vivo, tumors from each treatment group described above were examined for proliferation, apoptosis, and induction of DNA damage. PCNA IHC was performed and revealed no significant differences in percentage of PCNA positive cells, with approximately half of cells being positive in each treatment group (Figure 5A). A lack of effect on progression through the cell cycle and proliferation may explain why combination with taxanes was not synergistic with endoglin downregulation in vitro. TUNEL assay was performed to evaluate to detect differences in apoptosis between treatment groups. Control, carboplatin and anti-endoglin siRNA groups were not significantly different. However, the cohort receiving combination therapy had a significantly higher percent of apoptotic cells when compared to control (p<.001, Figure 5B). This increase, though statistically significant, is relatively small, which may be due to clearance of dead cells over the course of the 4-week experiment. To determine if DNA damage was still noted in the tumors collected at completion of therapy, fluorescent IHC was performed to evaluate for γ-H2AX as an indicator of in vivo DSB. A significantly higher amount of DNA damage was detected in both treatment groups receiving anti-endoglin treatment than either control or single-agent carboplatin treatment (Figure 5C). Additionally, 53BP1 is a mediator of DNA damage response and a tumor suppressor whose accumulation on damaged chromatin promotes DNA repair and enhances DNA damage response signaling. A significantly higher number of 53BP1-positive cells was noted in both cohorts that received anti-endoglin treatment when compared to either control or single-agent platinum (Figure 5D). These data are consistent with in vitro studies demonstrating that endoglin downregulation alone leads to DNA damage and apoptosis.
FIGURE 5.
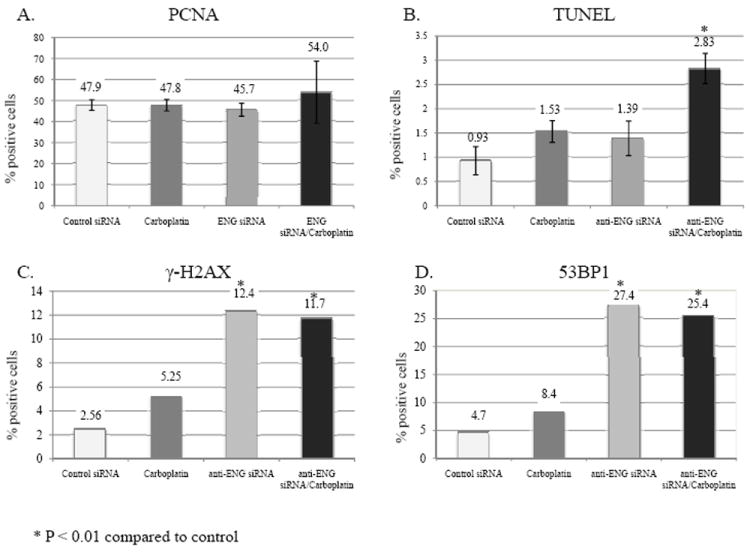
Tumors from each treatment group in our orthotopic mouse model were collected and analyzed by PCNA immunohistochemistry, TUNEL assay, γ-H2AX IHC and 53BP1 IHC. A) There were no significant differences in PCNA IHC, with approximately half of cells being positive. B) There was a significant increase in apoptosis in the cohort receiving combination therapy when compared to control as demonstrated by TUNEL assay. C) Fluorescent IHC was performed to evaluate for γ-H2AX as an indicator of DNA damage. There was a significantly higher amount of DNA damage in both treatment groups receiving anti endoglin treatment when compared to control or single-agent carboplatin. D) Lastly, 53BP1 is a key protein in the DNA damage checkpoint that was evaluated by IHC. A significantly higher amount of 53BP1 was noted in both cohorts that received anti-endoglin treatment when compared to either control or single agent carboplatin.
