

Abstract
Forensic analysis of low template (LT) DNA mixtures is particularly complicated when (1) LT components concur with high template components, (2) more than three contributors are present, or (3) contributors are related. In this study, we generated a set of such complex LT mixtures and examined two methods to assist in DNA profile analysis and interpretation: the “n/2” consensus method (Benschop et al. 2011) and the pool profile approach. N/2 consensus profiles include alleles that are reproducibly amplified in at least half of the replications. Pool profiles are generated by injecting a blend of independently amplified PCR products on a capillary electrophoresis instrument. Both approaches resulted in a similar increase in the percentage of detected alleles compared to individual profiles, and both rarely included drop-in alleles in case mixtures of pristine DNAs were used. Interestingly, the consensus and the pool profiles often showed differences for the actual alleles detected for the LT component(s). We estimated the number of contributors using different methods. Better approximations were obtained with data in the consensus and pool profiles compared to the data of the individual profiles. Consensus profiles contain allele calls only, while pool profiles consist of both allele calls and peak height information, which can be of use in (statistical) profile analysis. All advantages and limitations of the various types of profiles were assessed, and based on the results we infer that both consensus and pool profiles (or a combination thereof) are helpful in the interpretation of complex LT DNA mixtures.
Electronic supplementary material
The online version of this article (doi:10.1007/s00414-011-0647-5) contains supplementary material, which is available to authorized users.
Keywords: Forensic science, Complex mixtures, Low template STR typing, Consensus method, Pool, Profile, Next Generation Multiplex (NGM)
Introduction
Short tandem repeat (STR) typing is performed to obtain individualizing evidence of human biological evidentiary traces for court-going purposes. In the past decade, many technological developments have resulted in more sensitive DNA profiling [2–12]. With that, it has become possible to analyze minute amounts of DNA. Notwithstanding the increased sensitivity of DNA analysis, most samples with very low amounts of DNA result in incomplete STR profiles. Often, these profiles show low template (LT) amplification artifacts like allele drop-out, allele drop-in, increased stutter peaks, and severe peak imbalance. Moreover, evidential biological stains may contain cell material of several individuals. DNA mixtures complicate STR typing and profile interpretation, especially when there are unequal contributions of DNA from the various individuals. For these unequal DNA mixtures, the use of methods that sensitize STR typing meets with serious constraints as increased cycling or higher injection settings result in overamplified or overloaded STR profiles. The corresponding profiles are even more complex to interpret when DNA of three or more individuals is present, when sporadic contamination occurs, or when relatives are involved [13–15]. To establish profile interpretation, one can infer the genotype(s) by a consensus method [1, 5, 16, 17]. In Benschop et al. [1], we examined several consensus methods that varied for the number of PCR amplifications and the requested level of reproducibility. We reported that the most accurate consensus method is “n/2,” which corresponds to reporting the alleles that appear in half of the replicates (rounded up). In addition, this approach provided most optimal results when performing DNA database searches. We found three of four replicates the most advantageous n for the n/2 consensus method [1].
Caragine and co-workers [16] used a more conservative approach as additional criteria were set to the consensus approach; besides detection in at least two of three replicates, the peak heights of the alleles in the individual profiles need to stay within specified ratios and need to be present in a pooled sample [16]. A pooled sample, or pool profile, is generated by blending independently amplified PCR products (obtained from the same DNA extract) and injecting this blend on a capillary electrophoresis (CE) instrument [16]. While consensus profiles focus on reproduced allele calls and only contain qualitative information, pool profiles also contain quantitative information meaning the heights and areas of the peaks. Peak heights (or areas) are useful for mixture deconvolution, although peak heights often show imbalance when typing LT samples. According to Caragine et al., pool profiles contain improved peak height ratios (PHRs) compared to individual amplifications [16]. We infer that these pool profiles may be more expedient for mixture interpretation than profiles obtained from individual amplifications.
In this study, we compared the performance of the n/2 consensus profiles to the pool profiles for two sets of complex mixtures with LT components, using three and four Next Generation Multiplex (NGM) amplifications. We investigated the performance of n/2 consensus profiles, pool profiles and the individual profiles. Regarding the pool profiles, we realize that the implementation might be difficult in practice, especially when the independent amplifications are completed at different moments in time. To overcome this issue, we also generated virtual pool profiles. A virtual pool profile is based on the allele calls and their peak heights in a set of fully analyzed individual amplifications: when the average peak height reaches the detection threshold, alleles are assigned to a virtual pool profile. Comparisons of the different types of profiles in this study (Fig. 1) included: (1) the percentage of detected donor alleles and (2) the number and position of drop-in alleles. Thirdly, we inferred the number of contributors in the mixtures using three different methods: the maximum allele count, the maximum likelihood estimator, and the GeneMapper ID-X mixture analysis tool. All three methods are applicable to mixtures that result in complete and partial profiles, although the precision of all methods decreases when more drop-outs occur. As a fourth aspect, we estimated the mixture proportion, when appropriate (depending on type of mixture and type of profile). Finally, we explored to what extent consensus and pool profiles can be combined to facilitate the interpretation of complex LT DNA mixtures. Determining the evidential value by creating hypothesis-based calculations was not included in this study.
Fig. 1.
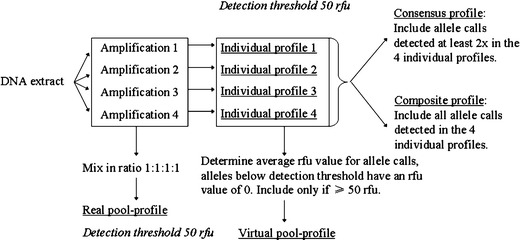
The various types of profiles used in this study when based on four independent amplifications per DNA extract, namely: individual profiles, real pool profile, virtual pool profile, consensus profile (n = 4 x = 2), and composite profile
Material and methods
DNA samples
The DNA samples consisted of commercially available pristine DNAs, DNAs extracted from buccal swabs (donated by four brothers with informed consent), and extracts from mimicked strangulation experiments. Three different pristine DNAs were used (all Applied Biosystems™ (AB), Nieuwerkerk aan den IJssel, The Netherlands), namely hDNA (200 ng/μl) that is the DNA standard in the Quantifiler Human kit, 9947A (10 ng/μl), which is the negative control of the Y-Filer kit, and DNA007 (0.1 ng/μl), which is the positive control of e.g. the NGM kit. For each pristine DNA we use a single batch. The four mimicked strangulation samples (the hands of donor A rubbed on the arm of donor B) were obtained from two different male–female couples after informed consent [18].
DNA was extracted using the QIAamp mini kit as described by the manufacturer (Qiagen, Venlo, The Netherlands) with minor adjustments for the buccal swabs (elution in 25% AE instead of 100% AE). The NGM genotypes of all donors involved in the experiments were known.
