

Abstract
Previous studies suggest that levels of the astrocyte-derived S100B protein, such as those occurring in brain extra-cellular spaces consequent to persistent astroglial activation, may have a pathogenetic role in Alzheimer's disease (AD). Although S100B was reported to promote β amyloid precursor protein overexpression, no clear mechanistic relationship between S100B and formation of neurofibrillary tangles (NFTs) is established. This in vitro study has been aimed at investigating whether S100B is able to disrupt Wnt pathway and lead to tau protein hyperphosphorylation. Utilizing Western blot, electrophoretic mobility shift assay, supershift and reverse transcriptase-polymerase chain reaction techniques, it has been demonstrated that micromolar S100B concentrations stimulate c-Jun N-terminal kinase (JNK) phosphorylation through the receptor for advanced glycation ending products, and subsequently activate nuclear AP-1/cJun transcription, in cultured human neural stem cells. In addition, as revealed by Western blot, small interfering RNA and immunofluorescence analysis, S100B-induced JNK activation increased expression of Dickopff-1 that, in turn, promoted glycogen synthase kinase 3β phosphorylation and β-catenin degradation, causing canonical Wnt pathway disruption and tau protein hyperphosphorylation. These findings propose a previously unrecognized link between S100B and tau hyperphosphorylation, suggesting S100B can contribute to NFT formation in AD and in all other conditions in which neuroinflammation may have a crucial role.
Keywords: Alzheimer's disease, reactive gliosis, S100B, Wnt pathway, Dickopff-1, glycogen synthase kinase 3β (GSK-3β), tau protein hyperphosphorylation, neurofibrillary tangles (NFTs)
Introduction
A hallmark of Alzheimer's disease (AD) are the neu-rofibrillary tangles (NFTs) seen in the pathologic brain [1]. NFTs are comprised of abnormal hyperphosphorylated tau microtubule associated protein, indicating that a dysregulation of tau phosphorylation may be responsible for the abnormal aggregation and the impaired function of this protein in the AD brain [2, 3]. Several studies suggest that glycogen synthase kinase 3β (GSK-3β), which phosphorylates serine/threonine and regulates the Wnt signalling pathway, likely contributes to tau hyperphosphorylation. Unlike most protein kinases, GSK-3β is normally constitutively active in all cells, and is primarily regulated through inhibition [4]. In general, GSK-3β phosphorylation is inhibited through the canonical Wnt signalling pathway [5]. Wnt, binding to frizzled receptor, recruits dishevelled protein, which in turn antagonizes GSK-3β activity. GSK-3β regulates tau by phosphorylation as well as regulates splice variance. Hyperphosphorylated tau has less affinity toward the microtubules, and readily aggregates into paired helical filaments (PHFs) and NFTs, disrupting microtubule stability. This leads to alterations in the synaptic plasticity and causes neurodegenerative changes [6]. However, also hyperphosphorylated tau itself appears to be neurotoxic and able per se to promote the neuronal injury and the cell death observed in AD brain [7].
Aberrant processing of β amyloid peptide (Aβ), which is thought to contribute to the development of AD, disrupts the Wnt signalling cascade, induces GSK-3β activation, stimulates tau hyperphosphorylation and provokes neuronal cell damage [8].
Aβ not only exhibits direct cytotoxic mechanisms that impact on neuronal survival but also promotes inflammatory processes that contribute to the AD phenotype [9]. While the initial glial activation due to Aβ beneficially contributes to the restoration of brain function, on the contrary a persistent reactive gliosis may result in maladaptative detrimental responses, thus substantially contributing to AD progression. For example, ongoing inflammation can trigger various cell stress-response pathways, including overexpression of the secreted glycoprotein Dickopff-1 (DKK-1). DKK-1 up-regulates GSK-3β activity, promotes tau hyper-phosphorylation, NFT formation and neuronal degeneration. Thus, DKK-1 inhibits Wnt signalling in a manner similar to Aβ, and thereby fosters a self-sustaining feedback loop resulting in cellular injury [8, 10, 11].
In this scenario S100B, a small soluble protein belonging to the large family of EF-related (EF helices) Ca++ and Zn++-binding proteins, primarily secreted by astrocytes [for review see 12], may contribute to the AD brain phenotype. While at nanomolar concentrations S100B provides a pro-survival effect on neurons and stimulates the neurite outgrowth, at higher micromolar concentrations the protein promotes neuroinflammatory processes and neuronal apoptosis [13]. S100B has been reported overexpressed in AD brain, especially where the neuritic plaques are concentrated. S100B, S100B mRNA and its specific activity were higher in AD patients than in age-matched controls, and the excess of S100B was localized to activated astrocytes surrounding the neuritic plaques. This suggests that elevate levels of S100B are closely associated with reactive gliosis and AD neuropathology [14], and this notion is substantiated by findings showing that S100B can directly increase the expression of amyloid precursor protein (APP) and APP mRNA in a time- and dose-dependent manner [15]. Lastly, detrimental effects of S100B are transduced by interactions with receptor for advanced glycation ending products (RAGE) [16], which in microglial cells has been reported to lead to c-Jun N-terminal kinase (JNK) activation [17]. Perturbations in the JNK sig-nalling cascade can regulate DKK-1 expression [18].
To date, no studies have investigated whether S100B is able to disrupt Wnt signalling and promotes tau protein hyperphosphorylation. The present study, using neural stem cells (NSCs), has been performed to explore this hypothesis. The findings propose a previously unrecognized link between S100B and tau hyperphosphorylation. Finally they reveal the biochemical mechanisms implicated in this effect and provide evidence that S100B contributes to NFT formation in AD.
Materials and methods
Cell culture
Derivation of foetal NSCs has previously been described [19]. NSCs were cultured in DMEM, 30% Ham's F-12, 1% antibiotic-antimycotic, 2% B27 (all from Invitrogen, Milan, Italy), 20 ng/ml EGF (Sigma, St Louis, MO, USA), 20 ng/ml FGF2 (R&D Systems Inc., Minneapolis, MN, USA) for 1 week. NSCs were maintained at a density of 5 × 105 cells/ml of medium, half of the medium being replaced with fresh medium every 3 days. For differentiation, cells were resuspended in plating media (DMEM/F-12, 2% B27), treated with 10-μM retinoic acid [20] (Sigma) and allowed to differentiate in plating media for 7–10 days. During differentiation, half of the plating medium was replaced every 2 days with fresh medium. After a brief incubation for 2–4 hrs in Neurobasal medium (Invitrogen), cells were supplemented with 1 mM glutamine, 2% B27 and 1% foetal bovine serum (FBS) for adhesion and treatments. SHSY5Y cells were cultured in DMEM supplemented with 5% foetal calf serum (FCS), 15% horse serum (HS), 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in 5% CO2/95% air and differentiated into neurons with 10 μM retinoic acid. Depending upon the experimental procedure, cells were plated on Petri dishes or on cover slips and fixed in 4% paraformaldehyde for immunofluorescence analysis.