Discussion
Endoglin is overexpressed in solid tumor vasculature and is a reliable marker of angiogenesis.5 Multiple anti-angiogenic therapies have been studied in ovarian cancer, and anti-endoglin therapy has been proposed for several cancers in which increased endothelial endoglin expression has been noted.23 However, to date, few studies address the expression of endoglin on tumor cells and its potential role in cancer progression. Building off our previous findings that Endoglin is increase in recurrent samples when compared to matched primary tumors11, we have demonstrated that endoglin expression is highly expressed in many ovarian cancer cell lines, and that downregulation results in induction of cell death through induction of DNA damage and a synergistic killing effect with platinum agents both in vitro and in vivo. These novel findings demonstrate that therapeutics targeting endoglin may affect both the vasculature and malignant cells within the tumor microenvironment.
The primary canonical role of endoglin is as a co-receptor for TGF-beta.24-26 As such, its expression on endothelial cells is primarily on the cell membrane.27 However, we interestingly found endoglin expression in ovarian cancer cells was predominantly cytoplasmic, and clustered together in the perinuclear region of the cell. This would suggest that endoglin either has a separate TGF-beta-independent function dependent on nuclear proximity, or trafficking to the cell membrane is an important component of its regulation. Only a small (5-6%), but well-defined population had surface expression. This distinct population would be consistent with a cancer stem cell-like population, as has been previously described in endoglin-positive renal cell carcinoma18. Endoglin-positive meningioma cells have similar increased tumorigenicity and capacity to differentiate into adipocytes and osteocytes.28
Henriksen et al. evaluated endoglin expression in primary ovarian cancer cells and found that high tumor cell endoglin staining correlated with short overall survival.29 Another group has shown that cells from cultured ascites that progressed towards a mesenchymal phenotype were high in endoglin.30 We identified endoglin as a potential target for therapeutics through a screen of stem cell pathways overexpressed in recurrent ovarian cancer samples. Among members of the TGF-β, Notch, Wnt, and Hedgehog pathways, endoglin was most significantly and consistently overexpressed in recurrent ovarian cancer samples when compared to their matched primaries, suggesting a role in chemoresistance.11 We specifically examined stem cell pathways to address the question of whether the cancer stem cell population may be responsible for surviving initial chemotherapy. Endoglin has previously been implicated in stem cell biology, having originally been described on hematopoietic progenitor cells31, and later demonstrated to identify precursor cells capable of tissue-specific differentiation32-33.
It makes sense that cells with prolonged survival, such as stem/progenitor cells, would reply on pathways to mediate DNA damage. Because of the association noted with increased endoglin expression in platinum (and taxane)-resistant recurrent ovarian cancers,11 and the contribution of enhanced DNA repair for platinum resistance,19 we further examined the contribution of endoglin to DNA repair. We have found a previously unknown contribution of endoglin to expression of numerous DNA repair genes. These encompass several subtypes of DNA repair, predominantly double stranded break repair (BARD1, H2AFX, NBN), but also nucleotide excision repair (SIRT1, NTHL1), and cell cycle arrest (DDIT3, PPP1R15A), which may be a reactionary process in order to accomplish DNA repair. Recently BARD1 has been implicated in ovarian cancer pathogenesis for its interaction with BRCA1 and 2. BARD1 and BRCA1 interact with each other through their amino terminal RING finger domains. This interaction is required for BRCA1 stability, as well as for nuclear localization. The BRCA1-BARD1 complex serves as an E3 ubiquitin ligase, which has been noted to have critical activity in both the cell cycle check point through H2AX, NPM and γ-tubulin and in DNA fragmentation.34-35 Additionally, patients with mutations of both BARD1 and BRCA2 have a substantially increased risk for development of both breast and ovarian cancer. While BARD 1 has been found to interact and co-localize with BRCA1 at the spindle poles in early mitosis, it also interacts with BRCA2 at late mitosis in the midbody. Therefore BARD1 has been found to sequentially link the function of these36 two proteins. In our analysis, BARD1 expression was reduced by 50-75% and H2AX expression was reduced 35-50% following endoglin knockdown. endoglin-mediated downregulation of BARD1 and its subsequent effects on BRCA1 and 2 and H2AX may therefore explain why we found substantial decreased cell viability, DNA damage and increased apoptosis.34
Silent Information Regulator Type 1 (SIRT1) is a nicotinamide adenine dinucleotide-dependent class III histone deacetylase (HDAC). SIRT1 has is associated with longevity and has been found to act primarily by inhibiting cellular senescence. SIRT1 is up-regulated in tumor cell lines and human tumors, and may be involved in tumorigenesis.36 It has also been found to be over-expressed in chemoresistant tumors of cancer patients. SIRT1 inhibition leads to decrease in MDR1 expression and increase in drug sensitivity in ovarian cancer cell lines.37 Our research suggests that Endogin knockdown was associated with a 30-50% reduction in SIRT1. This inhibition may help account for the increased platinum sensitivity we found with endoglin downregulation.