DNA quantification
Pristine and buccal DNA extracts were quantified on a RT7900HT apparatus using the Quantifiler Human kit as described by the manufacturer (AB). DNA extracts were diluted to 300, 150, 30, and 6 pg/μl, respectively, and quantified again to confirm the DNA concentration. The mock casework mixtures were not quantified as these were known to contain low amounts of DNA (samplings of contact traces on skin).
Mixture ratios
Two-, three- and four-person mixtures were made from pristine DNAs of unrelated individuals and from DNA extracts from buccal swabs of related individuals. For minor components, we used 30 pg of DNA per contributor. For major contributors, five or ten times higher contributions were added (150 or 300 pg, respectively). In addition, to mimic sporadic contamination, we admixed 6 pg (one diploid cell equivalent) of an unrelated contributor to some of the mixtures. In total, 37 different mixtures were generated (Table 1).
Table 1.
Overview of the 37 mixtures made from pristine DNA and/or DNA extracted from buccal swabs. In the mixture ratio column, 1, 5 and 10 correspond to 30 pg, 150 pg, and 300 pg, respectively. “+” represents a sporadic contamination of 6 pg of DNA. For all, but one, mixture types two samples were prepared, by using DNA of other contributors or by switching the major and minor component(s). The percentage of alleles shared between all donors (homozygotes counted as two) is presented per mixture
| Relatedness in mixture | Number of contributors | Mixture ratio | Relatedness of mixture components | Percentage of shared alleles | |
|---|---|---|---|---|---|
| Mixture 1 (%) | Mixture 2 (%) | ||||
| Unrelated (U) | 2 | 1:1 | U:U | 41 | 38 |
| 2+ | 1:1+ | U:U:U | 56 | 53 | |
| 3 | 1:1:1 | U:U:U | 56 | 53 | |
| 4 | 1:1:1:1 | U:U:U:U | 63 | 62 | |
| 2 | 5:1 | U:U | 41 | 38 | |
| 2+ | 5:1+ | U:U:U | 56 | 53 | |
| 3 | 5:1:1 | U:U:U | 56 | 53 | |
| 4 | 5:1:1:1 | U:U:U:U | 63 | 62 | |
| 2 | 10:1 | U:U | 41 | 38 | |
| 2+ | 10:1+ | U:U:U | 56 | 53 | |
| 3 | 10:1:1 | U:U:U | 56 | 53 | |
| 4 | 10:1:1:1 | U:U:U:U | 63 | 62 | |
| Related (R, brothers) | 2 | 1:1 | R:R | 59 | 77 |
| 2 | 5:1 | R:R | 59 | 77 | |
| 2 | 10:1 | R:R | 59 | 77 | |
| 3 | 5:1:1 | U:R:R | 61 | – | |
| 3 | 5:1:1 | R:R:U | 68 | 59 | |
| 3 | 5:1:1 | R:R:R | 84 | 84 | |
| 4 | 5:1:1:1 | R:R:R:U | 77 | 77 | |
| Total number of mixtures | 37 | ||||
Next generation multiplex PCR
Each DNA mixture was amplified in fourfold using the Next Generation Multiplex (NGM) kit (AB) at 29 cycles as recommended by the manufacturer. We used the same NGM batch within a set of amplifications—one NGM batch for pristine DNA mixtures (Table 1) and one batch for mimicked strangulation mixtures. For the mimicked strangulation mixtures, two different inputs (4 and 10 μl) of DNA extract were used. The negative control consisted of 10 μl dH2O and the positive control of 3 μl (300 pg) DNA007. All amplifications were performed on the same 9700 PCR apparatus (AB).
Capillary electrophoresis
All PCR products were run on the same ABI3130xl CE instrument. Pools were prepared by mixing equal amounts of three or four independent PCR amplifications obtained from the same DNA extract. CE mixtures contained 8.7 μl HiDi Formamide, 0.3 μl LIZ500, and 1 μl of individual or blended PCR products or 1 μl of NGM allelic ladder. CE injection settings were 3 kV 15 s for all individual and pooled PCR products. PCR products from mock casework mixtures were also subjected to higher CE injection settings (9 kV 10 s).
STR profile analysis
STR profiles were analyzed using GeneMapper ID-X version 1.1.1 software (AB). The analysis was performed using a detection threshold of 50 relative fluorescence units (rfu). We applied back stutter (−1 repeat unit) ratios specific for each locus as recommended by the manufacturer (AB). Forward stutter (+1 repeat unit) ratios were applied as determined during the in-house validation of the NGM kit (D22S1045, 7.36%; other loci, 2.5%).
Consensus profiles
Consensus profiles were made by the LCstat software (http://www.liacs.nl/∼hmeiland/projects/lcstat/), which uses the exported GeneMapper data. Alleles detected in at least half (rounded up) of the amplifications were assigned to the “n/2” consensus profile. Consensus profiles from three and four PCR amplifications were denoted n = 3 x = 2 and n = 4 x = 2, respectively [1]. In these designations, n represents the number of PCR amplifications and x represents the requested level of reproducibility of the alleles across the amplifications.
Virtual pool profiles
As the implementation of pool profiles may be difficult in practice, particularly when the replicates are produced with several weeks or even months time difference, we generated virtual pool profiles. For each locus, the peak heights of the detected alleles are averaged across the three and four replicates, and only the alleles with an average peak height above or equal to the detection threshold (50 rfu) are reported. An example of generating virtual pool profiles is shown in Table 2.
Table 2.
Example of allele calls in virtual pool profiles generated from amplifications one to four
| Allele call (rfu) | |||||
|---|---|---|---|---|---|
| 10 | 11 | 15 | 16 | 18 | |
| Amplification 1 | 100 | 80 | 200 | 300 | 55 |
| Amplification 2 | nd | nd | 150 | 150 | nd |
| Amplification 3 | 150 | nd | 300 | 200 | nd |
| Amplification 4 | 100 | 120 | 100 | 450 | nd |
| Average peak height | 88 | 50 | 188 | 275 | 14 |
| Allele calls in virtual pool | 10 | 11 | 15 | 16 | – |
nd allele not detected: not present or below the detection threshold of 50 rfu
Mixture analysis: number of contributors
Estimating the number of individuals that contributed to a DNA mixture is relevant in mixture deconvolution and in the calculation of likelihood ratios [19, 20]. In this study, we carried out estimations of the number of contributors in order to investigate the relative performance of individual, consensus, and pool profiles. We used three methods for this inference: (1) the allele counting method, also referred to as maximum allele count (MAC) [21]; (2) the maximum likelihood estimator (MLE, carried out using the Forensim package for the R statistical software) [22, 23] and (3) the GeneMapper ID-X mixture analysis tool (GMID-X).
The allele counting method simply relies on counting the maximum number of alleles observed throughout all loci: the locus showing the maximum number of different alleles determines the estimate for the number of contributors. For example, if the maximum number of different alleles observed at any locus of the profile is two, then the MAC yields an estimate of one for the number of contributors. If at most three or four alleles are observed, then the MAC gives an estimate of two.