Protein isolation and Western blot analysis
After treatments, cells were washed with ice-cold PBS and centrifuged at 180 ×g for 10 min. at 4°C. The cell pellet was resuspended in 100 μl of ice-cold hypotonic lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM PMSF, 1.5 μg/ml soybean trypsin inhibitor, 7 μg/ml pepstatin A, 5 μg/ml leupeptin, 0.1 mM benzamidine, 0.5 mM DTT) and incubated on ice for 15 min. Cell lysates were centrifuged at 13,000 ×g for 1 min. at 4°C. Protein concentration was determined, and equivalent amounts (100 μg) of each sample underwent SDS-PAGE electrophoresis. Proteins were then transferred onto nitrocellulose membranes according to the manufacturer's instructions (Bio-Rad, Hercules, CA, USA) and immunoblot was carried out with the following antibodies: anti-JNK (1:500; Santa Cruz Biotechnology, La Jolla, CA, USA), anti-DKK-1 (1:1000, Santa Cruz Biotechnology), anti-β-actin (1:2000, Santa Cruz Biotechnology), anti-GSK-3β (1:250, Lab Vision, Fremont, CA, USA), anti-β-catenin (1:250, Lab Vision), anti-Tau (1:1000, Neomarker, Freemont, CA, USA). The membranes were incubated with the proper secondary antibody coupled to peroxidase (1:1000; DAKO, Golstrup, Denmark) and the immunocomplexes were visualized by the ECL chemiluminescence method (Amersham, Milan, Italy). Protein relative expression was quantified by densitometric scanning of the X-ray films with a GS 700 Imaging Densitometer (Bio-Rad) and a computer programme (Molecular Analyst, IBM, Milan, Italy).
RNA isolation and reverse transcriptase-PCR analysis
The mRNA level was determined using the semiquantitative reverse transcriptase-polymerase chain reaction (RT-PCR) method (Invitrogen). Total mRNA was extracted from cells by use of an ultrapure TRIzol reagent (Gibco BRL, Milan, Italy) as directed by the manufacturer. The concentration and purity of total mRNA were determined from the A260/A280 ratio using a UV spectrophotometer (DU 40, Beckman, Fullerton, CA, USA). The primer sequences used for PCR amplification were: c-Jun sense 5′-AAC- CTTCTATGACGATGCCCTCA-3′ and antisense 5′-CCT-GCTCATCTGTCACGTTCTT-3′; GAPDH sense 5′-GAAG-GTGAAGGTCGGAGT-3′ and antisense 5′-GAAGATGGT-GATGGGATTTC-3′; DKK-1 sense 5′-ACCAGACCATTGA-CAACTAC-3′ and antisense 5′-GTGTCTAGCACAACA-CAATC-3′.1 μg of total RNA from each specimen was subjected to RT-PCR, carried out using a SuperScript TM One-Step RT-PCR with Platinum Taq Kit (Invitrogen) in a total reaction volume of 25 μl, containing 2x reaction mix, 25 μM sense primer, 25 μM antisense primer, RT-PCR platinum Taq mix, and autoclaved distilled water. Electrophoresis was performed on the amplification products using a 1% agarose gel and the bands were visualized by staining with ethidium bromide. The integrated density values of the bands representing amplified products were acquired and analysed by GS 700 Imaging Densitometer (Bio-Rad) and a computer programme (Molecular Analyst, IBM).
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts, prepared from treated NSCs, were obtained according the technique described by De Stefano et al.[21]. Samples were incubated with 10000 cpm of the 32P-labelled AP-1 (5′-CGCTTGATGAGTCAGCCGGAA-3′) oligonucleotide (Promega Corp., Madison, WI, USA). The binding assay was performed in the presence of poly(dI-dC) (Pharmacia Biosystem, Milton Keynes, UK) as a nonspecific competitor and 32P-labelled AP-1 for 30 min. at room temperature. After electrophoresis, the gels were dried and autoradiographed. In supershift and competition assays, 500 μl of AP-1 antibodies (anti-Fra-1, anti-c-Fos, anti-c-Jun and anti-Jun-D, Santa Cruz Biotechnologies) were preincubated with nuclear extracts for 30 min. before the addition of the labelled probe.
Immunofluorescence
NSCs were plated onto glass slide chambers coated with poly-D-lysine (BD, Milan, Italy) for several hours and were fixed with 4% paraformaldehyde in PBS. The cultures were placed in PBS blocking solution containing 10% FCS and 5% HS, and incubated with the following antibodies: anti-DKK-1 (1:100, Santa Cruz Biotechnology), anti-MAP-2 (1:300, Lab Vision), anti-tau (1:200, Neomarker). Following PBS washing, sections were incubated in the dark for 30 min. with Texas Red conjugated or fluorescein isothio-cyanate conjugated secondary antibody (1:200, AbCam, Cambridge, UK). Nuclei were stained with Hoechst (Sigma). Laser scanning confocal microscopy was thus performed by using a Zeiss LSM 510 laser scanning system and immunopositive cells quantified by a computer programme.
DKK-1 RNA interference
Small interfering RNA (siRNA) technique for DKK-1 was carried out on SHSY5Y cells according to protocol indicated by the manufacturer (Santa Cruz Biotechnology, see catalogue number sc-37082) and scrambled sequence for DKK-1 was used as a negative control. Specific silencing was confirmed by at least three independent immunoblot and RT-PCR experiments.
