

Abstract
The relevance of many BRCA2 variants of uncertain significance (VUS) to breast cancer has not been determined due to limited genetic information from families carrying these alterations. Here we classified six new variants as pathogenic or non-pathogenic by analysis of genetic information from families carrying 65 individual BRCA2 DBD missense mutations using a multifactorial likelihood model of cancer causality. Next, we evaluated the utility of a homology directed DNA break repair (HDR) functional assay as a method for inferring the clinical relevance of VUS in the DNA binding domain (DBD) of BRCA2 using 18 established non-pathogenic missense variants and all 13 established pathogenic missense mutations from the BRCA2 DBD. Compared to the known status of these variants based on the multifactorial likelihood model, the sensitivity of the HDR assay for pathogenic mutations was estimated at 100% (95%CI: 75.3%-100%), and specificity was estimated at 100% (95%CI: 81.5%-100%). A statistical classifier for predicting the probability of pathogenicity of BRCA2 DBD variants was developed using these functional results. When applied to 33 additional VUS, the classifier identified eight with >99% probability of non-pathogenicity and 18 with ≥ 99% probability of pathogenicity. Thus, in the absence of genetic evidence a cell based HDR assay can provide a probability of pathogenicity for all VUS in the BRCA2 DBD, suggesting that the assay can be used in combination with other information to determine the cancer relevance of BRCA2 VUS.
Keywords: Homology directed repair, missense, VUS, BRCA2, DNA binding domain
Introduction
The tumor suppressor gene BRCA2 encodes a large protein (3418 amino acids) involved in the repair of DNA Double Strand Breaks (DSB) (1, 2). Inactivating germ-line mutations in the BRCA2 gene predispose to breast, ovarian, pancreas and several other types of cancer (3) and confer an average cumulative lifetime risk to age 80 of 51% for breast cancer and 11-12% for ovarian cancer (4). Because mutations that truncate or inactivate BRCA2 are associated with an elevated risk of cancer, identification of germline mutations through clinical genetic testing is widely used to identify individuals who can benefit from risk management strategies.
The majority of known pathogenic mutations in BRCA2 result in protein truncation. However, hundreds of missense BRCA2 variants (5) influence on cancer risk have also been identified in the BRCA2 gene. These variants of uncertain significance (VUS) represent a significant challenge for cancer risk assessment. Characterization of the cancer relevance of VUS has relied on a combination of genetic and in silico approaches. Specifically, a multifactorial likelihood model has been developed that estimates an overall likelihood ratio (LR) of pathogenicity for each VUS based on co-segregation of variants with cancer in families, co-occurrence of variants with known pathogenic mutations, empirical evaluation of personal and family history of cancer, and tumor histopathology (6-9). The likelihood of pathogenicity can be combined with the prior probability of pathogenicity, based on cross-species sequence conservation and physico-chemical properties of mutated residues (Align GVGD (10)), to generate a posterior probability of pathogenicity (11, 12). The posterior probability of pathogenicity is used for categorizing VUS according to a five-tier classification system introduced by an IARC Working Group (12). Briefly, Class1 (posterior probability<0.001) and Class2 (0.001<posterior probability<0.049) VUS are non-pathogenic and likely non-pathogenic VUS. Class3 (0.05<posterior probability<0.949) VUS remain unclassified because of lack of sufficient family information for classification. Class4 (0.95<posterior probability<0.99) and Class5 (posterior probability >0.99) VUS are likely pathogenic and pathogenic, respectively. Many VUS in the BRCA1 and BRCA2 genes lack sufficient family information for classification. As a result, alternative methods are needed to interpret VUS pathogenicity. One approach is to use functional assays that assess the impact of genetic variants on the activity of the protein (13, 14). In this study, we evaluate the ability of a cell-based homology directed repair (HDR) assay to assess the pathogenicity of missense variants in the DNA binding domain (DBD) of BRCA2. While this assay has been used to identify missense mutations that inactivate BRCA2 (15, 16), the sensitivity and specificity of the assay for classified (12) pathogenic BRCA2 mutations has not been established. Here we identified a series of pathogenic and non-pathogenic BRCA2 DBD mutations and used these standards to define the sensitivity and specificity of the HDR assay. On the basis of the results we also developed a statistical classifier for prediction of the pathogenicity of all BRCA2 DBD variants and applied this model to 33 additional VUS from the BRCA2 DBD.
Materials and Methods
VUS selection
A total of 64 missense variants localized to the DBD of BRCA2 were selected for functional analysis by the HDR assay. All VUS were identified in patients who underwent clinical genetic testing for mutations in the BRCA1 and BRCA2 genes. Of these, 63 are reported in the BIC database (5). Details of selection criteria are provided in Results.
Posterior probability approach
LR of pathogenicity for each variant were estimated based on co-segregation of the variant with breast cancer in families of mutation carriers; co-occurrence (in trans) with known pathogenic mutations, and the family and personal history of cancer of individuals found to carry the variant (6, 9). Family information was obtained by Myriad Genetics Laboratories Inc. (Salt Lake City, Utah), cancer risk assessment clinics at the Mayo Clinic, the University of Pennsylvania, The Research Institute of the McGill University Health Center, the German Hereditary Breast Cancer Consortium (GC-HBOC), the Austrian Hereditary Breast and Ovarian Cancer Group and the Instituto Nazionale dei Tumori in Milan. Prior probabilities of pathogenicity based on sequence analysis were derived from Align GVGD (17). A prior probability of 0.01 (95% CI 0.00–0.06) was assigned to substitutions at evolutionary variable positions (18), located within the BRCA2 DBD functional domain (11).
HDR assay
The selected missense variants were cloned into a 3x FLAG-tagged full-length BRCA2 cDNA expression plasmid using the QuickChange site-directed mutagenesis kit XL (Stratagene) (13). Mutations were verified by DNA sequencing. Wild-type and mutant constructs were co-transfected with an I-Sce1–expressing pcBASce plasmid (13) into V-C8 cells containing the DR-GFP reporter plasmid. The BRCA2 deficient V-C8 hamster lung fibroblast cell line (XRCC11) displays chromosomal instability and abnormal centrosomes, reduced nuclear localization of the RAD51 protein that is central in the HDR processes, and sensitivity to cross-linking agents such as methyl methanesulfonate (MMS) (19). Cells expressing green fluorescent protein (GFP) following HDR-dependent repair of an I-Sce1 induced DNA double strand break in the DR-GFP reporter plasmid were quantified by fluorescent activated cell sorting (FACS) after 72 hrs. Transfection efficiency was evaluated by immunofluorescence (IF) based counting of GFP positive cells. Expression of wildtype and mutant BRCA2 was evaluated by immunoblotting with rabbit Anti-Flag antibody (1:1000 F7425 Sigma) of mouse Anti-Flag M2 (F1804 Sigma) immunoprecipitates from protein lysates.
