

Abstract

A branched flexible linker that incorporates a fluorescent dansyl moiety was synthesized and used to connect two high affinity NDP-α-MSH ligands or two low affinity MSH(4) ligands. The linker was incorporated into the conjugate by solid-phase synthesis. In vitro biological evaluations showed that potency of binding to the human melanocortin 4 receptor was not diminished for linker–ligand combinations relative to the corresponding ligand alone.
Introduction
Early detection of many human cancers depends on the availability of nontoxic reagents that can seek out and selectively bind to cancer cells and report their existence and location by noninvasive molecular imaging.1 We are engaged in a program to develop reagents that target human cancers that presently are difficult to detect, such as melanoma and pancreatic cancer. Our strategy involves linking together reporter moieties and two or more ligands that bind to cell surface receptors that are overexpressed in cancer cells.2 The resulting multivalent molecules could display enhanced affinity for targeted cells.3
Factors that must be considered in designing linkers for bioactive ligands include the distance that must be spanned, ease of use, biocompatibility, and value-added functionality. The distance that must be spanned between targeted receptors on the cell surface will vary and will often exceed the typical dimensions of small molecules.4 In such cases, several small molecule linkers connected in series will be necessary to span the distance between receptors.
For peptide ligands, linkers should terminate in an amine and a carboxylic acid to facilitate incorporation by solid-phase synthesis. Each linker employed in the assembly of compounds that contain multiple copies of one ligand, or ligands of multiple types, must be validated as noninterfering with the binding of the ligands of interest. Both rigid linkers5 and flexible linkers6 are in development. The use of water-soluble and biocompatible poly(ethylene oxide) oligomers (PEGs) as linkers is well established.7 While several linear PEGs that terminate in amine and carboxylic acid functional groups are commercially available,8 to the best of our knowledge, branched PEG oligomers such as 1 that are akin to lysine have not been reported.9 Such branching would afford the opportunity to attach, in addition to peptide ligands, fluorescent moieties for imaging or therapeutic moieties for targeted treatment of diseased cells. Linkers such as 1 are modular and can be connected together to bridge larger distances and/or to mix ligand, imaging, and therapeutic moieties on one chain. We report herein a synthesis of a fluorescent derivative of 1, attachment of “high-affinity” NDP-α-MSH or “low-affinity” MSH(4) variants of α-melanocyte stimulating hormone10 to produce compounds 2 and 3, as well as control compounds 4 and 5, and evaluation of the bioactivities of these peptides.

Results and Discussion
A synthesis of the fluorescent derivative 17 of linker 1 is depicted in Scheme 1. Known amine 611 was allowed to react with acrylonitrile in methanol, producing nitrile 7 in 80% yield. Separately, the sodium salt derived from tri(ethylene glycol) was allowed to react with known iodide 812 in THF, giving alcohol 9 in 65% yield. This alcohol was subjected to Swern oxidation to produce the unstable aldehyde 10. Reductive coupling of amine 7 and aldehyde 10 using sodium triacetoxyborohydride gave nitrile 11. This compound, which possesses the skeleton of linker 1, was subjected to partial hydrolysis of the ortho ester and subsequently to transesterification using potassium carbonate in dry methanol to give ester 12 in 57% yield from alcohol 9. Reductive ethylation of the nitrile moiety of 12 was accomplished by the method of Sajiki et al.13 Reaction of 13 with dansyl chloride afforded sulfonamide 14 in 47% yield from 12. Acid-catalyzed removal of the Boc group, saponification of the methyl ester, and protection of the free amine as the trifluoroacetamide (14 → 15 → 16 → 17) was effected in 93% yield over the three steps.
Scheme 1. Synthesis of Compound 17a.