Results
S100B induces JNK phosphorylation and AP-1/c-Jun activation through RAGE dependent interaction
Prior studies have shown that S100B interactions with RAGE in microglia are accompanied by increased oxidative stress signalling that is linked to the activation of different members of the stress-activated protein kinases (SAPKs) family, and subsequent induction of nuclear transcription factors [17, 18]. Initiation of this pathway is dependent upon exposure to micro-molar concentrations of exogenous S100B and interactions with the RAGE extracellular domain but is independent of RAGE transducing activity [17, 22]. To address whether a similar pathway might exist within a mixed human neuronal and glial cell population, we first investigated the effects of exogenous S100B challenge on phosphorylation of the stress-responsive JNK. Incubation of differentiated NSCs with increasing concentrations of purified S100B protein (0.05–5 μM) revealed a dose-dependent increase in JNK phosphorylation (pJNK) within 30 min. of S100B stimulation (Fig. 1). We then assessed the involvement of RAGE in mediating S100B activation of pJNK. S100B-treated cultures (maximal response obtained at 5 μM) were challenged with a specific RAGE blocking antibody at two dilutions (1:1000 or 1:10,000 v/v). The RAGE blocking antibody significantly reduced JNK phosphorylation in a concentration-dependent manner (Fig. 1). These experiments suggest that S100B interaction with RAGE leads to the activation of the stress-response kinase JNK in mixed human neuronal and glial cell populations.
1.
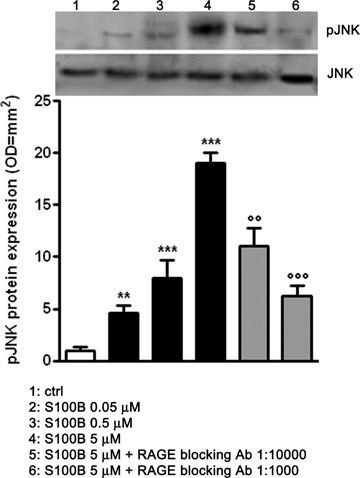
S100B induces JNK phopsphorylation through RAGE interaction in a dose-dependent fashion. pJNK protein expression was evaluated in lysates of NSCs 30 min. following exposure at increasing concentrations of S100B (0.05–5 μM), by Western blot (upper panel) and densitometric analysis of corresponding bands (lower panel). Statistics show a significant dose-dependent effect of S100B on JNK phosphorilation. Different concentrations of RAGE blocking antibody (1:1000 or 1:10,000) reverted the effect induced by the highest concentration of S100B. Results are the mean ±S.E.M. of three independent experiments. ***P < 0.001 and **P < 0.01 versus unchallenged cells (ctrl);°°°P < 0.001 and °°P < 0.01 versus 5 μM S100B challenged cells.
Phosphorylation of JNK as well as other SAPKs can lead to nuclear AP-1 complex up-regulation [23]. We therefore tested whether S100B exposure (5 μM) resulted in AP-1 complex induction by EMSA. S100B-induced AP-1 complex up-regulation peaked at 2 hrs after stimulus and progressively declined over 6 hrs after S100B exposure (Fig. 2A). Based on these results, we fixed the S100B exposure at 2 hrs for the subsequent investigations. Further EMSA analysis revealed that specific RAGE blocking antibody was able to decrease AP-1 complex up-regulation in a concentration-dependent manner (1:1000 or 1:10,000 v/v) within S100B-induced versus untreated differentiated NSCs. Blocking using an unrelated control antibody was ineffective and the addition of 100-fold molar excess of unlabelled AP-1 oligonucleotide completely abolished AP-1 DNA complex formation, showing the specificity of action by the RAGE antibody and S100B (Fig. 2B). Supershift studies were also performed on S100B challenged cultures (5 μM) to identify activation of specific AP-1 complex components, using antibodies directed against c-Fos, c-Jun, Fra-1 and Jun-D. A supershift in the c-Fos and c-Jun complexes was appreciated at 2 hrs after S100B treatment, while no significant effect was detected in the other tested complexes (Fig. 2C). Finally, RT-PCR analysis of the c-Jun/JNK pathway demonstrated that c-Jun mRNA expression was induced 6 hrs following cell stimulation with increasing concentrations of S100B (0.05–5 μM) in a concentration-dependent fashion, when compared with unchallenged cells. The S100B induction of c-Jun was attenuated by RAGE blocking antibody, while unrelated blocking antibody had no effect (Fig. 2D). Collectively, these studies demonstrate that the soluble, astroglial-derived S100B protein interacts with RAGE leading to the JNK phosphorylation and the pJNK-dependent up-regulation of c-Jun, a component of the AP-1 complex.
2.
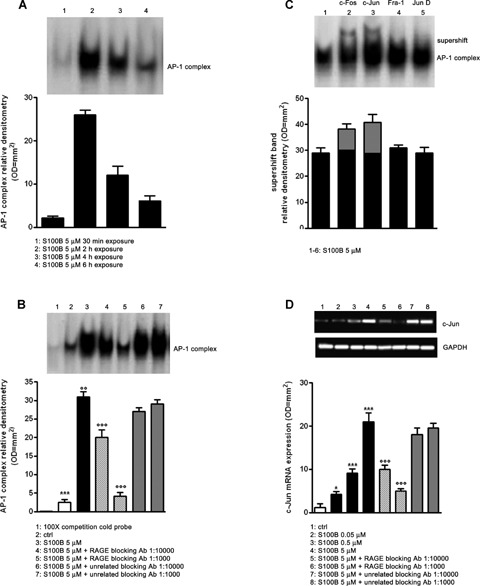
S100B induces AP-1/DNA complex activation through RAGE interaction. (A) AP-1/DNA complex activation was detected in NSCs at various time point (30 min. to 6 hrs) following the exposure to 5 μM of S100B. The time course of AP-1/DNA binding activity was evaluated by EMSA (upper panel) and densitometric analysis of corresponding bands (lower panel). (B) AP-1/DNA binding activity induced by 5 μM of S100B in NSCs in the presence or absence of two different concentrations of RAGE blocking antibody or unrelated blocking antibody (1:1000 or 1:10,000). Statistics show that S100B significantly enhanced the activation of AP-1/DNA complex and that the specific RAGE blocking antibody significantly blunted this effect, while the unrelated antibody failed to influence it. The AP-1/DNA binding activity was measured 2 hrs after S100B exposure by EMSA analysis (upper panel). The lower panel shows densitometric analysis of corresponding bands. (C) AP-1 antibodies (anti-Fra-1, anti-c-Fos, anti-c-Jun and anti-Jun-D) were preincubated with nuclear extracts from NSCs exposed to 5 μM of S100B. Results of supershift analysis (upper panel) demonstrate the effect of the antibodies on the changes in the relative mobility of AP-1 species 2 hrs following NSC stimulation with S100B. Densitometric analysis of corresponding bands is reported in the lower panel. (D) NSCs were challenged with increasing concentrations of S100B (0.05–5 μM) and lysed 6 hrs later. c-Jun mRNA expression was evaluated by RT-PCR (upper panel) and densitometric analysis of corresponding bands (lower panel). Statistics demonstrate significant and concentration-dependent effect of S100B on c-Jun mRNA expression. The figure also shows two different dilutions of RAGE blocking antibody (1:1000 or 1:10,000) were able to revert the effect of the highest concentration of S100B, whereas the same dilutions of an unrelated blocking antibody were ineffective. Results are the mean ±S.E.M. of three independent experiments.***P < 0.001, **P < 0.01, and *P < 0.05 versus unchallenged cells (ctrl);°°°P < 0.001 and °°P < 0.01 versus 5 μM S100B challenged cells.