Unlike allele counting, the MLE method makes explicit use of allele frequencies for the target population. The MLE method searches for the number of contributors that maximizes the probability of the data conditioned on the number of contributors. The MLE estimations are carried out using a population database for which we used the allele frequencies obtained from 2,085 randomly sampled Dutch individuals that were analyzed with various STR typing kits (Titia Sijen and Peter de Knijff, under preparation).
The GMID-X module relies both on allele counting and peak height information, and the underlying model is based on Gill et al. [24] (GeneMapper ID-X version 1.1.1, User’s Manual, AB). While the number of alleles per locus can be used to indicate the minimum number of contributors, the peak heights may point to an additional contributor in case of severe peak unbalance. Peak unbalance is measured by the peak height ratio, and the accepted peak balance for different ranges of the peak heights is used as input parameters for the mixture module. The mixture analysis parameters that we used for GMID-X analysis are based on single-source profiles produced during the in-house validation of the NGM kit. The mixture interpretation threshold was set at 50 rfu, and the PHR thresholds were as follows: PHR = 0.60 for rfu range 50 to 400, PHR = 0.67 for rfu range 401 to 1,000, and PHR = 0.86 for rfu range 1,001 to 8,000. With GMID-X, a sample is considered to originate from one contributor if no or just one locus contains at most three allele calls and no loci fail the set PHR thresholds. A sample is considered a potential mixture of two contributors if two or more loci contain three or more allele calls, with the maximum number of alleles not exceeding four or in case of two-peak loci failing the PHR thresholds. A sample is considered to contain a minimum of three contributors if there are more than four called alleles at any locus (GeneMapper ID-X version 1.1.1, User’s Manual, AB). GMID-X does not differentiate above three contributors. GMID-X runs GeneMapper profiles and is therefore applicable to individual profiles and pool profiles only, while MAC and MLE can be applied to consensus profiles as well. Although previous studies showed that the precision of the estimations decreases when allelic drop-out occurs [23], and that the true number of contributors to an evidentiary trace is never known with absolute certainty [15], we used incomplete profiles to carry out the estimations of the number of contributors as these are often obtained in casework analysis. We used different methods, different profile types, and various LT samples, as it is informative when assessing the relative performance of the methods and profile types.
Mixture analysis: ratio observed vs. expected peak height
A calculation based on observed peak heights was used to compare the ratio observed to expected peak heights of the pool and individual profiles. Only two-person mixtures were analyzed. Importantly, the genotypes of the contributors are known. The peak height (PH; rfu) of each allele is divided by the expected PH of that allele. Examples of the calculated expected PH and the ratio observed to expected PH are shown in Table 3. The effects of alleles residing at stutter position were not taken into account; we did not apply a correction for the stutter portion that may have accompanied an allele. Allele drop-ins were not taken into consideration either.
Table 3.
Four examples (A–D) of calculated ratios of observed PH to expected PH for the donor alleles at a given locus
| A | Genotype 30-pg contributor 1 | 9 | 10 | C | Genotype 300-pg contributor | 14 | 16 | ||||||||
| Genotype 30-pg contributor 2 | 8 | 11 | Genotype 30-pg contributor | 16 | 19 | ||||||||||
| Expected contribution | 1 | : | 1 | : | 1 | : | 1 | Expected contribution | 10 | : | 11 | : | 1 | ||
| Observed PH (rfu) | 55 | 82 | 55 | 84 | Observed PH (rfu) | 1,204 | 1,290 | 105 | |||||||
| Sum of PHs | 276 | Sum of PHs | 2,599 | ||||||||||||
| Expected PHa | 69 | 69 | 69 | 69 | Expected PHb | 1,181 | 1,300 | 118 | |||||||
| Ratio observed PH/expected PH | 0.80 | 1.19 | 0.80 | 1.22 | Ratio observed PH/expected PH | 1.02 | 0.99 | 0.89 | |||||||
| B | Genotype 300-pg contributor | 9 | 10 | D | Genotype 150-pg contributor | 22 | |||||||||
| Genotype 30-pg contributor | 8 | 11 | Genotype 30-pg contributor | 20 | 23 | ||||||||||
| Expected contribution | 1 | : | 10 | : | 10 | : | 1 | Expected contribution | 1 | : | 10 | : | 1 | ||
| Observed PH (rfu) | 199 | 1,098 | 1,213 | 195 | Observed PH (rfu) | nd | 1,193 | 65 | |||||||
| Sum of PHs | 2,705 | Sum of PHs | 1,258 | ||||||||||||
| Expected PHb | 123 | 1,230 | 1,230 | 123 | Expected PHc | 105 | 1,048 | 105 | |||||||
| Ratio observed PH/expected PH | 1.62 | 0.89 | 0.99 | 1.59 | Ratio observed PH/expected PH | 0.00 | 1.14 | 0.62 | |||||||
nd allele not detected using a detection threshold of 50 rfu
a(Sum of PHs/4) × expected contribution
b(Sum of PHs/22) × expected contribution
c(Sum of PHs/12) × expected contribution
Mixture analysis: mixture proportion
The mixture proportion was estimated with GMID-X that uses peak height information for this feature as based on Gill et al. [24]. The tool can only be applied to mixtures assigned to have two contributors. The software gives the average minor contributor mixture proportion (M x) for each sample which is calculated from four-peak and three-peak loci that have passed the PHR thresholds. Loci that fail the PHR threshold or loci that contain one or two peaks are excluded from the estimation (GeneMapper ID-X version 1.1.1, User’s Manual, AB).
Results and discussion
Comparing the performance of consensus and pool profiles
Percentage of detected alleles and number of drop-in alleles
For 37 mixtures (Table 1), four independent amplifications (individual profiles) were obtained. For the 300-pg components, full NGM profiles were obtained in all amplifications. For the 150-pg components, most profiles (33 of the 37) were complete, and only four amplifications showed one or two drop-out alleles. Full profiles were never observed for the 30-pg components. From these individual profiles, consensus profiles (both n = 3 x = 2 and n = 4 x = 2), real pool profiles (by blending three or four PCR mixtures), and virtual pool profiles (from averaging the results of three or four amplifications) were generated (Fig. 1). We studied the 30-pg LT contributors in the various types of profiles by determining the percentage of detected non-shared alleles. In concordance with earlier findings [1], the lowest percentage of detected alleles is obtained for individual profiles (Table 4). The use of four independent amplifications for generating a consensus or pool profile results in a higher percentage of detected alleles compared to three amplifications. These findings hold irrespective of the variables that reside within our set of complex mixtures (Table 1) such as different mixture proportions, different numbers of contributors, or related or unrelated donors (data not shown). Virtual pool profiles show a lower percentage of detected alleles than real pool profiles. This result can be explained by the procedure by which these profiles are obtained (Fig. 1); for virtual pool profiles, the analyzed GeneMapper profiles are used, while real pool profiles are based on the mixed PCR products. Consequently, peaks below the detection threshold (50 rfu for all types of profiles) are not taken into account in virtual pool profiles, while they do contribute to real pool profiles. The n = 4 x = 2 consensus profiles and real pool profiles of four amplifications show a similar average percentage of detected alleles (Table 4). For 32 of the 37 mixtures, the consensus and pool profiles from the same mixture and the same number of amplifications contain differences for the actual allele calls as shown in the example in Fig. 2. The maximum number of different allele calls per mixture is 9 and the total number of different allele calls for the 32 mixtures is 121. Of these alleles, 56% (68 alleles) are detected in the consensus, but not in the corresponding pool profile, and 44% (53 alleles) are detected in the pool profile, but not in the corresponding consensus. These results show that consensus profiles obtained from complex LT mixtures contain true allele calls that might be absent in the accompanying pool profile and vice versa.