In regards to therapeutic development in cancer patients, delivery of siRNA constructs has the potential to offer long duration of target inhibition as well as reduced toxicity compared other approaches.16, 20, 38-44 However, development of a delivery modality for siRNA constructs remains the rate-limiting step in translational research. Early delivery modalities included delivery of “naked” siRNA. Later attempts included high-pressure siRNA injections and intratumoral injections, neither of which has demonstrated substantial success. The development of chitosan encapsulation and nanoliposomes to deliver siRNA has become widely accepted in translational studies and is and promising as a therapeutic modality as modifications to enhance in vivo delivery progress.22 SiRNA mediated therapeutics are being used in ongoing trials with patients with macular degeneration, AIDS, malignant melanoma, acute renal failure, hepatitis B, and now in cancer patients, where phase I trials are in development. One particular advantage of siRNA-based therapeutics over conventional treatment modalities would apply to endoglin-based targeting. If indeed the cytoplasmic portion of endoglin is important to chemoresistance, downregulation of production at the mRNA level may be more effective than antibody-based targeting currently aimed at inhibiting angiogenesis.45-46
Because of the rarity of endoglin expression in normal tissues, anti-endoglin therapy has the potential to offer tumor-directed therapy in addition to anti-angiogenic therapy. Anti-endoglin therapy is being explored as a therapeutic in several cancers as an anti-angiogenic agent. In ovarian cancer, endoglin-targeted therapies may offer the additional advantage of targeting tumor cells overexpressing endoglin, including platinum-resistant tumors. Its effects on BRCA1 and 2 and H2AX through BARD1 downregulation, and its association with SIRT1 downregulation contribute to DNA damage repair and enhancement of platinum sensitivity. Our data strongly suggest that endoglin-targeted therapy has the potential to improve platinum sensitivity through induction of DNA damage and should be actively pursued as a potential therapy in the treatment of ovarian cancer.
Supplementary Material
Statement of Translational Relevance.
Ovarian cancer remains the most lethal gynecologic malignancy, largely due to its high rate of chemoresistant recurrence. Endoglin (CD105) is overexpressed on tumor-associated endothelial cells and is a target for anti-angiogenic therapy, but expression on tumor cells has only been recently demonstrated. In the current study, we demonstrate that endoglin is actually predominantly expressed in the cytoplasm of malignant cells, and downregulating endoglin promotes apoptosis, induces DNA damage, and sensitizes cells to platinum therapy in vitro and in vivo. This occurs through effects on numerous DNA repair genes, most prominently BARD1. The novel demonstration of efficacy in targeting tumor cells themselves, in addition to the previously-recognized effects of targeting vasculature, make this therapeutic an attractive mechanism to target both compartments of the tumor microenvironment.