S100B induces DKK-1 protein expression
Among the different c-Jun molecular targets, DKK-1 is a stress stimuli induced protein which behaves as a potent endogenous Wnt pathway disruptor [10]. In order to assess whether S100B might affect DKK-1 expression, we performed immunoblot analysis on differentiated human neuronal and glial cultures, challenged with increasing concentrations of purified S100B (0.05–5 μM). This treatment caused a concentration-dependent rise of DKK-1 protein expression 12 hrs after stimulation (Fig. 3). Under these same experimental conditions, S100B (5 μM) challenged cells were treated with RAGE blocking antibody (1:1000 or 1:10,000 v/v) in the presence or absence of the specific JNK phosphorylation inhibitor SP600125 (1 or 10 μM). S100B induction of DKK-1 up-regulation was blocked by RAGE blocking antibody in a dose-dependent fashion with almost complete inhibition of DKK-1 induction seen at the higher RAGE blocking antibody titre (1:1000 v/v) or in the presence of SP600125 (Fig. 3). These data thus support a pivotal role for JNK in mediating the molecular signalling mechanisms downstream of S100B stimulation, leading to DKK-1 up-regulation.
3.
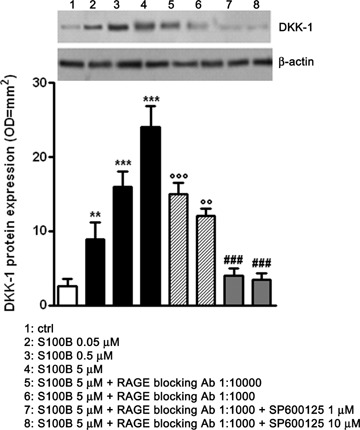
S100B induces DKK-1 protein expression through RAGE and JNK involvement in a dose-dependent fashion. NSCs were challenged with increasing concentrations of S100B (0.05–5 μM) and 12 hrs later they were lysed and DKK-1 protein expression was evaluated by Western blot (upper panel) and densitometric analysis of corresponding bands (lower panel). β-actin served as a loading control. Statistics indicate that S100B significantly promoted DKK-1 protein expression in a dose-dependent fashion. Moreover two different dilutions of RAGE blocking antibody (1:1000 or 1:10,000) were able to revert in a dose-dependent manner the effect of the highest concentration of S100B. This effect was strengthened by the specific JNK phosphorylation inhibitor SP600125 (1 or 10 μM). Results are the mean ± S.E.M. of three independent experiments. ***P < 0.001 and **P < 0.01 versus unchallenged cells (ctrl);°°°P < 0.001 and °°P < 0.01 versus 5 μM S100B challenged cells; ###P < 0.001 versus 5 μM S100B plus RAGE blocking antibody (1:1000) challenged cells.
S100B leads to tau protein hyperphosphorylation by inducing GSK-3β phosphorylation and causing β-catenin degradation in the Wnt pathway
Activation of the canonical Wnt pathway leads to two main molecular mechanisms, represented by the inhibition of GSK-3β and the relative accumulation/degradation of β-catenin in the cytoplasm [5, 8]. Different stress-related stimuli such as DKK-1 can also disrupt the Wnt pathway, inhibiting GSK-3β suppression and inducing tau protein hyperphosphorylation. Given our prior observation of DKK-1 induction by S100B, we investigated whether the GSK-3β and β-catenin expression was altered 24 hrs following cell stimulation with S100B (0.05–5 μM) by immunoblot analysis. S100B caused a concentration-dependent increase in GSK-3β phosphorylation (as shown by increased immunoreactivity of p-Ser-9 bands, Fig. 4A), as well as a parallel degradation in β-catenin expression in treated versus untreated cells (Fig. 4B). Both of these effects on S100B (5 μM) challenged cells were reduced by RAGE blocking antibody (1:1000 or 1:10,000 v/v), suggesting that S100B functions as a disruptor of the canonical Wnt pathway through a RAGE-dependent interaction.
4.
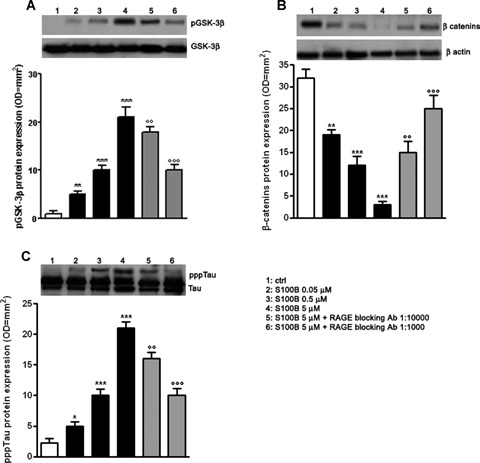
S100B induces tau protein hyperphosphorylation through Wnt pathway disruption. (A) NSCs were challenged with increasing concentrations of S100B (0.05–5 μM). The pGSK-3β protein expression was evaluated 24 hrs following treatment by Western blot (upper panel) and densitometric analysis of corresponding bands (lower panel). GSK-3β served as a loading control. Statistics demonstrate significant and concentration-dependent effect of S100B on pGSK-3β protein expression. Two different dilutions of RAGE blocking antibody (1:1000 or 1:10,000) were able to concentration-dependently antagonize the effect of the highest concentration of S100B. (B) β-catenin protein expression was evaluated 24 hrs following treatment by Western blot (upper panel) and densitometric analysis of corresponding bands (lower panel). β-actin served as a loading control. Statistics show that S100B significantly and concentration-dependently affected β-catenin protein expression. Two different dilutions of RAGE blocking antibody (1:1000 or 1:10,000) were able to revert the effect of the highest concentration of S100B in a concentration-dependent manner. (C) pppTau protein expression was evaluated 48 hrs following treatment by Western blot (upper panel) and densitometric analysis of corresponding bands (lower panel). Tau served as a loading control. Statistics indicate that S100B was able to concentration-dependently and significantly promote pppTau protein expression. Two different dilutions of RAGE blocking antibody (1:1000 or 1:10,000) antagonized the effect of the highest concentration of S100B in a concentration-dependent fashion. Results are the mean ±S.E.M. of three separated experiments.***P < 0.001, **P < 0.01, and *P < 0.05 versus unchallenged cells (ctrl);°°°P < 0.001 and °°P < 0.01 versus 5 μM S100B challenged cells.