Table 4.
Average percentages (and standard deviations) of detected non-shared alleles of the 30-pg contributor(s) in complex LT mixtures
| Percentage of detected alleles of the 30-pg contributor(s) (%) | ||
|---|---|---|
| Individual profiles (n = 296) | 63 ± 21 | |
| 3 amplifications | 4 amplifications | |
| Consensus profiles n/2 (n = 74) | 64 ± 22 | 78 ± 19 |
| Real pool profiles (n = 74) | 71 ± 20 | 78 ± 18 |
| Virtual pool profiles (n = 74) | 62 ± 23 | 65 ± 24 |
Fig. 2.
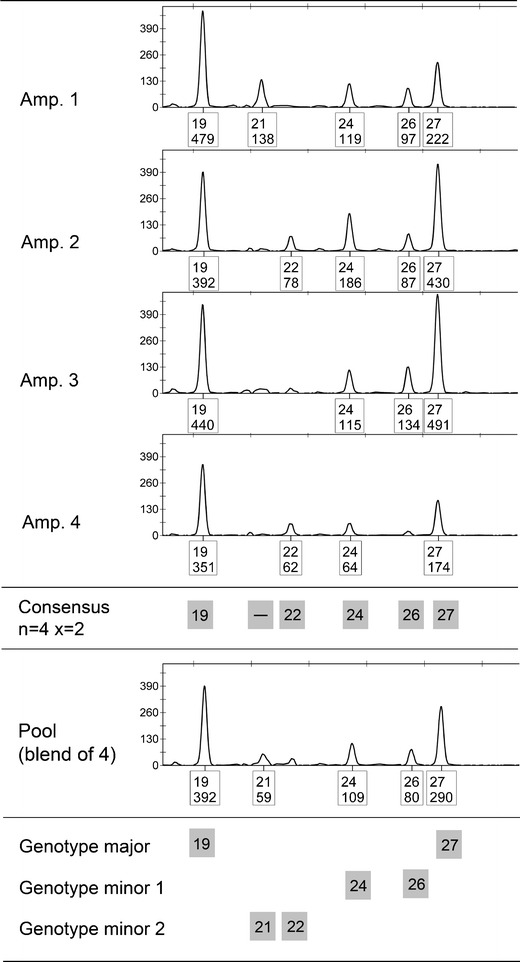
Example of the differences observed in the alleles included in the n = 4 x = 2 consensus and the pool profile (blend of the four amplifications). For this three-person mixture, two differences are obtained for locus FGA: in the consensus allele 21 of minor 2 is missing and in the pool profile allele 22 of minor 2 is absent
Next to studying the detection of donor alleles, we assayed the number of drop-in alleles that occur in the various types of profiles. As expected [1], individual profiles show the highest number of allele drop-ins (Table 5). Most of these appear to be amplification artifacts common with LT amplification. The majority are located at the −1/+1 stutter position between two true alleles that differ two repeat units in length. None of the consensus profiles contained a drop-in allele, and only one drop-in was observed in the pool profiles (Table 5). The infrequent occurrence of drop-in alleles in these 37 mixtures is most likely due to the use of pristine DNA [1].
Table 5.
Number and position of drop-in alleles observed in NGM profiles obtained from complex LT mixtures of pristine DNA. The virtual and real pool profiles that contain a drop-in allele are obtained from the same one mixture
| Number of profiles | Total number of drop-ins | Drop-in position | ||||
|---|---|---|---|---|---|---|
| Back (−1 repeat unit) | Forward (+1 repeat unit) | Between true alleles | Unrelated | |||
| Individual profiles | 148 | 17 | 4 | 2 | 10 | 1 |
| Consensus profiles | 74 | 0 | 0 | 0 | 0 | 0 |
| Real pool profiles | 74 | 2 1 | 0 | 0 | 2 | 0 |
| Virtual pool profiles | 74 | 1 2 | 0 | 0 | 1 | 0 |
For all further analyses described in this study, we selected the n = 4 x = 2 consensus profiles and the real pool profiles blended from four amplifications as these show higher percentages of detected alleles compared to the consensus/pool profiles generated from three amplifications and the virtual pools.
Mixture analysis
Determination of the number of contributors
An important step in mixture deconvolution is assessment of the potential number of contributors [14, 15, 21, 25]. The number of alleles per locus, the ratios of the peak heights, and the occurrence of alleles in relation to their population frequencies are useful indicators [20]. We applied different estimation methods to the individual profiles, consensus profiles (n = 4 x = 2), and pool profiles (blend of four) of the 37 mixtures (Table 1). The different methods comprise (1) counting the maximum number of alleles per locus (MAC), (2) using the MLE [23], and (3) applying the GMID-X mixture analysis tool (not for consensus profiles). All three methods are developed for complete profiles. In this study, we carry out estimations of the number of contributors on partial profiles. Although the inference is likely to be less precise than for full profiles (with no missing data), we believe that the estimates provide useful information for a comparison between different profile types, and between the different methods employed to carry out the estimations. When comparing the three methods, correct estimations were more often obtained when using the MLE method; with MAC, 132 of 222 profiles have a correct estimation; with MLE, 135 of 222; and with GMID-X, 67 of 145 (the four-person mixtures are excluded here as GMID-X assigns these as ≥3 contributors; Table 6). The slightly higher percentage of correctly estimated numbers of contributors when using MLE is probably because MLE accommodates allele sharing by taking into account the frequencies of the genotypes within a population [23]. For only one individual profile, we obtained an overestimated number of contributors with all three methods (results not shown). This overestimation was due to allele drop-in. Underestimations were more frequently obtained (results not shown) which is probably due to the partial profiles that were obtained for the LT contributors. This occurs for predominantly three types of mixtures. The first type has a very low (6 pg) additional contributor which is more often missed than recognized with all three methods (GMID-X, MAC, and MLE, Table 6). The extreme drop-out rate for this 6-pg contributor provokes the underestimation. The second type involves the mixtures of four contributors as these were designed to contain three or four LT components (Table 1). The third type comprises mixtures of three brothers, and the underestimations for these mixtures appear due to the genetic overlap between the donors. Since at most four alleles are present per locus, peak imbalance is the only indicator for a mixture of three relatives, and consequently, MAC an MLE will fail to recognize three contributors. GMID-X uses peak height information, but GMID-X did neither recognize that these mixtures have three contributors, which is probably due to the LT nature of the samples.