Acknowledgments
Funding support provided in part by the University of Alabama at Birmingham Center for Clinical and Translational Science (5UL1RR025777), the Reproductive Scientist Development Program through the Ovarian Cancer Research Fund and the National Institutes of Health (K12 HD00849), and the Department of Defense Ovarian Cancer Research Academy (OC093443), the Ovarian Cancer Research Fund, U54 CA151668, P50 CA083639, and the RGK Foundation.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: A Cancer Journal for Clinicians. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Gomez E, Del Castillo G, Juan Francisco S, Lopez-Novoa JM, Bernabeu C, Quintanilla M. The role of the TGF-beta coreceptor endoglin in cancer. ScientificWorldJournal. 2010;10:2367–84. doi: 10.1100/tsw.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marioni G, Staffieri A, Manzato E, Ralli G, Lionello M, Giacomelli L, et al. A higher CD105-assessed microvessel density and worse prognosis in elderly patients with laryngeal carcinoma. Arch Otolaryngol Head Neck Surg. 2011 Feb;137(2):175–80. doi: 10.1001/archoto.2010.244. [DOI] [PubMed] [Google Scholar]
- 4.Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999 Jan 8;274(2):584–94. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- 5.Dallas NA, Samuel S, Xia L, Fan F, Gray MJ, Lim SJ, et al. Endoglin (CD105): a marker of tumor vasculature and potential target for therapy. Clin Cancer Res. 2008 Apr 1;14(7):1931–7. doi: 10.1158/1078-0432.CCR-07-4478. [DOI] [PubMed] [Google Scholar]
- 6.Fonsatti E, Altomonte M, Arslan P, Maio M. Endoglin (CD105): a target for antiangiogenetic cancer therapy. Curr Drug Targets. 2003 May;4(4):291–6. doi: 10.2174/1389450033491073. [DOI] [PubMed] [Google Scholar]
- 7.Gromova P, Rubin BP, Thys A, Cullus P, Erneux C, Vanderwinden JM. ENDOGLIN/CD105 is expressed in KIT positive cells in the gut and in gastrointestinal stromal tumors. J Cell Mol Med. 2011 Mar 24; doi: 10.1111/j.1582-4934.2011.01315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davidson B, Stavnes HT, Forsund M, Berner A, Staff AC. CD105 (Endoglin) expression in breast carcinoma effusions is a marker of poor survival. Breast. 2010 Dec;19(6):493–8. doi: 10.1016/j.breast.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Bock AJ, Tuft Stavnes H, Kaern J, Berner A, Staff AC, Davidson B. Endoglin (CD105) expression in ovarian serous carcinoma effusions is related to chemotherapy status. Tumour Biol. 2011 Feb 26; doi: 10.1007/s13277-011-0157-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taskiran C, Erdem O, Onan A, Arisoy O, Acar A, Vural C, et al. The prognostic value of endoglin (CD105) expression in ovarian carcinoma. Int J Gynecol Cancer. 2006 Sep-Oct;16(5):1789–93. doi: 10.1111/j.1525-1438.2006.00658.x. [DOI] [PubMed] [Google Scholar]
- 11.Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res. 2012 Feb 1;18(3):869–81. doi: 10.1158/1078-0432.CCR-11-2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Yang ES, Jiang J, Nowsheen S, Xia F. DNA damage-induced cytotoxicity is dissociated from BRCA1’s DNA repair function but is dependent on its cytosolic accumulation. Cancer Res. 2010 Aug 1;70(15):6258–67. doi: 10.1158/0008-5472.CAN-09-4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang ES, Wang H, Jiang G, Nowsheen S, Fu A, Hallahan DE, et al. Lithium-mediated protection of hippocampal cells involves enhancement of DNA-PK-dependent repair in mice. J Clin Invest. 2009 May;119(5):1124–35. doi: 10.1172/JCI34051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.SABiosciences. DNA Damage Signaling Pathway PCR Array. 2011 cited; Available from: http://www.sabiosciences.com/rt_pcr_product/HTML/PAHS-029Z.html.