Tau hyperphosphorylation is an expected consequence of Wnt pathway disruption. We therefore examined whether S100B treatment (0.05–5 μM) altered tau protein phosphorylation levels in the mixed neuronal and glial cultures. S100B exposure induced tau protein hyperphosphorylation at 48 hrs following stimulation in treated versus untreated cells, as shown by the immunoreactive pppTau bands (Fig. 4C). Similar to RAGE antibody inhibition of S100B-induced GSK-3β phosphorylation and β-catenin degradation, RAGE antibody (1:1000 or 1:10,000 v/v) blockade of interactions with S100B resulted in a concentration-dependent reduction in tau protein hyperphosphorylation (Fig. 4C).
DKK-1 inhibition abolishes S100B-induced tau protein hyperphosphorylation
The current experiments show that blocking S100B and RAGE interactions inhibits JNK phosphorylation, activation of the AP-1/cJUN complex, DKK-1 expression, components of the canonical Wnt pathway and tau microtubule associated protein phosphorylation in mixed human neuronal and glial cultures. Moreover, inhibitors of JNK phosphorylation block DKK-1 expression, and prior studies have shown that DKK-1 overex-pression promotes GSK-3β activity and tau hyperphosphorylation. To address whether S100B mediates DKK-1 protein expression and tau protein phosphorylation in neurons, we used immunoflurorescence analysis to identify neuronal staining for these proteins following S100B exposure. Cultures were exposed to S100B (5 μM) in the presence or absence of RAGE blocking antibody (1:1000 v/v), stained for the neuronal marker microtubule associated protein-2 (MAP2), DKK-1 and hyperphosphorylated tau protein (pppTau), and then visualized 12 or 48 hrs after stimulation. Consistent with the results obtained by immunoblot analysis, both DKK-1 and pppTau immunopositive neurons were increased in S100B treated versus untreated cells. As expected, this increase in immunoreactivity was also significantly blocked by RAGE antibody (Fig. 5A, B).
5.
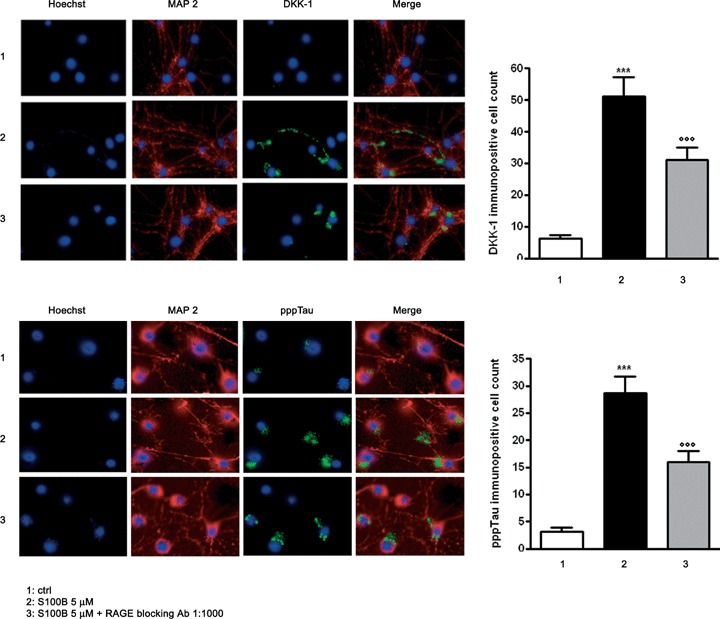
S100B induces DKK-1 expression and tau protein hyperphosphorylation through RAGE interaction. (A) Left panel: representative photomicrographs of NSCs showing DKK-1 immunostaining (green). NSC cultures were exposed to S100B 5 μM in the presence or absence of RAGE blocking antibody (1:1000), and DKK-1 protein expression was evaluated 12 hrs later by immunofluorescence analysis. Right panel: quantification of immunoreactivity expressed as the number of DKK-1 immunopositive neuronal cells. Statistics indicate that RAGE blocking antibody significantly reverted the effect of S100B on DKK-1 protein expression. (B) Left panel: representative photomicrographs of NSCs showing pppTau immunostaining (green). NSC cultures were exposed to S100B 5 μM in the presence or absence of RAGE blocking antibody (1:1000), and pppTau protein expression was evaluated 48 hrs later by immunofluorescence analysis. Right panel: quantification of immunoreactivity expressed as the number of pppTau immunopositive neuronal cells. Statistics indicate that RAGE blocking antibody significantly reverted the effect of S100B on pppTau protein expression. Anti-MAP-2 antibody was used as a neuronal marker (red), and nuclei were stained with Hoechst 33258 (blue). Scale bar = 20 μm. Data are shown as mean ± S.E.M. of five experiments.***P < 0.001 versus unchallenged cells (ctrl);°°°P < 0.001 versus 5 μM S100B challenged cells.