Statistical analysis
Data normalization
Mutants were evaluated by the HDR assay in batches. In each batch, wild-type BRCA2 and the D2723H known pathogenic mutant were included as positive and negative controls. The basal HDR activity due to the transfection of the vector alone was also measured. All experiments were performed in duplicate. The mean fold change in GFP positive cells for each mutant relative to vector alone was calculated. These values were normalized relative to the ratio of D2723H to wildtype BRCA2 in each experiment and rescaled to reference fold-change values of 5.0 for wild-type and 1.0 for D2723H, which reflected the pre-normalization HDR fold changes, while providing simple integer values as anchors for known non-pathogenic or pathogenic variants, respectively. Using these normalized values the average normalized Ln (HDR) value and standard error for each variant were computed, while pooling data across all replicated experiments. Because the HDR assay results on the fold-difference scale are positively skewed, the standard error increases as a function of the mean HDR fold-difference. This relationship is not evident when computing the summary statistics from Ln-transformed data. Therefore, all statistical analyses were performed after transforming the fold-difference measurements to the Ln scale. A linear mixed model was also used to analyze all data available for each variant and to extract model-based estimates of the mean and standard error of the HDR activity for each VUS.
Sensitivity and Specificity
A ROC analysis (20) of the normalized mean HDR assay results of the known pathogenic and non-pathogenic variants was performed to identify an HDR assay cut-off that simultaneously maximized both the sensitivity and specificity of the assay for pathogenic variants. The midpoint between the extremes of the ranges of HDR assay values for pathogenic and non-pathogenic variants was selected. The sensitivity and specificity of the assay was calculated together with exact 95% confidence intervals based on this cut-point. In order to determine the degree to which variability in the assay might influence the results, the sensitivity and specificity of the HDR assay was also estimated using the most extreme assay reading for each variant: the highest observed values for the pathogenic variants and the lowest observed measurements for the non-pathogenic variants.
VUS Classification via the HDR Assay
A statistical classifier for determining the clinical relevance of BRCA2 variants was developed using the HDR data from variants already classified as pathogenic or non-pathogenic by means of the multifactorial likelihood method. A discriminant analysis approach was used to develop the classifier (21, 22). After verifying that the distribution of the normalized and log-transformed HDR assay results were well approximated by Gaussian distributions, the data observed for the known pathogenic and non-pathogenic variants were used to estimate the parameters of the two Gaussian distributions that describe the HDR activity within each of these two classes of variant. Linear mixed models which incorporated random per-variant and per-experiment intercepts to allow for the presence of repeated measurements were used to estimate means and variance components that quantified the behavior of the HDR assay when applied to known pathogenic and non-pathogenic variants. Separate models were fit to the normalized and loge-transformed data from the known pathogenic and non-pathogenic variants, and the model parameters were extracted. The estimated Gaussian distribution of the non-pathogenic variants, fN(x), had a mean of 1.57 and a variance equal to the sum of the variance components from the linear mixed model, or 0.06. The equivalent distribution of the pathogenic variants, fP(x), had a mean of 0.05 and a variance of 0.07.
Given the distinct Gaussian distributions for the non-pathogenic and pathogenic variants, fN(x) and fP(x) respectively, the posterior probability that a new variant, with HDR assay result equal to exp(x), is pathogenic is:
where pP is the prior probability that the VUS is pathogenic. It is possible to use external estimates of prior probability in this equation, such as prior probabilities based on the Align GVGD model (17). However, for this study we chose to set equal prior probabilities that a variant was either pathogenic or non-pathogenic because the results obtained with the HDR-based classifications are meant to be independent of other data. Consequently it is possible to compare the HDR-based posterior probabilities to the posterior probabilities obtained using other methods, such as the multifactorial likelihood model or its components. Assuming equal prior probabilities, we computed posterior probabilities that each variant was pathogenic based on the HDR assay result. We elected to consider the assay result as inconclusive unless the posterior probability of pathogenicity was either ≥0.99 or ≤0.01. To assess the influence of the assumed prior probabilities on this model, we also estimated posterior probabilities of pathogenicity using prior probabilities from the Align GVGD model.
Results
Identification of pathogenic and non-pathogenic variants using a multifactorial likelihood model
In this study we focused on variants in the C-terminal DBD of BRCA2 because the region has been implicated in the homologous recombination (HDR) activity of BRCA2 and contains all of the known pathogenic BRCA2 missense mutations (18, 23). In addition, many of the highly conserved amino acids in the region, that are likely important for function, have been found to contain missense mutations in individuals with a family history of breast and/or ovarian cancer. Likelihood ratios of pathogenicity for many variants in the BRCA2 DBD have been estimated based on a large collection of family data provided by Myriad Genetics Laboratories (6). Posterior probabilities of pathogenicity have also been estimated by combining the likelihood ratios with prior probabilities of pathogenicity based on Align GVGD modeling of protein sequences (18, 23).
Collection of family information for BRCA2 DBD Class 3 VUS with posterior probabilities of pathogenicity approaching Class 2 (<0.05) or Class 4 (>0.95) thresholds yielded 18 pedigrees that were informative for segregation analysis. Re-estimation of the posterior probability of pathogenicity for the associated variants resulted in classification of three variants (S3020C, S2414L and T3013I) as Class 2 (likely non-pathogenic) and three (G2609D, L2688P and N3124I) as Class 4 (likely pathogenic). When these Class 4 missense substitutions were combined with all known Class 4 and Class 5 missense mutations (18, 23), a total of 13 known pathogenic missense mutations from the BRCA2 DBD were available for further study (Table 1). Of these, eight were Class 5 and five were Class 4. In addition, we selected the three new Class 2 variants, 13 previously classified Class 1 and Class 2 variants (23) and the polymorphisms A2466V (24) and A2951T (25, 26) as non-pathogenic standards for our HDR studies (Table 1).
Table 1.