- 15.Steg A, Wang W, Blanquicett C, Grunda JM, Eltoum IA, Wang K, et al. Multiple gene expression analyses in paraffin-embedded tissues by TaqMan low-density array: Application to hedgehog and Wnt pathway analysis in ovarian endometrioid adenocarcinoma. J Mol Diagn. 2006 Feb;8(1):76–83. doi: 10.2353/jmoldx.2006.040402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han HD, Mangala LS, Lee JW, Shahzad MM, Kim HS, Shen D, et al. Targeted gene silencing using RGD-labeled chitosan nanoparticles. Clin Cancer Res. 2010 Aug 1;16(15):3910–22. doi: 10.1158/1078-0432.CCR-10-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010 Aug 9;18(2):185–97. doi: 10.1016/j.ccr.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008 Oct;22(10):3696–705. doi: 10.1096/fj.08-102590. [DOI] [PubMed] [Google Scholar]
- 19.Shahzad MM, Lopez-Berestein G, Sood AK. Novel strategies for reversing platinum resistance. Drug Resist Updat. 2009 Dec;12(6):148–52. doi: 10.1016/j.drup.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steg AD, Katre AA, Goodman BW, Han HD, Nick AM, Stone RL, et al. Targeting the Notch Ligand Jagged1 in Both Tumor Cells and Stroma in Ovarian Cancer. Clin Cancer Res. 2011 Jul 13; doi: 10.1158/1078-0432.CCR-11-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nick AM, Stone RL, Armaiz-Pena G, Ozpolat B, Tekedereli I, Graybill WS, et al. Silencing of p130cas in ovarian carcinoma: a novel mechanism for tumor cell death. J Natl Cancer Inst. 2011 Nov 2;103(21):1596–612. doi: 10.1093/jnci/djr372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011 Jan;11(1):59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11(1):79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 24.Koleva RI, Conley BA, Romero D, Riley KS, Marto JA, Lux A, et al. Endoglin structure and function: Determinants of endoglin phosphorylation by transforming growth factor-beta receptors. J Biol Chem. 2006 Sep 1;281(35):25110–23. doi: 10.1074/jbc.M601288200. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez-Barbero A, Obreo J, Alvarez-Munoz P, Pandiella A, Bernabeu C, Lopez-Novoa JM. Endoglin modulation of TGF-beta1-induced collagen synthesis is dependent on ERK1/2 MAPK activation. Cell Physiol Biochem. 2006;18(1-3):135–42. doi: 10.1159/000095181. [DOI] [PubMed] [Google Scholar]
- 26.She X, Matsuno F, Harada N, Tsai H, Seon BK. Synergy between anti-endoglin (CD105) monoclonal antibodies and TGF-beta in suppression of growth of human endothelial cells. Int J Cancer. 2004 Jan 10;108(2):251–7. doi: 10.1002/ijc.11551. [DOI] [PubMed] [Google Scholar]
- 27.Yoshitomi H, Kobayashi S, Ohtsuka M, Kimura F, Shimizu H, Yoshidome H, et al. Specific expression of endoglin (CD105) in endothelial cells of intratumoral blood and lymphatic vessels in pancreatic cancer. Pancreas. 2008 Oct;37(3):275–81. doi: 10.1097/mpa.0b013e3181690b97. [DOI] [PubMed] [Google Scholar]
- 28.Hu D, Wang X, Mao Y, Zhou L. Identification of CD105 (endoglin)-positive stem-like cells in rhabdoid meningioma. J Neurooncol. 2012 Feb;106(3):505–17. doi: 10.1007/s11060-011-0705-3. [DOI] [PubMed] [Google Scholar]
- 29.Henriksen R, Gobl A, Wilander E, Oberg K, Miyazono K, Funa K. Expression and prognostic significance of TGF-beta isotypes, latent TGF-beta 1 binding protein, TGF-beta type I and type II receptors, and endoglin in normal ovary and ovarian neoplasms. Lab Invest. 1995 Aug;73(2):213–20. [PubMed] [Google Scholar]
- 30.Ho CM, Chang SF, Hsiao CC, Chien TY, Shih DT. Isolation and characterization of stromal progenitor cells from ascites of patients with epithelial ovarian adenocarcinoma. J Biomed Sci. 2012;19:23. doi: 10.1186/1423-0127-19-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pierelli L, Bonanno G, Rutella S, Marone M, Scambia G, Leone G. CD105 (endoglin) expression on hematopoietic stem/progenitor cells. Leuk Lymphoma. 2001 Nov-Dec;42(6):1195–206. doi: 10.3109/10428190109097744. [DOI] [PubMed] [Google Scholar]