Table 6.
Percentages of profiles with correct estimation of the number of contributors using MAC, MLE and GMID-X. a) Mixtures containing DNA of unrelated individuals. b) Mixtures containing DNA of two or three brothers
| A | Number of contributors | |||||||||||||||
| 2 | 3 (of which one 6 pg) | 3 | 4 | |||||||||||||
| Number | MAC (%) | MLE (%) | GMID-X (%) | Number | MAC (%) | MLE (%) | GMID-X (%) | Number | MAC (%) | MLE (%) | GMID-X (%) | Number | MAC (%) | MLE (%) | GMID-X (%) | |
| Individual profiles | n = 24 | 96 | 96 | 96 | n = 24 | 33 | 33 | 0 | n = 24 | 67 | 67 | 67 | n = 24 | 8 | 13 | na |
| Consensus (n = 4 x = 2) | n = 6 | 100 | 100 | naa | n = 6 | 33 | 33 | na | n = 6 | 83 | 83 | na | n = 6 | 50 | 67 | na |
| Pool profiles (blend of four) | n = 6 | 100 | 100 | 100 | n = 6 | 33 | 33 | 0 | n = 6 | 83 | 100 | 83 | n = 6 | 67 | 67 | na |
| B | Number of contributors | |||||||||||||||
| 2 (of which 2 brothers) | 3 (of which 2 brothers) | 3 (of which three brothers) | 4 (of which three brothers) | |||||||||||||
| Number | MAC (%) | MLE (%) | GMID-X (%) | Number | MAC (%) | MLE (%) | GMID-X (%) | Number | MAC (%) | MLE (%) | GMID-X (%) | Number | MAC (%) | MLE (%) | GMIDX (%) | |
| Individual profiles | n = 24 | 100 | 100 | 100 | n = 12 | 58 | 58 | 58 | n = 8 | 0 | 0 | 0 | n = 8 | 13 | 13 | na |
| Consensus (n = 4 x = 2) | n = 6 | 100 | 100 | na | n = 3 | 100 | 100 | na | n = 2 | 0 | 0 | na | n = 2 | 0 | 0 | na |
| Pool profiles (blend of 4) | n = 6 | 100 | 100 | 100 | n = 3 | 100 | 100 | 100 | n = 2 | 0 | 0 | 0 | n = 2 | 0 | 0 | na |
A mixtures containing DNA of unrelated individuals, B mixtures containing DNA of two or three brothers
aNot applicable as GMID-X denotes four-contributor mixtures ≥3-contributor mixtures
These results show that better approximations of the number of contributors are obtained with consensus and pool profiles than with individual profiles, which may be due to the higher percentage of detected alleles for these profiles (Table 4). MAC, MLE, and GMID-X can be used to estimate the number of contributors, albeit with reduced efficacy in case of a high number of drop-outs or related donors (high rate of allele sharing).
Ratio observed vs. expected peak height
In mixed STR profiles, peak heights can be used to estimate the mixture ratio and assign alleles to a major or minor contributor. With LT STR profiles, alleles often show peak height imbalance or peaks remain below the detection threshold. According to Caragine et al. [16], the PHR is improved in pool profiles compared to individual profiles (since consensus profiles only contain allele calls PHR does not apply here). We examined the ratio observed to expected PH for the 30, 150, and 300-pg contributors’ alleles in two-person mixtures. The pool profiles (blend of four) and the corresponding individual profiles were examined. As expected, the ratio improves (the ratio is closer to 1) with increasing DNA inputs (Fig. 3). Interestingly, pool profiles show a better ratio observed to expected PH than individual profiles, although the ratio is still not perfect (a perfect ratio = 1).
Fig. 3.
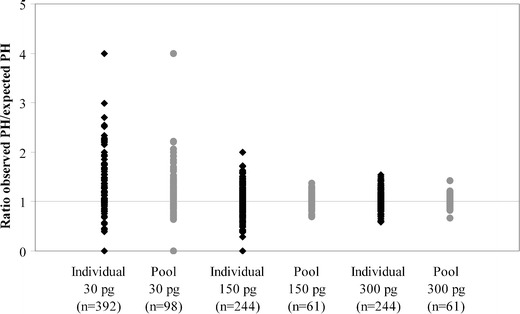
Ratio of observed PH to expected PH for the 30-, 150-, and 300-pg contributors’ alleles in individual profiles and pool profiles (blend of four) obtained from two-person mixtures with ratios 1:1, 1:5, and 1:10. The perfect ratio of observed to expected PH is 1, which is indicated by a horizontal line
Mixture proportion
Mixture deconvolution is supported by information on the proportions that individuals have in the mixture. Improved estimations of the mixture proportion may be obtained with pool profiles as they contain an improved observed to expected PH ratio (Fig. 3). To examine this aspect, we applied the GMID-X mixture analysis tool to the pool profiles and individual profiles obtained from the two-person mixtures and compared the results. Table 7 shows that the true mixture proportion is well approached for mixtures with ratios 1:5 (30 to 150 pg) and 1:10 (30 to 300 pg) for both individual and pool profiles. A 1:1 ratio (30 to 30 pg) is estimated less well, probably because the DNA quantities of both contributors are low level, resulting in imbalanced peaks for both donors. Approximately similar mixture proportions are estimated for mixtures of related and unrelated individuals (data not shown), though the relatives have a higher percentage of shared alleles (Table 1). Apparently, estimations of the mixture proportion by GMID-X are (at least under the settings we used) not very sensitive to changes in the peak height ratios that occur when pool profiles of mixtures of relatives are used or when pool profiles instead of individual profiles are used.
Table 7.