Classification of BRCA2 DBD variants as pathogenic and non-pathogenic by a multifactorial probability based model
| VUS | AlignGVGD* | Likelihood ratios derived from each component |
combined odds in favor of causality | posteriorP ‡ | IARC class ¥ | ||
|---|---|---|---|---|---|---|---|
| co-segregation odds | co-occurence odds | family history odds | |||||
| K2411T | 0.01 | 1.00 | 0.06 | 0.15 | 0.01 | 8.49×10-5 | 1 |
| S2414L | 0.01 | 1.00 | 1.02 | 2.79 | 2.86 | 0.03 | 2 |
| A2466V | 0.01 | / | / | / | / | / | / |
| P2589H | 0.01 | 1.00 | 1.02 | 1.44 | 1.48 | 0.01 | 2 |
| G2609D | 0.81 | 1.00 | 1.02 | 4.02 | 4.12 | 0.95 | 4 |
| W2626C | 0.81 | 1.00 | 2.13 | 22.65 | 48.14 | 1.00 | 5 |
| I2627F | 0.29 | 2.01 | 1.28 | 407.62 | 1.05×103 | 1.00 | 5 |
| L2647P | 0.81 | 1.00 | 1.19 | 3.86 | 4.58 | 0.95 | 4 |
| L2653P | 0.81 | 0.58 | 1.16 | 35.96 | 24.06 | 0.99 | 5 |
| D2665G | 0.81 | 0.01 | 0.13 | 0.24 | 3.34×10-4 | 1.42×10-3 | 2 |
| M2676T | 0.01 | 1.00 | 1.16 | 0.83 | 0.96 | 0.01 | 2 |
| L2688P | 0.81 | 1.00 | 1.19 | 3.44 | 4.08 | 0.95 | 4 |
| T2722R | 0.81 | 10.80 | 1.24 | 6.95 | 93.47 | 1.00 | 5 |
| D2723G | 0.81 | 1.00 | 1.59 | 75.96 | 120.57 | 1.00 | 5 |
| D2723H | 0.81 | 1.38×104 | 4.17 | 9.77×106 | 5.62×1011 | 1.00 | 5 |
| K2729N | 0.01 | 0.04 | 6.35 | 0.01 | 1.63×10-3 | 1.65×10-5 | 1 |
| G2748D | 0.81 | 6.73 | 1.44 | 257.49 | 2.49×103 | 1.00 | 5 |
| R2787H | 0.01 | 1.00 | 1.05 | 1.25 | 1.31 | 0.01 | 2 |
| E2856A | 0.01 | NA | NA | 3.89×10-11 | 3.89×10-11 | 3.93×10-13 | 1 |
| V2908G | 0.66 | 1.00 | 0.06 | 0.07 | 3.91×10-3 | 0.01 | 2 |
| A2951T | 0.01 | / | / | / | / | / | / |
| D2965H | 0.01 | 1.00 | 1.08 | 1.07 | 1.15 | 0.01 | 2 |
| R2973C | 0.01 | 2.36×10-3 | 1.84 | 0.04 | 1.76×10-4 | 1.78×10-6 | 1 |
| T3013I | 0.01 | 1.82 | NA | NA | 1.82 | 0.02 | 2 |
| S3020C | 0.01 | 1.00 | 1.02 | 1.94 | 1.98 | 0.02 | 2 |
| R3052W | 0.81 | 3.98×103 | 1.48 | 1.61 | 9.55×103 | 0.99 | 5 |
| P3063S | 0.01 | 1.00 | 1.10 | 0.67 | 0.73 | 0.01 | 2 |
| Y3092C | 0.81 | 1.02 | 1.28 | 4.62×10-3 | 0.01 | 0.03 | 2 |
| D3095E | 0.66 | 0.43 | 1.67 | 31.82 | 22.59 | 0.98 | 4 |
| Y3098H | 0.01 | 5.03 | 0.19 | 3.54×10-4 | 3.46×10-4 | 3.49×10-6 | 1 |
| N3124I | 0.81 | 0.04 | 2.07 | 81.64 | 7.23 | 0.97 | 4 |
GenBank Reference BRCA2 NM_000059.3
/not applicable because the variant is a well-defined polymorphism. NA not available.
Prior probability based on Align GVGD (17)
Posterior Probability = Posterior Odds/(Posterior Odds + 1), where the Posterior Odds = Odds for causality X [Prior Probability/(1-prior probability)]).
Class1/2: non-pathogenic/likely non-pathogenic; Class4/5: pathogenic/likely pathogenic (12)
A further 27 VUS from the BRCA2 DBD were selected for evaluation based on high cross-species sequence conservation of the parental residues (AlignGVGD-based prior probability of 0.81). Six other VUS with prior probabilities ranging from 0.01 to 0.61 were included as an initial investigation of the correlation between probabilities derived from AlignGVGD and the HDR assay. Family data were available for 22 of these 33 variants, but the information was insufficient for classification of the VUS as pathogenic or non-pathogenic using the multifactorial likelihood model. Thus, the 22 variants with family data and the 11 without data remained classified as Class 3 VUS (Table 2).
Table 2.