- 32.Aslan H, Zilberman Y, Kandel L, Liebergall M, Oskouian RJ, Gazit D, et al. Osteogenic differentiation of noncultured immunoisolated bone marrow-derived CD105+ cells. Stem Cells. 2006 Jul;24(7):1728–37. doi: 10.1634/stemcells.2005-0546. [DOI] [PubMed] [Google Scholar]
- 33.Jiang T, Liu W, Lv X, Sun H, Zhang L, Liu Y, et al. Potent in vitro chondrogenesis of CD105 enriched human adipose-derived stem cells. Biomaterials. 2010 May;31(13):3564–71. doi: 10.1016/j.biomaterials.2010.01.050. [DOI] [PubMed] [Google Scholar]
- 34.Irminger-Finger I. BARD1, a possible biomarker for breast and ovarian cancer. Gynecol Oncol. 2010 May;117(2):211–5. doi: 10.1016/j.ygyno.2009.10.079. [DOI] [PubMed] [Google Scholar]
- 35.Irminger-Finger I, Busquets S, Calabrio F, Lopez-Soriano FJ, Argiles JM. BARD1 content correlates with increased DNA fragmentation associated with muscle wasting in tumour-bearing rats. Oncol Rep. 2006 Jun;15(6):1425–8. [PubMed] [Google Scholar]
- 36.Jang KY, Kim KS, Hwang SH, Kwon KS, Kim KR, Park HS, et al. Expression and prognostic significance of SIRT1 in ovarian epithelial tumours. Pathology. 2009;41(4):366–71. doi: 10.1080/00313020902884451. [DOI] [PubMed] [Google Scholar]
- 37.Chu F, Chou PM, Zheng X, Mirkin BL, Rebbaa A. Control of multidrug resistance gene mdr1 and cancer resistance to chemotherapy by the longevity gene sirt1. Cancer Res. 2005 Nov 15;65(22):10183–7. doi: 10.1158/0008-5472.CAN-05-2002. [DOI] [PubMed] [Google Scholar]
- 38.Salva E, Kabasakal L, Eren F, Ozkan N, Cakalagaoglu F, Akbuga J. Local delivery of chitosan/VEGF siRNA nanoplexes reduces angiogenesis and growth of breast cancer in vivo. Nucleic Acid Ther. 2012 Feb;22(1):40–8. doi: 10.1089/nat.2011.0312. [DOI] [PubMed] [Google Scholar]
- 39.Han HD, Mora EM, Roh JW, Nishimura M, Lee SJ, Stone RL, et al. Chitosan hydrogel for localized gene silencing. Cancer Biol Ther. 2011 May 1;11(9):839–45. doi: 10.4161/cbt.11.9.15185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rudzinski WE, Aminabhavi TM. Chitosan as a carrier for targeted delivery of small interfering RNA. Int J Pharm. 2010 Oct 31;399(1-2):1–11. doi: 10.1016/j.ijpharm.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 41.Mao S, Sun W, Kissel T. Chitosan-based formulations for delivery of DNA and siRNA. Adv Drug Deliv Rev. 2010 Jan 31;62(1):12–27. doi: 10.1016/j.addr.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 42.Feng S, Agoulnik IU, Truong A, Li Z, Creighton CJ, Kaftanovskaya EM, et al. Suppression of relaxin receptor RXFP1 decreases prostate cancer growth and metastasis. Endocr Relat Cancer. 2010;17(4):1021–33. doi: 10.1677/ERC-10-0073. [DOI] [PubMed] [Google Scholar]
- 43.Dass CR, Choong PF. The use of chitosan formulations in cancer therapy. J Microencapsul. 2008 Jun;25(4):275–9. doi: 10.1080/02652040801970461. [DOI] [PubMed] [Google Scholar]
- 44.Howard KA, Rahbek UL, Liu X, Damgaard CK, Glud SZ, Andersen MO, et al. RNA interference in vitro and in vivo using a novel chitosan/siRNA nanoparticle system. Mol Ther. 2006 Oct;14(4):476–84. doi: 10.1016/j.ymthe.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 45.Rosen LS, Hurwitz HI, Wong MK, Goldman J, Mendelson DS, Figg WD, et al. A Phase I First-in-Human Study of TRC105 (Anti-Endoglin Antibody) in Patients with Advanced Cancer. Clin Cancer Res. 2012 Sep 1;18(17):4820–9. doi: 10.1158/1078-0432.CCR-12-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsujie M, Tsujie T, Toi H, Uneda S, Shiozaki K, Tsai H, et al. Anti-tumor activity of an anti-endoglin monoclonal antibody is enhanced in immunocompetent mice. Int J Cancer. 2008 May 15;122(10):2266–73. doi: 10.1002/ijc.23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.