Average mixture proportions (M x) ± the standard deviations determined using the GMID-X mixture analysis tool for individual profiles and pool profiles (blend of four) from mixtures of two unrelated persons
| Mixture ratio | Expected M x | Average M x obtained by GMID-X | |
|---|---|---|---|
| Individual profiles (n = 16) | Pool profiles (n = 4) | ||
| 1:1 | 0.5 | 0.34 ± 0.08 | 0.38 ± 0.08 |
| 1:5 | 0.2 | 0.23 ± 0.05 | 0.17 ± 0.06 |
| 1:10 | 0.1 | 0.11 ± 0.04 | 0.08 ± 0.00 |
Performance of the consensus and the pool profiles obtained from mock casework mixtures
So far, we have compared the performance of the consensus to the pool profile approach on mixtures of pristine and diluted high template DNAs, with well-established amounts of DNA per contributor (Table 1). These mixtures show very few drop-in alleles (Table 5). In order to investigate situations that are more relevant to forensic casework, we also studied samplings from mimicked strangulation experiments [18]. These contact traces are prone to contamination as both volunteers did not wash their hands prior to contact. To establish a mock casework set ranging from very LT to moderately LT samples, different inputs and settings were used: amplifications were carried out using both 4 and 10 μl of each DNA extract, and all amplifications were analyzed using both standard and sensitized CE injection settings. These mock casework samples show trends similar to the mixtures of pristine DNAs: an increase in the percentage of contributor’s alleles and a decrease in the number of drop-in/additional alleles for consensus and pool profiles compared to individual profiles (Tables 4, 5, and 8). The pool profiles show slightly less drop-in alleles than the consensus profiles (Table 8). As expected, a higher number of drop-in/additional alleles is observed for larger DNA inputs or upon sensitized analysis (Table 8 and Supplementary Table 1). In contrast to the results with the mixtures of pristine DNAs (Table 5), these drop-in alleles regularly occur at random positions (e.g., 23% of the drop-ins found in 10 μl increased CE individual profiles are at non-stutter positions, Supplementary Table 1), suggesting the presence of sporadic contamination or additional low-level contributor(s) in these samplings.
Table 8.
Results for mock casework mixtures (contact traces with two donors). Amplification was carried out using 4 μl and 10 μl of each DNA extract, and CE analysis was performed using standard and higher CE injection settings. The average percentages of detected alleles and the average numbers of drop-in alleles are shown per individual profile, consensus profile (n = 4 x = 2) and pool profile (blend of four)
| 4-μl PCR input | 10-μl PCR input | |||||||
|---|---|---|---|---|---|---|---|---|
| Standard CE | Increased CE | Standard CE | Increased CE | |||||
| Percentage of detected alleles (%) | Number of drop-in alleles/profile | Percentage of detected alleles (%) | Number of drop-in alleles/profile | Percentage of detected alleles (%) | Number of drop-in alleles/profile | Percentage of detected alleles (%) | Number of drop-in alleles/profile | |
| Individual profiles (n = 16) | 65 ± 11 | 0.5 ± 1.0 | 81 ± 9 | 2.9 ± 2.2 | 86 ± 9 | 2.1 ± 2.0 | 93 ± 6 | 5.1 ± 2.5 |
| Consensus profiles (n = 4) | 72 ± 13 | 0.0 ± 0.0 | 91 ± 6 | 1.8 ± 2.4 | 90 ± 8 | 1.8 ± 2.1 | 97 ± 4 | 5.0 ± 3.2 |
| Pool profiles (n = 4) | 67 ± 13 | 0.0 ± 0.0 | 87 ± 8 | 1.5 ± 1.7 | 90 ± 7 | 0.5 ± 1.0 | 96 ± 5 | 3.0 ± 2.9 |
As with the mixtures of pristine DNAs, the mock casework mixtures contain differences in the allele calls in the consensus and pool profiles. For the mixtures analyzed using standard CE settings (n = 8), the number of different allele calls varies from 1 to 9 per profile and reaches a total of 19; 15 alleles are observed in the consensus profile, but not in the accompanying pool profile, and four alleles are observed in the pool profile, but not in the accompanying consensus profile.
We estimated the number of contributors using MLE since this approach appeared to be the most informative with the pristine DNA mixtures (Table 6). As the mock casework mixtures were obtained after mimicked strangulation between two individuals, we expected to obtain estimations of at least two contributors, although sporadic contamination may provoke estimates of more than two contributors. One underestimation (just one contributor) was obtained for an individual profile; for all other profiles, the method estimated two or three contributors (Supplementary Table 2). Estimations of three contributors were obtained with profiles containing many extra alleles (such as the 10 μl inputs or after sensitized CE; Table 8, Supplementary Table 1).
Strategies to use consensus and/or pool profiles
For both the pristine DNA and the mock casework mixtures, differences occur for the specific alleles that are present in the consensus and pool profiles. This can be explained from the manner by which these profiles were generated (Fig. 1): consensus profiles include alleles that are reproducibly amplified above detection threshold; pool profiles contain alleles that have sufficient peak height for detection in a blend of independent amplifications. Consequently, peaks below detection threshold have a weigh in pool profiles, but not in the consensus approach. Both strategies appear to be sound approaches, which is confirmed by the similar detection rates and low drop-in levels for both methods (Tables 4, 5, and 8). Accordingly, combining the results of consensus and pool profiles may be advantageous. We examined several strategies that combine genotyping results of consensus and pool profiles. The most stringent approach requires that an allele is called in both the consensus and the pool profile. A more permissive combination is to include alleles detected in either the consensus or the pool profile. We compared these two combined approaches to various other strategies so that in total six approaches are compared: (1) individuals profiles, (2) the composite profile of all four amplifications (n = 4 x = 1), (3) the consensus profile (n = 4 x = 2), (4) the pool profile (blend of four), (5) both the consensus and the pool profile, and (6) either the consensus or the pool profile (Fig. 2). The least conservative strategy (strategy 2, the composite method) shows, as expected and presented in [1], the highest percentage of detected alleles but also an unacceptable high number of drop-in alleles, especially for contact trace mixtures that are prone to have low level DNA contamination from other individuals (Table 9). The most conservative strategy (strategy 5), in which alleles need to be confirmed in both the consensus and the pool profile, shows the lowest number of drop-in alleles but also a low percentage of detected alleles (Table 9). Since the majority of consensus and pool profiles contain different alleles from the LT component(s), a combination of these profiles (strategy 6) results in a higher percentage of detected alleles than solely the consensus or pool profile (Table 9). For all cases where a drop-in allele occurred in both the consensus and the pool profile for these mixtures, this was the same allele. Therefore, the number of drop-in alleles in the combined profile did not increase compared to only the consensus or only the pool profile. As a higher percentage of the contributors’ alleles is detected when combining the consensus and the pool profile, a more accurate estimate on the number of contributors may be obtained. With either the consensus or the pool profile approach (strategy 6, Table 9), 30 of the 37 mixtures of pristine DNAs were correctly estimated by MLE, while 28 were correctly estimated using MAC. This is an improvement over the results with only consensus or pool profiles (strategy 3 or 4, Table 9): the numbers of correct estimations are 25 of 37 for consensus profiles with MAC, 26 of 37 for consensus profiles with MLE, 26 of 37 for pool profiles with MAC, and 27 of 37 for pool profiles with MLE (Table 6). These results imply that combining the consensus and pool profiles results can have added value.
Table 9.