Genetic and functional characterization of 64 BRCA2 DBD variants
| Multifactorial likelihood Model | Functional Model | |||||||
|---|---|---|---|---|---|---|---|---|
| VUS | AlignGVGD* | posteriorP ‡ | IARC class ¥ | VUS | HDR normalized | HDR SE‡ | Pr(deleterious)± | Pr(neutral) ± |
| T3013I | 0.01 | 0.02 | 2 | T3013I | 5.96 | 0.48 | 1.37 ×10-7 | 1 |
| K2411T | 0.01 | 8.49 ×10-5 | 1 | K2411T | 5.74 | 0.43 | 2.25 ×10-7 | 1 |
| M2676T | 0.01 | 0.01 | 2 | M2676T | 5.68 | 0.43 | 2.67 ×10-7 | 1 |
| S2414L | 0.01 | 0.03 | 2 | S2414L | 5.58 | 0.52 | 5.55 ×10-7 | 1 |
| E2856A | 0.01 | 3.93 ×10-13 | 1 | E2856A | 5.54 | 0.42 | 3.99 ×10-7 | 1 |
| Y3098H | 0.01 | 3.49 ×10-6 | 1 | Y3098H | 5.53 | 0.16 | 1.84 ×10-7 | 1 |
| D2665G | 0.81 | 1.42 ×10-3 | 2 | D2665G | 5.30 | 0.65 | 2.99 ×10-6 | 1 |
| D2965H | 0.01 | 0.01 | 2 | D2965H | 5.23 | 0.39 | 1.01 ×10-6 | 1 |
| A2466V | 0.01 | / | / | A2466V | 4.68 | 0.43 | 8.64 ×10-6 | 1 |
| P3063S | 0.01 | 0.01 | 2 | P3063S | 4.43 | 0.21 | 9.96 ×10-6 | 1 |
| P2589H | 0.01 | 0.01 | 2 | P2589H | 4.35 | 0.39 | 2.67 ×10-6 | 1 |
| Y3092C | 0.81 | 0.03 | 2 | Y3092C | 4.29 | 0.36 | 2.94 ×10-5 | 1 |
| A2951T | 0.01 | / | / | A2951T | 4.14 | 0.38 | 6.02 ×10-5 | 1 |
| V2908G | 0.66 | 0.01 | 2 | V2908G | 3.94 | 0.50 | 2.60 ×10-4 | 1 |
| S3020C | 0.01 | 0.02 | 2 | S3020C | 3.69 | 0.24 | 2.79 ×10-4 | 1 |
| R2787H | 0.01 | 0.01 | 2 | R2787H | 3.61 | 0.27 | 4.55 ×10-4 | 1 |
| K2729N | 0.01 | 1.65 ×10-5 | 1 | K2729N | 3.30 | 0.42 | 3.7 ×10-3 | 1 |
| R2973C | 0.01 | 1.78 ×10-6 | 1 | R2973C | 3.15 | 0.40 | 7.57 ×10-3 | 0.99 |
| F2406L | 0.01 | NA | 3 | F2406L | 6.58 | 0.60 | 4.05 ×10-8 | 1 |
| L2865V | 0.29 | 0.25 | 3 | L2865V | 4.96 | 0.64 | 9.27 ×10-6 | 1 |
| G2813E | 0.81 | 0.77 | 3 | G2813E | 4.66 | 0.42 | 9.02 ×10-6 | 1 |
| H2440R | 0.01 | NA | 3 | H2440R | 4.30 | 0.34 | 2.72 ×10-5 | 1 |
| D2679G | 0.81 | 0.73 | 3 | D2679G | 4.25 | 0.38 | 3.84 ×10-5 | 1 |
| D2679Y | 0.81 | 0.83 | 3 | D2679Y | 4.09 | 0.25 | 4.64 ×10-5 | 1 |
| S2807L | 0.81 | NA | 3 | S2807L | 3.84 | 0.35 | 2.01 ×10-4 | 1 |
| G2901D | 0.81 | 0.73 | 3 | G2901D | 3.48 | 0.23 | 7.43 ×10-4 | 1 |
| T3033I | 0.81 | 0.9 | 3 | T3033I | 2.80 | 0.21 | 0.03 | 0.97 |
| R2842L | 0.81 | NA | 3 | R2842L | 2.78 | 0.21 | 0.04 | 0.96 |
| E3002D | 0.66 | 0.75 | 3 | E3002D | 2.69 | 0.25 | 0.06 | 0.94 |
| Y3092S | 0.81 | 0.64 | 3 | Y3092S | 2.51 | 0.12 | 0.17 | 0.83 |
| G2812E | 0.81 | 0.75 | 3 | G2812E | 2.51 | 0.18 | 0.17 | 0.83 |
| P2800S | 0.81 | NA | 3 | P2800S | 2.43 | 0.18 | 0.27 | 0.73 |
| P2800R | 0.81 | NA | 3 | P2800R | 2.43 | 0.15 | 0.27 | 0.73 |
| R2842C | 0.81 | 0.84 | 3 | R2842C | 1.76 | 0.16 | 0.99 | 0.01 |
| Y2726C | 0.81 | 0.76 | 3 | Y2726C | 1.70 | 0.16 | 0.99 | 6.33 ×10-3 |
| D2611G | 0.81 | NA | 3 | D2611G | 1.67 | 0.15 | 1 | 4.39 ×10-3 |
| D3073G | 0.81 | 0.86 | 3 | D3073G | 1.52 | 0.14 | 1 | 8.93 ×10-4 |
| L2654P | 0.81 | 0.79 | 3 | L2654P | 1.45 | 0.13 | 1 | 4.06 ×10-4 |
| G2793E | 0.81 | 0.75 | 3 | G2793E | 1.32 | 0.12 | 1 | 7.78 ×10-5 |
| Y2660D | 0.81 | NA | 3 | Y2660D | 1.30 | 0.10 | 1 | 4.54 ×10-5 |
| G2585R | 0.81 | NA | 3 | G2585R | 1.26 | 0.12 | 1 | 3.55 ×10-5 |
| G2793R | 0.81 | 0.61 | 3 | G2793R | 1.22 | 0.11 | 1 | 1.94 ×10-5 |
| L2792P | 0.81 | 0.92 | 3 | L2792P | 1.20 | 0.11 | 1 | 1.35 ×10-5 |
| R2659G | 0.81 | NA | 3 | R2659G | 1.12 | 0.07 | 1 | 2.51 ×10-6 |
| R2784Q | 0.66 | 0.12 | 3 | R2784Q | 1.11 | 0.10 | 1 | 3.97 ×10-6 |
| G3076V | 0.81 | 0.94 | 3 | G3076V | 1.09 | 0.10 | 1 | 2.80 ×10-6 |
| E3002K | 0.66 | 0.66 | 3 | E3002K | 1.05 | 0.10 | 1 | 1.61 ×10-6 |
| G3076E | 0.81 | 0.92 | 3 | G3076E | 1.04 | 0.09 | 1 | 1.14 ×10-6 |
| L2510P | 0.81 | NA | 3 | L2510P | 1.04 | 0.10 | 1 | 1.16 ×10-6 |
| D2723A | 0.81 | 0.85 | 3 | D2723A | 0.88 | 0.08 | 1 | 6.40 ×10-6 |
| R2784W | 0.81 | 0.83 | 3 | R2784W | 0.83 | 0.08 | 1 | 2.42 ×10-8 |
| W2626C | 0.81 | 1 | 5 | W2626C | 1.30 | 0.12 | 1 | 6.13 ×10-5 |
| G2609D | 0.81 | 0.95 | 4 | G2609D | 1.28 | 0.08 | 1 | 2.91 ×10-5 |
| T2722R | 0.81 | 1 | 5 | T2722R | 1.26 | 0.12 | 1 | 3.55 ×10-5 |
| I2627F | 0.29 | 1 | 5 | I2627F | 1.09 | 0.10 | 1 | 2.79 ×10-6 |
| D2723G | 0.81 | 1 | 5 | D2723G | 1.03 | 0.10 | 1 | 1.00 ×10-6 |
| D3095E | 0.66 | 0.98 | 4 | D3095E | 1.02 | 0.09 | 1 | 8.42 ×10-7 |
| L2688P | 0.81 | 0.95 | 4 | L2688P | 1.01 | 0.09 | 1 | 6.90 ×10-7 |
| D2723H | 0.81 | 1 | 5 | D2723H | 1.00 | 0.02 | 1 | 1.39 ×10-7 |
| L2647P | 0.81 | 0.95 | 4 | L2647P | 0.99 | 0.09 | 1 | 5.19 ×10-7 |
| N3124I | 0.81 | 0.97 | 4 | N3124I | 0.96 | 0.10 | 1 | 3.91 ×10-7 |
| L2653P | 0.81 | 0.99 | 5 | L2653P | 0.96 | 0.09 | 1 | 2.77 ×10-7 |
| G2748D | 0.81 | 1 | 5 | G2748D | 0.83 | 0.08 | 1 | 2.32 ×10-8 |
| R3052W | 0.81 | 0.99 | 5 | R3052W | 0.81 | 0.07 | 1 | 1.38 ×10-8 |
GenBank Reference BRCA2 NM_000059.3
/not applicable because the variant is a well-defined polymorphism. NA not available.