S100B induction of both DKK-1 and pppTau expression in neurons raises the possibility that S100B promotes DKK-1 expression, and thereby disrupts the canonical Wnt pathway (increased GSK-3β phosphorylation and enhanced β-catenin degradation), leading to tau protein hyperphosphorylation. To address this possibility, we used an mRNA silencing (siRNA) approach to selectively inhibit mRNA and protein expression of DKK-1 in SHSY5Y human neuroblastoma cells. No significant changes in DKK-1 expression were seen on the transcriptional and translational level following control siRNA (Cyclophin B) or untransfected cells, 12 and 24 hrs after transfection, respectively (Fig. 6). As observed for differentiated NSCs, 5 μM S100B exposure significantly increased GSK-3β phosphorylation and β-catenin degradation by 24 hrs after stimulation. S100B pre-treatment also resulted in significant tau protein hyperphosphorylation at 48 hrs after exposure, in comparison with untreated SHSY5Y cells. Under these same experimental conditions, DKK-1 siRNA almost completely abolished GSK-3β phosphorylation, β-catenin degradation, tau protein hyperphosphorylation (Fig. 7A, B, C). Finally, these same changes in the canonical Wnt pathway were seen within SHSY5Y neuroblastoma cells transfected with DKK-1 siRNA (Fig. 8). Taken together, these findings demonstrate that S100B functions through DKK-1 in disrupting the Wnt pathway and causing tau protein hyperphosphorylation.
6.
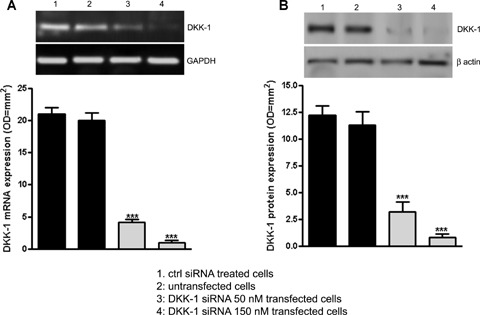
DKK-1 siRNA in SHSY5Y cell cultures.(A) SHSY5Y cells were transfected with DKK-1 siRNA (50 or 150 nM) and 12 hrs later DKK-1 mRNA knockdown was evaluated by RT-PCR (upper panel). Lower panel reports the densito-metric analysis of corresponding bands. GAPDH served as a loading control.(B) DKK-1 protein expression was evaluated 24 hrs following transfection by Western blot (upper panel). In the lower panel densitometric analysis of corresponding bands is reported. β-actin served as a loading control. Untransfected and control siRNA treated cells were used as internal controls to verify the specificity of the treatment. Statistics demonstrate that DKK-1 siRNA significantly and dose-dependently induced DKK-1 mRNA knockdown in SHSY5Y cells. Results are the mean ± S.E.M. of three separated experiments.***P < 0.001 versus internal controls.
7.
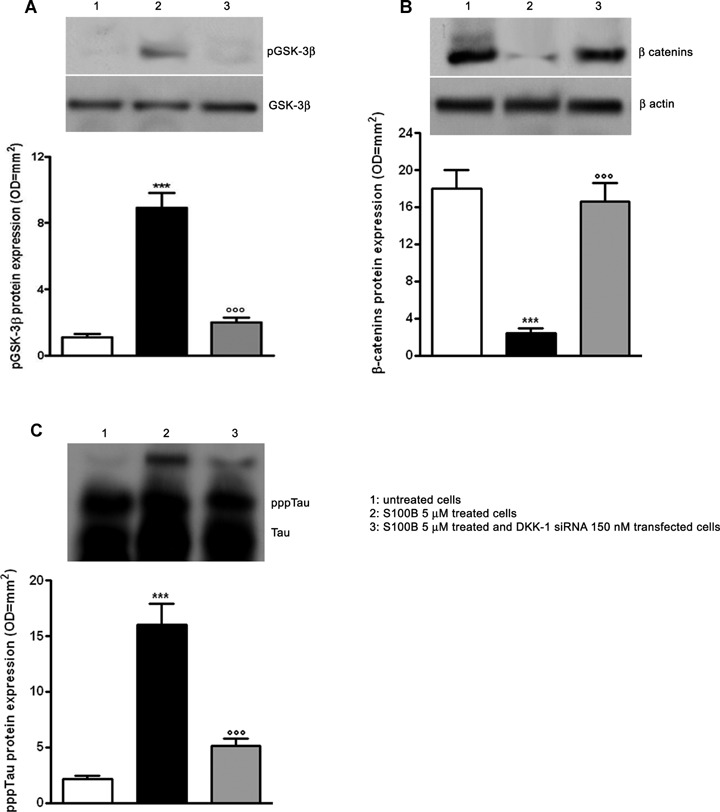
S100B requires DKK-1 activation to induce Wnt pathway disruption and tau protein hyperphosphorylation. (A) SHSY5Y cells were transfected with 150 nM DKK-1 siRNA and treated with S100B 5 μM. The pGSK-3β protein expression was evaluated 24 hrs following treatment by Western blot (upper panel) and densitometric analysis of corresponding bands (lower panel). GSK-3β served as a loading control.(B) β-catenin protein expression was evaluated 24 hrs following treatment by Western blot (upper panel) and densitometric analysis of corresponding bands (lower panel). β-actin served as a loading control.(C) pppTau protein expression was evaluated 48 hrs following treatment by Western blot (upper panel) and densitometric analysis of corresponding bands (lower panel). Tau served as a loading control. Statistics show that S100B required DKK-1 activation to reduce β-catenin protein expression and to promote pGSK-3β and pppTau protein expression. Untransfected and untreated cells were used as internal controls. Results are the mean ± S.E.M. of three independent experiments. ***P < 0.001 versus untreated cells; °°°P < 0.001 versus untrans-fected cells treated with S100B 5 μM.
8.
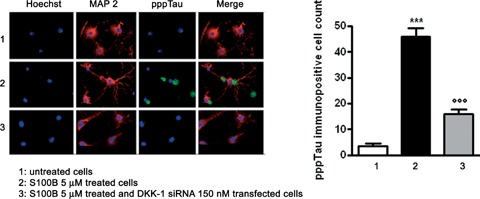
DKK-1 siRNA abolishes S100B-induced tau protein hyperphosphorylation. SHSY5Y cells were transfected with 150 nM DKK-1 siRNA and treated with S100B 5 μM. pppTau protein expression was evaluated 48 hrs later by immuno-fluorescence analysis (left panel) and subsequent immunopositive cell count (right panel). Anti-MAP-2 antibody was used as a neuronal marker (red), while nuclei were stained with Hoechst 33258 (blue). Scale bar = 20 μm. Untransfected and untreated cells were used as internal controls. Results are the mean ±S.E.M. of three independent experiments.***P < 0.001 versus untreated cells;°°°P < 0.001 versus untransfected cells treated with S100B 5 μM.