Average percentages of detected alleles (pristine DNAs, non-shared 30-pg contributor alleles; mock casework mixtures, alleles of both donors) and average numbers of drop-in alleles for six strategies
| Strategy | Requirement for allele assignment; allele detected in at least: | Pristine DNA mixtures (n = 37) | Mock casework mixtures | ||||
|---|---|---|---|---|---|---|---|
| 3 kV 15 s CE (n = 8)a | 9 kV 10 s CE (n = 8)a | ||||||
| Percentage of detected alleles (%) | Number of drop-in alleles | Percentage of detected alleles (%) | Number of drop-in alleles | Percentage of detected alleles (%) | Number of drop-in alleles | ||
| 1 | Individual profile | 63 ± 21 | 0.11 ± 0.3 | 76 ± 15 | 1.3 ± 1.4 | 87 ± 10 | 4.0 ± 2.6 |
| 2 | Composite profile (n = 4 x = 1) | 95 ± 8 | 0.46 ± 0.7 | 92 ± 8 | 4.0 ± 3.7 | 98 ± 2 | 11.8 ± 5.3 |
| 3 | Consensus profile (n = 4 x = 2) | 78 ± 19 | 0.00 ± 0.0 | 81 ± 14 | 0.9 ± 1.6 | 94 ± 6 | 3.4 ± 3.1 |
| 4 | Pool profile (blend of four) | 78 ± 18 | 0.03 ± 0.2 | 79 ± 16 | 0.3 ± 0.7 | 92 ± 8 | 2.3 ± 2.4 |
| 5 | Consensus and pool profile | 72 ± 22 | 0.00 ± 0.0 | 78 ± 17 | 0.3 ± 0.7 | 91 ± 9 | 2.1 ± 2.4 |
| 6 | Consensus or pool profile | 83 ± 15 | 0.03 ± 0.2 | 83 ± 13 | 0.9 ± 1.6 | 95 ± 5 | 3.5 ± 3.1 |
aResults of 4- and 10-μl input combined
Concluding remarks
In this study, we examined the suitability of the “n/2” consensus method for complex LT DNA mixtures amplified by the NGM kit and considered the use of pool profiles. From pristine DNAs, a set of LT mixtures was prepared that had up to four contributors, which were present in various ratios (1:1, 1:5, and 1:10), with different quantities (6, 30, 150, or 300 pg), and with and without DNA of related donors. In addition, we analyzed two-donor mock casework samples (contact traces) that appeared to contain sporadic contamination.
For these complex LT mixtures, similar results were obtained as with the LT samples described in [1]: in n/2 consensus profiles, the percentages of detected alleles increase and the number of (stutter) drop-in alleles decreases compared to individual profiles. In addition, a consensus profile based on four amplifications is more informative than a consensus profile based on three amplifications, as on average 14% more of the alleles of a minor contributor are detected. Pool profiles were found to achieve a similar increase in the percentage of detected alleles as the consensus approach, especially when real pools (and not virtual pools) are made by blending four amplifications. Interestingly, the consensus and pool profiles can contain differences in the actual allele calls of a LT component. Therefore, an approach that includes alleles detected in either the consensus or the pool profile resulted in a higher percentage of detected alleles while, importantly, the number of drop-in alleles remained similar. Still, for contact trace samples, such as the mimicked strangulation mixtures used here [18], additional alleles may arise as a result of sporadic contamination or an additional low-level contributor. As apparent from Table 9, these extra alleles may end up in the deduced profile even when using a very conservative strategy (e.g., detection in both the consensus and the pool profile).
It is a common practice in many forensic laboratories to use a consensus approach to infer the genotypes from LT STR profiles. However, for this method only the qualitative data of an STR profile are used and only qualitative data will be available for further analyses. Hence, for consensus profiles, statistical approaches to weigh the DNA evidence that involve the use of quantitative data (e.g., peak height information) cannot be applied. Contrarily, pool profiles contain both qualitative and quantitative data. Pool profiles show improved peak height ratios compared to individual profiles [16], and may therefore be preferred in statistical models dedicated to the interpretation of evidentiary traces. Three methods (MAC, MLE, and GMID-X) were tested to estimate the number of contributors in the mixtures, and the MLE method was found to be the most informative which may be due to the fact that MLE takes allele sharing into account by using population allele frequencies.
With this study, it becomes evident that both consensus and pool profiles (and combinations thereof) have value for the analysis of DNA evidence. What method is best applied depends (among others) on the workflow within a forensic laboratory, and Table 10 provides an overview of the advantages and limitations of the various approaches.
Table 10.
Overall characteristics of various types of profiles or combinations of profile types
| Type of profile | Lab procedure | Detection of LT donor alleles (% for pristine DNA mixturesa)b | Occurrence of drop-ins (# for pristine DNA mixturesa)b | Applicability of statistical tools | Overall |
|---|---|---|---|---|---|
| Individual profile | Simple: 1 amplification | Lowest (63%) | Some (0.11/profile) | All | Low % detected donor alleles |
| Composite profile | From independent amplifications. Only allele calls needed; simply created on paper/Excel | Highest increase (+32%) | Increased (+0.35/profile) | Limitedc | Increase in drop-in alleles |
| Real pool profile | Mix the independent amplifications; PCRs preferably executed simultaneously; additional CE analysis | Increased (+15%) | Decreased (−0.08/profile) | All | Informative and suited for all tools but additional lab work |
| Virtual pool profile | Data from independent amplifications. Two aspects needed: (1) allele calls, (2) peak heights; created on paper/Excel | Slightly increased (+2%) | Decreased (−0.11/profile) | Limitedc | Little information gained |
| Consensus profile (n/2) | Data from independent amplifications. Only allele calls needed; simply created on paper/Excel | Increased (+15%) | Decreased (−0.11/profile) | Limitedc | Informative, little additional work |
| Combination: consensus and pool-profile | Both additional lab work and additional excel analyses needed | Increased (+9%) | Decreased (−0.11/profile) | Limitedc | Little information gained (very conservative) and laborious |
| Combination: consensus or pool profile | Both additional lab work and additional paper/Excel analyses needed | Higher increase (+20%) | Decreased (−0.08/profile) | Limitedc | Informative but laborious |
a Similar trends observed for mock casework mixtures
b The values found for individual profiles are taken as reference to determine increase or decrease
c MAC and MLE can be applied, while tools that make use of quantitative data (like GMID-X) cannot be applied
Electronic supplementary materials
Below is the link to the electronic supplementary material.
(PDF 14 kb)
(PDF 13 kb)
Acknowledgments
The authors are grateful to the donors for providing the buccal swabs, to Saskia Verheij for providing the contact trace extracts from mimicked strangulation samples, and to Ankie van Gorp for critically reading the manuscript. This study was supported by a grant from the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research (NWO) within the framework of the Forensic Genomics Consortium Netherlands.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Corina Benschop, Email: c.benschop@nfi.minjus.nl.
Hinda Haned, Email: h.haned@nfi.minjus.nl.
Titia Sijen, Email: t.sijen@nfi.minjus.nl.