Prior probability based on Align GVGD (17)
Posterior Probability = Posterior Odds/(Posterior Odds + 1), where the Posterior Odds = Odds for causality X [Prior Probability/(1-prior probability)]).
Class1/2:non-pathogenic/likely non-pathogenic; Class3:unclassified; Class4/5:pathogenic/likely pathogenic (12)
Standard Error
Model-based estimates of conditional probabilities in favor or against pathogenicity for VUS based on HDR activity.
Sensitivity and specificity of the HDR assay
To assess the sensitivity and specificity of the HDR assay for pathogenic mutations, the influence of the 13 known pathogenic and 18 known non-pathogenic mutations on the HDR activity of BRCA2 was evaluated. Wild-type and mutant forms of BRCA2 were co-expressed with I-Sce1 in BRCA2 deficient V-C8 cells containing the DR-GFP reporter. The BRCA2 constructs exhibited similar transfection efficiencies and produced similar levels of full-length BRCA2 protein (Supplementary Fig. S1) (13). Cells expressing GFP after BRCA2 dependent repair of I-Sce1 DNA double strand breaks were quantified by flow cytometry. Normalized fold changes in GFP positive cells, which equate to HDR activity, are shown in Figure 1 for the known pathogenic and non-pathogenic variants. Importantly, the HDR activity of the Class1 and 2 variants did not overlap with the HDR activity of Class 4 or 5 mutations (Figure 1). For the purpose of assessing sensitivity and specificity of the HDR assay the midpoint between the mean of the normalized fold change for Class 1/2 variants and the mean for the Class 4/5 variants was selected as a cut-point for discriminating between pathogenic and non-pathogenic variants. Using this threshold of 2.25, the 13 pathogenic mutations and 18 non-pathogenic variants were correctly classified. Sensitivity was estimated at 100% (95% CI: 75.3% – 100%), and specificity was estimated at 100% (95% CI: 81.5% - 100%). Importantly, these estimates were not altered when considering the lowest observed HDR assay values for the non-pathogenic variants and the highest observed values for the pathogenic variants instead of mean values. Furthermore, when restricting to the seven Class 1 and eight Class 5 variants, the sensitivity (100% (95% CI: 63.1% – 100%)) and the specificity (100% (95% CI: 71.5% - 100%)) of the assay were again estimated at 100%. Likewise the five Class 4 variants and 11 Class 2 variants yielded a sensitivity of 100% (95% CI: 47.8% - 100%) and specificity of 100% (95% CI: 71.5% - 100%). Given, these results we combined the Class 1 and Class 2 variants as non-pathogenic and the Class 4 and Class 5 variants as pathogenic for the purpose of developing a HDR-based probability model of pathogenicity.
Fig 1.

Homology Directed Repair (HDR) activity of 31 classified non-pathogenic variants and pathogenic mutations. The normalized HDR fold increase with standard error (SE) is displayed on a logarithmic scale for the combined Class1/Class2 variants and combined Class4/Class5 mutations.
Evaluation of VUS with the HDR assay
Given the high sensitivity and specificity of the HDR assay for pathogenic BRCA2 DBD mutations, we evaluated the influence of 33 Class 3 VUS from the BRCA2 DBD on BRCA2 HDR activity in an effort to determine the functional and disease relevance of these VUS. The transfection efficiencies and expression levels of the mutants were similar to wild-type BRCA2 (Supplementary Fig. S1) (13). The normalized HDR results for each VUS are shown in Supplementary Table S1. Thirteen VUS displayed low HDR activity within the range of the known pathogenic mutations (Table 2, Figure 2). In addition, eight VUS displayed high HDR activity within the range of activity of the known non-pathogenic mutations (Table 2, Figure 2). A further five VUS displayed activity that was marginally above the activity of known pathogenic mutations. However, seven VUS appeared to have intermediate HDR activity outside the ranges of the known pathogenic or non-pathogenic mutations (Figure 2). Re-cloning and revaluation of these variants did not alter these results.
Fig 2.

Homology Directed Repair (HDR) activity of 64 BRCA2 DNA binding domain mutants. The discriminant model-based normalized HDR fold change with standard error (SE) is displayed on a logarithmic scale for the analyzed non-pathogenic, unclassified and pathogenic variants. Solid lines represent 99.9% and 0.1% probability of pathogenicity and dotted lines represent 99% and 1% probability of pathogenicity. The SE is included only as a measure of the reproducibility of the HDR assay for each variant.
We also evaluated possible influences of the VUS on mRNA splicing. Here we calculated consensus values of potential splice sites using the MaxEnt (27) algorithm and Human Splicing Finder (9, 28). The c.8035G>T mutation (p.D2679Y) had an HSF score of 87.91 (wild type 61.07) and a MaxEnt score of 9.11 (wild type 1.46) and a moderate probability in favor of a de novo donor site. Aberrant in vivo splicing was not assessed due to lack of specimens. In addition, the D2723G variant was previously shown to cause aberrant splicing (deletion of 163 bp from exon 18) of a small proportion of the mutant allele (9). In contrast, we found low probabilities of de novo splice acceptor or donor formation for all other VUS in this study. This suggested that the variants evaluated in this study likely do not influence breast cancer risk through aberrant splicing.