Discussion
The current studies extend upon known mechanisms of neuronal injury in AD. While aberrant processing of Aβ in neurons intrinsically leads to disruption of the canonical Wnt pathway [24], NFT formation and neuronal cell death, these same processes can invoke an extrinsically mediated glial inflammatory response. Here we show that elevated levels of the astrocyte-released S100B protein can similarly disrupt the Wnt signalling pathway, through interactions with RAGE and up-regulation of the DKK-1 glycoprotein. The activation of this molecular cascade leads to GSK-3β activation and tau protein hyperphosphorylation. The dysregulation of tau phosphorylation is thought to be responsible for NFT formation and neuronal death in the AD brain [1, 25].
Several lines of evidence support a role for reactive gliosis in mediating AD progression. Senile plaques (SPs) and NFTs are co-localized with clusters of activated microglia and associated with a broad variety of astrocyte-derived inflammation-related proteins. In addition, both in vitro and in vivo studies have shown that Aβ peptide fragments induce a prominent neuroinflammatory response, responsible for the synthesis of different cytokines and pro-inflammatory mediators, including nitric oxide, prostaglandins, interleukins and specific glial-derived molecules such as S100B protein [26, 27]. Finally, reactive gliosis can induce oxidative stressors and Wnt disruption, and both factors result in amyloidogenesis and tauopathy [28].
S100B expression is associated with the pathology seen in the aging AD brain [29]. Mice that exhibit premature aging express higher concentrations of S100B than can otherwise be explained by mice age alone. S100B protein is also increased both in post mortem AD brain and in the cerebrospinal fluid of AD patients in the early stages of the disease [30]. Similarly, elevated S100B levels correlate with the early onset AD seen in trisomy 21 (Down's syndrome) [31, 32], while transgenic mice that overexpress APP also display increased numbers of activated astro-cytes and tissue concentrations of S100B, several months before the appearance of Aβ deposits and SP formation [33]. Finally, S100B is overexpressed in activated astrocytes surrounding SPs, where the degree of astrocyte response correlates with the degree of the neuritic pathology [29, 34].
Several pathological mechanisms can be triggered from the overexpression of S100B. Elevated S100B levels exert damaging effects on neurons through pro-inflammatory cytokines and inflammatory stress-related transcription factors [35–38].
In addition, S100B can directly increase APP expression [15], which in turn can activate astrocytes and raise S100B levels, thereby fostering a self-sustaining feedback loop that drives the progressive pathology seen in AD. Finally, the current studies demonstrate that S100B can disrupt the Wnt pathway through increases in the glycoprotein DKK-1. DKK-1 promotes activation of GSK-3β, a key member of Wnt pathway implicated in tauopathy.
A central role for DKK-1 in mediating Wnt disruption and neuronal degeneration in AD brain has previously been demonstrated [8, 10, 11]. In cortical neurons, APP induces the expression of the secreted DKK-1, which negatively modulates the canonical Wnt signalling pathway and thus activates the tauphosphorylating enzyme GSK-3β. DKK-1 is also expressed by degenerating neurons in AD brain, and co-localizes with NFTs and distrophic neurites. Results from this work now reveal that exposure to elevated levels of S100B protein (likely from reactive astrogliosis) can similarly disrupt Wnt signalling through DKK-1. In this manner, both neuronal (APP) and glial (S100B) factors may promote the same cellular mechanisms that are implicated in the pathogenesis of neurodegenerative disorders.
The current study emphasizes the key role of reactive gliosis in AD progression and reinforces the need to identify factors responsible for glial-maintained neuroinflammation. Glial inflammation leads to elevated expression of astrocyte-derived S100B. While S100B can promote APP expression and APP-dependent neuronal injury, we now show that this soluble protein can also disrupt Wnt signalling through DKK-1 and promote tau hyperphosphorylation. Tau hyperphosphorylation is associated with NFT formation and AD progression. These findings suggest that reagents that inhibit S100B function may serve a neuroprotective function and provide an effective strategy to delay the progression of AD.
Acknowledgments
This work was supported by Cofin Ministero dell' Università e della Ricerca Scientifica e Tecnologica 2004 (to Luca Steardo). Volney Sheen is supported by grants from the Julian and Carol Cohen, and is a Beckman Young Investigator and Doris Duke Clinical Scientist Development Award recipient.