References
- 1.Benschop CC, van der Beek CP, Meiland HC, van Gorp AG, Westen AA, Sijen T. Low template STR typing: effect of replicate number and consensus method on genotyping reliability and DNA database search results. Forensic Sci Int Genet. 2011;5:316–328. doi: 10.1016/j.fsigen.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Matai A, Harteveld J, Sijen T (2010) The Next Generation Multiplex (NGM™ Kit) in a forensic setting. Forensic News, July
- 3.Tucker VC, Hopwood AJ, Sprecher CJ, McLaren RS, Rabbach DR, Ensenberger MG, Thompson JM, Storts DR. Developmental validation of the PowerPlex((R)) ESI 16 and PowerPlex((R)) ESI 17 Systems: STR multiplexes for the new European standard. Forensic Sci Int Genet. 2010;5:257–258. doi: 10.1016/j.fsigen.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Tucker VC, Hopwood AJ, Sprecher CJ, McLaren RS, Rabbach DR, Ensenberger MG, Thompson JM, Storts DR (2011) Developmental validation of the PowerPlex((R)) ESX 16 and PowerPlex((R)) ESX 17 Systems. Forensic Sci Int Genet. doi:10.1016/j.fsigen.2011.03.009 [DOI] [PubMed]
- 5.Gill P, Whitaker J, Flaxman C, Brown N, Buckleton J. An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci Int. 2000;112:17–40. doi: 10.1016/S0379-0738(00)00158-4. [DOI] [PubMed] [Google Scholar]
- 6.Whitaker JP, Cotton EA, Gill P. A comparison of the characteristics of profiles produced with the AMPFlSTR SGM Plus multiplex system for both standard and low copy number (LCN) STR DNA analysis. Forensic Sci Int. 2001;123:215–223. doi: 10.1016/S0379-0738(01)00557-6. [DOI] [PubMed] [Google Scholar]
- 7.Kloosterman AD, Kersbergen P. Efficacy and limits of genotyping low copy number (LCN) DNA samples by multiplex PCR of STR loci. J Soc Biol. 2003;197:351–359. doi: 10.1051/jbio/2003197040351. [DOI] [PubMed] [Google Scholar]
- 8.Gaines ML, Wojtkiewicz PW, Valentine JA, Brown CL. Reduced volume PCR amplification reactions using the AmpFlSTR Profiler Plus kit. J Forensic Sci. 2002;47:1224–1237. [PubMed] [Google Scholar]
- 9.Weiler NE, Matai AS, Sijen T (2011) Extended PCR conditions to reduce drop-out frequencies in low template STR typing including unequal mixtures. Forensic Sci Int Genet. doi:10.1016/j.fsigen.2011.03.002 [DOI] [PubMed]
- 10.Forster L, Thomson J, Kutranov S. Direct comparison of post-28-cycle PCR purification and modified capillary electrophoresis methods with the 34-cycle “low copy number” (LCN) method for analysis of trace forensic DNA samples. Forensic Sci Int Genet. 2008;2:318–328. doi: 10.1016/j.fsigen.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Westen AA, Nagel JH, Benschop CC, Weiler NE, de Jong BJ, Sijen T. Higher capillary electrophoresis injection settings as an efficient approach to increase the sensitivity of STR typing. J Forensic Sci. 2009;54:591–598. doi: 10.1111/j.1556-4029.2009.01022.x. [DOI] [PubMed] [Google Scholar]
- 12.van Oorschot RA, Ballantyne KN, Mitchell RJ. Forensic trace DNA: a review. Investig Genet. 2010;1:14. doi: 10.1186/2041-2223-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torres Y, Flores I, Prieto V, Lopez-Soto M, Farfan MJ, Carracedo A, Sanz P. DNA mixtures in forensic casework: a 4-year retrospective study. Forensic Sci Int. 2003;134:180–186. doi: 10.1016/S0379-0738(03)00161-0. [DOI] [PubMed] [Google Scholar]
- 14.Clayton TM, Whitaker JP, Sparkes R, Gill P. Analysis and interpretation of mixed forensic stains using DNA STR profiling. Forensic Sci Int. 1998;91:55–70. doi: 10.1016/S0379-0738(97)00175-8. [DOI] [PubMed] [Google Scholar]
- 15.Buckleton JS, Curran JM, Gill P. Towards understanding the effect of uncertainty in the number of contributors to DNA stains. Forensic Sci Int Genet. 2007;1:20–28. doi: 10.1016/j.fsigen.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Caragine T, Mikulasovich R, Tamariz J, Bajda E, Sebestyen J, Baum H, Prinz M. Validation of testing and interpretation protocols for low template DNA samples using AmpFlSTR Identifiler. Croat Med J. 2009;50:250–267. doi: 10.3325/cmj.2009.50.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowen S, Debenham P, Dixon A, Kutranov A, Thomson J, Way K. An investigation of the robustness of the consensus method of interpreting low-template DNA profiles. Forensic Sci Int Genet. 2010;5:400–406. doi: 10.1016/j.fsigen.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 18.de Bruin KG, Verheij SM, Veenhoven M, Sijen T (2011) Comparison of stubbing and the double swab method for collecting offender epithelial material from a victim’s skin. Forensic Sci Int Genet. doi:10.1016/j.fsigen.2011.04.019 [DOI] [PubMed]
- 19.Gill P, Brenner CH, Buckleton JS, Carracedo A, Krawczak M, Mayr WR, Morling N, Prinz M, Schneider PM, Weir BS. DNA commission of the International Society of Forensic Genetics: recommendations on the interpretation of mixtures. Forensic Sci Int. 2006;160:90–101. doi: 10.1016/j.forsciint.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Buckleton J, Triggs CM, Walsh SJ. Forensic DNA evidence interpretation. Boca Raton: CRC Press; 2005. [Google Scholar]
- 21.Paoletti DR, Doom TE, Krane CM, Raymer ML, Krane DE. Empirical analysis of the STR profiles resulting from conceptual mixtures. J Forensic Sci. 2005;50:1361–1366. doi: 10.1520/JFS2004475. [DOI] [PubMed] [Google Scholar]
- 22.Haned H. Forensim: an open-source initiative for the evaluation of statistical methods in forensic genetics. Forensic Sci Int Genet. 2011;5:265–268. doi: 10.1016/j.fsigen.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 23.Haned H, Pene L, Lobry JR, Dufour AB, Pontier D. Estimating the number of contributors to forensic DNA mixtures: does maximum likelihood perform better than maximum allele count? J Forensic Sci. 2011;56:23–28. doi: 10.1111/j.1556-4029.2010.01550.x. [DOI] [PubMed] [Google Scholar]
- 24.Gill P, Sparkes R, Pinchin R, Clayton T, Whitaker J, Buckleton J. Interpreting simple STR mixtures using allele peak areas. Forensic Sci Int. 1998;91:41–53. doi: 10.1016/S0379-0738(97)00174-6. [DOI] [PubMed] [Google Scholar]
- 25.Egeland T, Dalen I, Mostad PF. Estimating the number of contributors to a DNA profile. Int J Legal Med. 2003;117:271–275. doi: 10.1007/s00414-003-0382-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 14 kb)
(PDF 13 kb)