A statistical classifier for BRCA2 DBD VUS based on the HDR assay
Although the HDR assay results for the 33 VUS suggested that a number of the VUS were functionally relevant, a quantitative model that accounted for the variance in the assay was needed to make predictions of pathogenicity or non-pathogenicity. Therefore, we developed a statistical classifier to estimate the probability of pathogenicity for any BRCA2 DBD evaluated by the HDR assay. Specifically, we estimated the mean and variances of the distributions of the HDR assay results for the known pathogenic and non-pathogenic mutation groups and used these results to compute the probability of each tested VUS being pathogenic, conditional on the observed HDR assay result (Table 2). Variants with ≥ 99% probability of pathogenicity were classified as pathogenic and variants with ≤ 1% probability of pathogenicity were classified as non-pathogenic. Thus, 18 unclassified variants were considered pathogenic, eight were considered non-pathogenic and seven VUS remained unclassified.
Since each variant displays a unique degree of variability in the HDR assay, the final posterior probability that any specific variant is pathogenic or non-pathogenic will depend on the results of individual assays. However, we also estimated cut-points in HDR activity that could be used for visual inspection of the probability and classification of any DBD variant. Specifically, the minimum values of the HDR assay that grouped all variants with probabilities of at least 99% (≥3.02) and 99.9% (≥3.48) of non-pathogenicity were identified (Figure 2). Similarly, the maximum values of the HDR assay that grouped all variants with probabilities of 99% (≤1.75) and 99.9% (≤1.53) of pathogenicity were identified (Figure 2). Importantly, the standard error associated with each variant is only shown to provide information on the reproducibility of the HDR results for each variant, because the variability of the mean HDR result for each variant is incorporated into the computation of the selected cut-points. Therefore, a variant is considered classified even when the standard error of the normalized fold change in HDR activity crosses these visual cutpoints.
Structural interpretation of predicted pathogenic variants
Thirteen BRCA2 DBD variants were classified as pathogenic using the multifactorial likelihood model and a further 18 variants were classified as pathogenic by the HDR assay alone. In an attempt to understand the mechanism by which these variants altered BRCA2 function the location of the mutated residues in the predicted three dimensional structure of the mouse BRCA2-DSS1-ssDNA domain (29) was determined and the impact of the mutations on the local protein structure was assessed (Figure 3). The missense variants mapped to four regions of the BRCA2 DBD, region 1: Helical domain (Figure 3A); region 2: Helical-OB1-DSS1 interface (Figure 3B); region 3: OB1-OB2-DSS1 interface (Figure 3C) and region 4: OB2-OB3 interface (Figure 3D). In region 1, several mutated residues were associated with the terminal helix of the helical domain. Residues Y2581 (human Y2660), L2574 (human L2653) and L2575 (human L2654) are located on one face of the helix, confined to the helical domain, while R2580 (human R2659) is located on the opposite face of the helix that interacts with OB1 and DSS1. Thus Y2581, L2575, L2576 are likely more important for the stability of the helix and the helical domain, while R2580 may influence the interaction of the helical domain with the OB1 domain and DSS1. This is supported by the observation that R2580 is potentially involved in hydrogen bond contacts with H2544 (human H2623) within the helical domain and G2645 (human G2724) of the OB1 domain. In region 2, a cluster of residues containing likely pathogenic variants is connected through a network of hydrogen-bonds. Disruption of this network by mutations is potentially detrimental to the integrity of the helical-OB1 interface. Specifically, W2547C (human W2626C) in the helical domain may disrupt a direct hydrogen bond to the backbone of W2646 (human W2725) in the OB1 domain. Similarly, Y2647C (human Y2726C) in OB1 may disrupt the hydrogen bonding to H2544 (human H2623) and E2584 (human E2663) in the helical domain. In addition, the T2643R mutation (human T2722R) in OB1 potentially leads to a steric clash with W2547 in the helical domain. Furthermore, OB1 residue D2644 (human D2723) associated with the human D2723H, D2723G and D2723A mutations, is hydrogen bonded to the OB1 residues S2591 (human S2670), the L2593 (human I2672) and the W2646 (human W2725), suggesting that these mutations may disrupt the helical-OB1 interface. Separately, disruption of the loop residue G2669 (human G2748) by the G2748D mutation may result in steric clashes with the DSS1 residues Q55 and A58, and destabilization of the DSS1-OB1 interaction. In region 3, the R2705 (human R2784) in OB1 forms hydrogen bonds with the side chains of N2702 (human N2781) in OB1, S2728 (human S2807) in OB2, and the D41 side chain of DSS1. This critical residue serves to clamp three flexible loops through polar interactions. The R2784W and R2784Q mutation of this residue may affect tertiary interactions between OB1 and OB2 and the interaction between BRCA2 and DSS1. In region 4, R2971 (human R3052) in OB3 is positioned at the junction of the OB2 and OB3 folds (Figure 2 D). Remarkably, this residue appears to interact with residues E2921 (human E3002) and V2746 (human A2825) in OB2 and with D2992 (human D3073) and N3042 (human N3124) in OB3. Thus, the R3052W, E3002K, D3073G and N3124I pathogenic mutations likely disrupt this interaction network and the structure and stability of the OB2-OB3 interface.
Fig 3.