References
- 1.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science. 1991;251:675–8. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 2.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. 1986;83:4044–8. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Ferrari GV, Inestrosa NC. Wnt signaling function in Alzheimer's disease. Brain Res Brain Res Rev. 2000;33:1–12. doi: 10.1016/s0165-0173(00)00021-7. [DOI] [PubMed] [Google Scholar]
- 6.Arendt T. Neurodegeneration and plasticity. Int J Dev Neurosci. 2004;22:507–14. doi: 10.1016/j.ijdevneu.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Fath T, Eidenmuller J, Brandt R. Tau-mediated cyto-toxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J Neurosci. 2002;22:9733–41. doi: 10.1523/JNEUROSCI.22-22-09733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caricasole A, Copani A, Caruso A, Caraci F, Iacovelli L, Sortino MA, Terstappen GC, Nicoletti F. The Wnt pathway, cell-cycle activation and beta-amyloid: novel therapeutic strategies in Alzheimer's disease? Trends Pharmacol Sci. 2003;24:233–8. doi: 10.1016/s0165-6147(03)00100-7. [DOI] [PubMed] [Google Scholar]
- 9.McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging. 2001;22:799–809. doi: 10.1016/s0197-4580(01)00289-5. [DOI] [PubMed] [Google Scholar]
- 10.Scali C, Caraci F, Gianfriddo M, Diodato E, Roncarati R, Pollio G, Gaviraghi G, Copani A, Nicoletti F, Terstappen GC, Caricasole A. Inhibition of Wnt signaling, modulation of tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol Dis. 2006;24:254–65. doi: 10.1016/j.nbd.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, Storto M, Gaviraghi G, Terstappen GC, Nicoletti F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer's brain. J Neurosci. 2004;24:6021–7. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmer DB, Cornwall EH, Landar A, Song W. The S100 protein family:history, function, and expression. Brain Res Bull. 1995;37:417–29. doi: 10.1016/0361-9230(95)00040-2. [DOI] [PubMed] [Google Scholar]
- 13.Van Eldik LJ, Wainwright MS. The Janus face of glial-derived S100B: beneficial and detrimental functions in the brain. Restor Neurol Neurosci. 2003;21:97–108. [PubMed] [Google Scholar]
- 14.Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–54. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Wang J, Sheng JG, Liu L, Barger SW, Jones RA, Van Eldik LJ, Mrak RE, Griffin WS. S100 beta increases levels of beta-amyloid precursor protein and its encoding mRNA in rat neuronal cultures. J Neurochem. 1998;71:1421–8. doi: 10.1046/j.1471-4159.1998.71041421.x. [DOI] [PubMed] [Google Scholar]
- 16.Adami C, Bianchi R, Pula G, Donato R. S100B-stimulated NO production by BV-2 microglia is independent of RAGE transducing activity but dependent on RAGE extracellular domain. Biochim Biophys Acta. 2004;1742:169–77. doi: 10.1016/j.bbamcr.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Bianchi R, Adami C, Giambanco I, Donato R. S100B binding to RAGE in microglia stimulates COX-2 expression. J Leukoc Biol. 2007;81:108–18. doi: 10.1189/jlb.0306198. [DOI] [PubMed] [Google Scholar]
- 18.Grotewold L, Ruther U. The Wnt antagonist Dickkopf-1 is regulated by Bmp signaling and c-Jun and modulates programmed cell death. EMBO J. 2002;21:966–75. doi: 10.1093/emboj/21.5.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheen VL, Ferland RJ, Harney M, Hill RS, Neal J, Banham AH, Brown P, Chenn A, Corbo J, Hecht J, Folkerth R, Walsh CA. Impaired proliferation and migration in human Miller-Dieker neural precursors. Ann Neurol. 2006;60:137–44. doi: 10.1002/ana.20843. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs S, Lie DC, De Cicco KL, Shi Y, De Luca LM, Gage FH, Evans RM. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc Natl Acad Sci USA. 2006;103:3902–7. doi: 10.1073/pnas.0511294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Stefano D, Maiuri MC, Iovine B, Ialenti A, Bevilacqua MA, Carnuccio R. The role of NF-kappaB, IRF-1, and STAT-1alpha transcription factors in the iNOS gene induction by gliadin and IFN-gamma in RAW 264.7 macrophages. J Mol Med. 2006;84:65–74. doi: 10.1007/s00109-005-0713-x. [DOI] [PubMed] [Google Scholar]
- 22.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274:19919–24. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 23.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 24.De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- 25.Dickson DW. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: cause or effect? J Clin Invest. 2004;114:23–7. doi: 10.1172/JCI22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mrak RE, Sheng JG, Griffin WS. Glial cytokines in Alzheimer's disease: review and pathogenic implications. Hum Pathol. 1995;26:816–23. doi: 10.1016/0046-8177(95)90001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mudher A, Lovestone S. Alzheimer's disease – do tauists and baptists finally shake hands? Trends Neurosci. 2002;25:22–6. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- 29.Mrak RE, Griffin WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2001;22:915–22. doi: 10.1016/s0197-4580(01)00293-7. [DOI] [PubMed] [Google Scholar]
- 30.Peskind ER, Griffin WS, Akama KT, Raskind MA, Van Eldik LJ. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer's disease. Neurochem Int. 2001;39:409–13. doi: 10.1016/s0197-0186(01)00048-1. [DOI] [PubMed] [Google Scholar]
- 31.Royston MC, McKenzie JE, Gentleman SM, Sheng JG, Mann DM, Griffin WS, Mrak RE. Overexpression of s100beta in Down's syndrome: correlation with patient age and with beta-amyloid deposition. Neuropathol Appl Neurobiol. 1999;25:387–93. doi: 10.1046/j.1365-2990.1999.00196.x. [DOI] [PubMed] [Google Scholar]
- 32.Griffin WS, Sheng JG, McKenzie JE, Royston MC, Gentleman SM, Brumback RA, Cork LC, Del Bigio MR, Roberts GW, Mrak RE. Lifelong overexpression of S100beta in Down's syndrome: implications for Alzheimer pathogenesis. Neurobiol Aging. 1998;19:401–5. doi: 10.1016/s0197-4580(98)00074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheng JG, Mrak RE, Bales KR, Cordell B, Paul SM, Jones RA, Woodward S, Zhou XQ, McGinness JM, Griffin WS. Overexpression of the neuritotrophic cytokine S100beta precedes the appearance of neuritic beta-amyloid plaques in APPV717F mice. J Neurochem. 2000;74:295–301. doi: 10.1046/j.1471-4159.2000.0740295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mrak RE, Sheng JG, Griffin WS. Correlation of astrocytic S100 beta expression with dystrophic neurites in amyloid plaques of Alzheimer's disease. J Neuropathol Exp Neurol. 1996;55:273–9. doi: 10.1097/00005072-199603000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu J, Van Eldik LJ. S100 beta induces apoptotic cell death in cultured astrocytes via a nitric oxide-dependent pathway. Biochim Biophys Acta. 1996;1313:239–45. doi: 10.1016/0167-4889(96)00095-x. [DOI] [PubMed] [Google Scholar]
- 36.Hu J, Ferreira A, Van Eldik LJ. S100beta induces neuronal cell death through nitric oxide release from astrocytes. J Neurochem. 1997;69:2294–301. doi: 10.1046/j.1471-4159.1997.69062294.x. [DOI] [PubMed] [Google Scholar]
- 37.Lam AG, Koppal T, Akama KT, Guo L, Craft JM, Samy B, Schavocky JP, Watterson DM, Van Eldik LJ. Mechanism of glial activation by S100B: involvement of the transcription factor NFkappaB. Neurobiol Aging. 2001;22:765–72. doi: 10.1016/s0197-4580(01)00233-0. [DOI] [PubMed] [Google Scholar]
- 38.Liu L, Li Y, Van Eldik LJ, Griffin WS, Barger SW. S100B-induced microglial and neuronal IL-1 expression is mediated by cell type-specific transcription factors. J Neurochem. 2005;92:546–53. doi: 10.1111/j.1471-4159.2004.02909.x. [DOI] [PubMed] [Google Scholar]