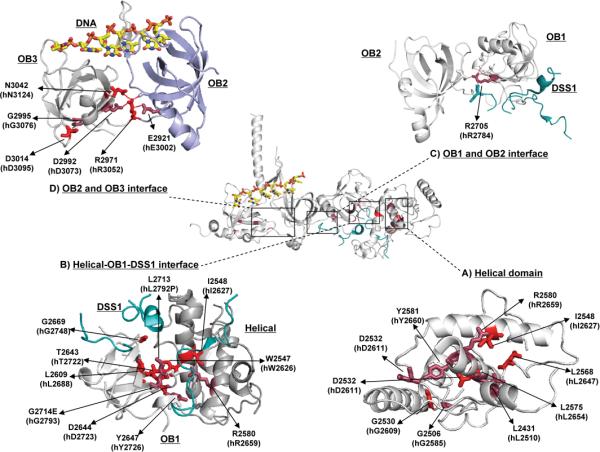
Diagram of the three dimensional structure of the BRCA2 DNA binding domain and the four regions containing HDR-inactivating variants. Variants classified as pathogenic by both the multifactorial likelihood model and the HDR assay are shown in Red. Variants classified as pathogenic by the HDR assay but unclassified by the multifactorial likelihood model because of lack of genetic evidence are shown in purple. A) Helical domain of BRCA2; B) Helical-OB1-DSS1 interface; C) OB1-OB2-DSS1 interface; D) OB2-OB3 interface. The mouse BRCA2-DSS1-ssDNA complex structure (pdb ID: 1MJE) served as the model for analysis of the variants. All figures were generated using Pymol Molecular Graphics version 1.3
Discussion
Due to the paucity of genetic data that can be used for the classification of BRCA1 and BRCA2 VUS (23), alternative methods that can predict the clinical relevance of these mutations are needed. Since disruption or depletion of BRCA2 causes defective HDR mediated DNA double-strand break repair, we evaluated the utility of a cell-based HDR assay for classification of VUS in the DBD of BRCA2. Once established, we asked if the HDR-based functional assay could independently validate 31 variants previously classified as pathogenic or non-pathogenic. The HDR assay clearly segregated these mutations into two groups, with all of the known pathogenic mutations defined by genetic studies displaying impaired HDR activity, suggesting that the assay could be utilized for characterization of germline variants in the BRCA2 DBD.
In an effort to gain insight into the clinical relevance of variants that could not be classified by genetic studies due to the limited availability of family data, we developed a model for estimating the probability of pathogenicity of BRCA2 DBD VUS. This classifier was based on the HDR assay results for known pathological and non-pathological variants. Although the number of variants in each of the known classes was limited, the degree of separation observed in the HDR assay between the two classes was very large relative to the variability of the assay. Therefore we were able to estimate with sufficient accuracy the parameters used in the model to discriminate between pathogenic and non-pathogenic variants. To test the robustness of the model, we also performed a sensitivity analysis that perturbed the parameters relative to the standard error of the measurements and verified that the overall conclusions were unchanged. The resulting estimates of the conditional probabilities that a variant is pathogenic accurately classified the 13 known pathogenic and 18 known non-pathogenic mutations. Furthermore, using probability of pathogenicity thresholds of 99% and 1%, the model predicted 18 other VUS as pathogenic and eight as non-pathogenic. In total, the multifactorial likelihood model and the HDR assay based probability model used in this study reclassified 21 VUS from the BRCA2 DBD as pathogenic or likely pathogenic. Given that only 10 BRCA2 DBD missense variants were previously identified as pathogenic, this study represents a substantial improvement in VUS classification. The large proportion of predicted pathogenic variants in the DBD is consistent with previous findings implicating the domain in the tumor suppression function of BRCA2. Indeed, Easton and colleagues estimated that 35% of missense mutations in the BRCA2 DBD were pathogenic (6). Overall our report suggests that in the absence of detailed genetic background needed for the multifactorial likelihood model, the HDR assay presents a viable alternative for assessing the clinical relevance of VUS in the BRCA2 DBD.
While the HDR-based classifier effectively identified 18 VUS as pathogenic and suggested that eight other VUS were non-pathogenic, seven VUS displayed intermediate HDR activity. Whether additional VUS in the BRCA2 DBD also exhibit intermediate HDR activity remains to be determined through additional studies. Because of limited family data, it is not known if these VUS with intermediate activity influence breast cancer risk. To investigate whether intermediate function equates to intermediate risk it may be necessary to identify all intermediate function VUS in the DBD, collect family information for all of these variants, and combine the information for the entire group in order to have sufficient data for estimation of risk and/or penetrance. The recently established ENIGMA consortium (Evidence-based Network for the Interpretation of Germline Mutant Alleles (30)) for large-scale collaborative studies of BRCA1 and BRCA2 sequence variants has begun this process.
We also evaluated the influence of A-GVGD-based prior probabilities of pathogenicity rather than equal prior probabilities for each variant on the posterior probability of pathogenicity. The results were similar, with a correlation of 0.987 (95% CI 0.979-0.992) between the posterior probabilities from the two models. However, VUS with high (0.81) and low (0.01) A-GVGD-based prior probabilities had slightly higher and lower posterior probabilities of pathogenicity, respectively, than those obtained from the equal prior probability model (Supplementary Table S2).Importantly, the seven VUS with intermediate HDR activity remained unclassified with either prior probability model (1% and 99% thresholds).
To ascertain if a structure-function correlation exists for the pathogenic missense mutations, we mapped the variants on the predicted three dimensional structure of a murine BRCA2-DSS1 complex. We found that many of the parental residues of the pathogenic missense mutants were located at the inter-domain interfaces of the BRCA2 DBD. These data suggest that the preservation of these inter-domain interactions are important for retention of BRCA2 HDR activity. However, five non-pathogenic variants were also found to be located at inter-domain interfaces (Supplementary Fig. S2). Specifically, R2787H (mouse R2708) is at the OB1-DSS1 interface, S3020C (mouse F2939) is at the OB2-OB3 and OB2-DNA interface, and T3013I (mouse V2932) is at the OB1-OB2 interface. Furthermore, the S2807L (mouse S2728) and G2813E (mouse G2734) variants, classified as non-pathogenic by the HDR assay but not by the multifactorial likelihood model because of lack of genetic evidence, are both at the OB1-OB2 interface. In addition, four of seven intermediate activity variants were located at interfaces (Supplementary Fig. S2). Specifically P2800R/S (mouse P2721) and G2812E (mouse G2733) are located at the OB1-OB2 interface and E3002D (mouse E2921) is at the OB2-OB3 interface. However, P2800R/S and G2812E are solvent exposed and can be accommodated in the structure. Additionally, while the pathogenic E3002K substitutes a negatively charged amino acid with a positively charged amino acid and is predicted to induce a dramatic change in the stability of the structure, the E3002D variant at the same residue maintains the negative charge and only affects hydrogen bonds with surrounding residues. The remaining intermediate activity VUS R2842C/L (mouse R2763), T3033I (mouse T2952) and Y3092S (mouse Y3011S) are not located at interdomain interfaces but may influence DNA binding. Overall these results demonstrate that not all residues at inter-domain interfaces are required for BRCA2 HDR activity. Mutation of certain interface residues may not affect the function because the specific change can be tolerated within the DBD structure or because the parental residues are not important for maintenance of inter-domain interactions. These findings suggest that the location of mutant residues alone is insufficient for predicting effects of VUS on protein function or on cancer risk.
We chose a cell-based HDR assay rather than an in vitro biochemical assay to ensure that the in vivo effects of VUS on HDR activity were measured. In addition, we based the HDR assay on the use of the full-length BRCA2 protein, in order to understand how BRCA2 VUS affect the HDR function of BRCA2 in the context of all other functional domains. Since several functional properties have been attributed to BRCA2, it is important to account for the effects of these characteristics, some of which have unknown relevance to cancer risk, on BRCA2 HDR activity. The use of full-length protein also limits the potential for misinterpretation of effects observed in partial proteins. Importantly, even though the assay is conducted using a full-length protein, only variants in domains directed implicated in HDR repair should be evaluated. Here we have focused on the BRCA2 DBD, but it is possible that variants in other domains, such as the N-terminal PALB2 interacting domain, can also be effectively evaluated. It is also possible that there are other functions of BRCA2 associated with the DNA binding domain of BRCA2 that are not detected by the HDR assay. If these alternative functions influence breast cancer risk then VUS with no effect or limited influence on HDR activity may actually influence breast cancer risk. For this reason, VUS with wild-type HDR activity should be treated with caution.
Supplementary Material
Acknowledgements
Grant Support
This research was supported by an NIH specialized program of research excellence (SPORE) in breast cancer award to the Mayo Clinic (CA116201) NIH grants R01 CA116167 and R01 CA128978. LG is supported by a fellowship from the Komen Foundation for the Cure. SMD is supported by funding from the Komen Foundation for the Cure. FJC and KLN are supported by funding from the Breast Cancer Research Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 2.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–83. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Easton DF, Steele L, Fields P, Ormiston W, Averill D, Daly PA, et al. Cancer risks in two large breast cancer families linked to BRCA2 on chromosome 13q12-13. Am J Hum Genet. 1997;61:120–8. doi: 10.1086/513891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antoniou AC, Cunningham AP, Peto J, Evans DG, Lalloo F, Narod SA, et al. The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br J Cancer. 2008;98:1457–66. doi: 10.1038/sj.bjc.6604305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The Breast Cancer Information Core [1996];An Open Access On-Line Breast Cancer Mutation Data Base. 1996 Available from: http://research.nhgri.nih.gov/bic/
- 6.Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–83. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–44. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chenevix-Trench G, Healey S, Lakhani S, Waring P, Cummings M, Brinkworth R, et al. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006;66:2019–27. doi: 10.1158/0008-5472.CAN-05-3546. [DOI] [PubMed] [Google Scholar]
- 9.Walker LC, Whiley PJ, Couch FJ, Farrugia DJ, Healey S, Eccles DM, et al. Detection of splicing aberrations caused by BRCA1 and BRCA2 sequence variants encoding missense substitutions: implications for prediction of pathogenicity. Hum Mutat. 2010;31:E1484–505. doi: 10.1002/humu.21267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. Available from: http://agvgd.iarc.fr/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB. In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat. 2008;29:1327–36. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–91. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farrugia DJ, Agarwal MK, Pankratz VS, Deffenbaugh AM, Pruss D, Frye C, et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008;68:3523–31. doi: 10.1158/0008-5472.CAN-07-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MS, Green R, Marsillac SM, Coquelle N, Williams RS, Yeung T, et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010;70:4880–90. doi: 10.1158/0008-5472.CAN-09-4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu K, Hinson SR, Ohashi A, Farrugia D, Wendt P, Tavtigian SV, et al. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005;65:417–26. [PubMed] [Google Scholar]
- 16.Couch FJ, Rasmussen LJ, Hofstra R, Monteiro AN, Greenblatt MS, de Wind N. Assessment of functional effects of unclassified genetic variants. Hum Mutat. 2008;29:1314–26. doi: 10.1002/humu.20899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29:1342–54. doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallee MP, Francy TC, Judkins MK, Babikyan D, Lesueur F, Gammon A, et al. Classification of missense substitutions in the BRCA genes: a database dedicated to Ex-UVs. Hum Mutat. 2012;33:22–8. doi: 10.1002/humu.21629. Available from: http://brca.iarc.fr/LOVD/home.php?select_db=BRCA2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraakman-van der Zwet M, Overkamp WJ, van Lange RE, Essers J, van Duijn-Goedhart A, Wiggers I, et al. Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol Cell Biol. 2002;22:669–79. doi: 10.1128/MCB.22.2.669-679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fawcett T. An introduction to ROC analysis. Pattern Recognition Letters. 2006;27:861–74. [Google Scholar]
- 21.McLachlan GJ. Discriminant analysis and statistical pattern recognition. Wiley; New York: 1992. [Google Scholar]
- 22.Fraley C, Raftery AE. Model-based clustering, discriminant analysis, and density estimation. Journal of the American Statistical Association. 2002;97:611–31. [Google Scholar]
- 23.Lindor NM, Guidugli L, Wang X, Vallee MP, Monteiro AN, Tavtigian S, et al. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat. 2012;33:8–21. doi: 10.1002/humu.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freedman ML, Penney KL, Stram DO, Le Marchand L, Hirschhorn JN, Kolonel LN, et al. Common variation in BRCA2 and breast cancer risk: a haplotype-based analysis in the Multiethnic Cohort. Hum Mol Genet. 2004;13:2431–41. doi: 10.1093/hmg/ddh270. [DOI] [PubMed] [Google Scholar]
- 25.Deffenbaugh AM, Frank TS, Hoffman M, Cannon-Albright L, Neuhausen SL. Characterization of common BRCA1 and BRCA2 variants. Genet Test. 2002;6:119–21. doi: 10.1089/10906570260199375. [DOI] [PubMed] [Google Scholar]
- 26.Wagner TM, Hirtenlehner K, Shen P, Moeslinger R, Muhr D, Fleischmann E, et al. Global sequence diversity of BRCA2: analysis of 71 breast cancer families and 95 control individuals of worldwide populations. Hum Mol Genet. 1999;8:413–23. doi: 10.1093/hmg/8.3.413. [DOI] [PubMed] [Google Scholar]
- 27.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–94. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 28.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. Available from: http://www.umd.be/HSF/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–48. doi: 10.1126/science.297.5588.1837. Available from: http://www.rcsb.org/pdb/explore.do?structureId=1iyj. [DOI] [PubMed] [Google Scholar]
- 30.Spurdle AB, Healey S, Devereau A, Hogervorst FB, Monteiro AN, Nathanson KL, et al. ENIGMA--evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33:2–7. doi: 10.1002/humu.21628. Available from: http://enigmaconsortium.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.