a Reagents and conditions: (a) acrylonitrile, CH3OH; (b) NaH, THF; (c) DMSO, (COCl)2, Et3N, CH2Cl2; (d) NaBH(OAc)3, ClCH2CH2Cl; (e) TsOH, aq CH3OH; (f) K2CO3, CH3OH; (g) H2, Rh/C, EtNH2, CH3OH; (h) dansyl chloride, Et3N, CH2Cl2; (i) HCl, Et2O, EtOAc, CH3OH; (j) NaOH, aq CH3OH, THF; (k) CF3CO2Et, CH3OH, Et3N.
Compound 17 incorporates a γ-alkoxy acid terminus instead of a β-alkoxy acid moiety7,8 to preclude unwanted β-elimination processes. N-Ethylation prior to introduction of the dansyl moiety clears the linker of an acidic proton that could interfere with solid-phase synthesis and/or make the fluorescence properties of the molecule pH sensitive.14
The solid-phase synthesis of compound 2 consisting of two NDP-α-MSH ligands connected in a head-to-tail fashion by the fluorescent linker is depicted in Scheme 2 and is presented below. Also shown in Scheme 2 and presented is the synthesis of control compound 4 consisting of one NDP-α-MSH ligand connected at the peptide N-terminus through the carboxylate of the fluorescent linker.
Scheme 2. Solid-Phase Synthesis of Compounds 2 and 4a.
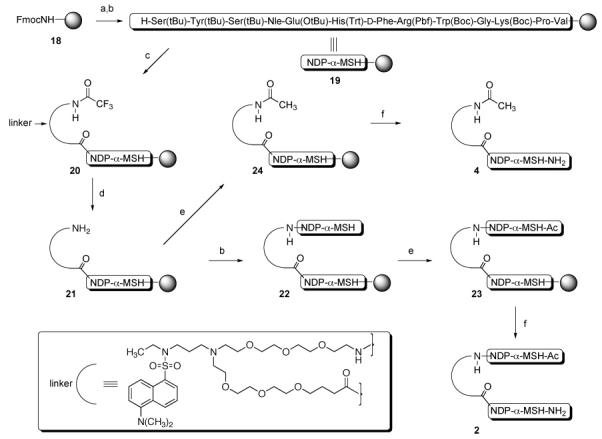
a Reagents and conditions: (a) piperidine; (b) Fmoc/t-Bu solid-phase synthesis (ref 15); (c) N-protected linker, HCTU, i-Pr2EtN, DMF; (d) THF/MeOH/N2H4 (70/15/15); (e) pyridine/Ac2O (90/10); (f) TFA/HSCH2CH2SH/PhSMe/H2O (91/3/3/3).
NDP-α-MSH10 was constructed on Rink amide Tentagel S resin (18, initial loading 0.17 mmol/g).15 The product resin 19 retained all side-chain protecting groups. Acid 17 was coupled to the N-terminus of 19 to give resin 20. The N-trifluoroacetamide protecting group was cleaved by treatment with 15% hydrazine and 15% methanol in THF, and the resulting resin 21 was split into two portions. For the synthesis of 2, the free amine group of resin 21 was coupled with Fmoc-valine, and solid-phase synthesis continued to completion of the second NDP-α-MSH ligand, giving resin 22. The N-terminus was then capped by N-acetylation, producing resin 23. For the synthesis of control 4, resin 21 was terminally N-acetylated to give resin 24. Simultaneous side-chain deprotection and cleavage of the peptides from resins 23 and 24 was effected using a mixture of trifluoroacetic acid, 1,2-ethanedithiol, thioanisole, and water (91/3/3/3), producing the desired compounds 2 and 4. Compounds 3 and 5 incorporating the MSH(4) ligand10 were similarly prepared.
Compounds 2–5 were purified by reversed-phase C18 preparative HPLC and were characterized by ESI-MS and MALDI-TOF. Mass spectral and HPLC characterization data are given in Table 1. Ligand binding was evaluated using a previously described lanthanide-based binding assay (see the Supporting Information).16 HEK293 cells overexpressing the human melanocortin 4 receptor (hMC4R) were used to assess ligand binding.17 Table 2 lists the IC50 values (averaged over n experiments) for the ligands NDP-α-MSH and MSH(4), as well as for compounds 2–5. Compound 2 bound with a 2-fold higher affinity as compared to 4, possibly a statistical effect as 2 contains two NDP-α-MSH ligands to one for 4. Synergistic affects are absent because the ligands in 2 are too far apart for enhanced activity from a proximity affect,2,5c but not far enough apart for enhanced binding due to receptor cross-linking. However, this statistical effect disappeared for compounds 3 and 5 that contain the weaker binding MSH(4) ligand. Compounds 2–5 were all slightly more potent binders at the hMC4 receptor when compared with the corresponding parental ligands. Thus, it appears that this linker does not interfere with binding of either the NDP-α-MSH ligand or the MSH(4) ligand at the hMC4 receptor.
TABLE 1.
Mass Spectral and HPLC Characterization Data of Compounds 2–5
| compd | formula | calcd mass [ion] |
mass found |
tR (% purity) |
K ′ |
|---|---|---|---|---|---|
| 2 | C189H275N45O46S | 789.6135 [(M + 5)5+] |
789.6113 | 19a (91) |
5.2 |
| 3 | C101H139N25O18S | 1012.0304 [(M + 2)2+] |
1012.0296 | 18a (93) |
4.8 |
| 4 | C113H169N25O28S | 590.3158 [(M + 4)4+] |
590.3174 | 18.4a (96) |
5.1 |
| 5 | C69H101N15O14S | 698.8765 [(M + 2)2+] |
698.8730 | 25.1b (95) |
6.6 |
Linear gradient of from 20 to 60% CH3CN in 0.1% aqueous TFA over 50 min.
Linear gradient of from 10 to 60% CH3CN in 0.1% aqueous TFA over 50 min.
TABLE 2.
Competitive Binding of NDP-α-MSH, MSH(4), and Compounds 2–5 to hMC4Ra
| compd | IC50 | n |
|---|---|---|
| NDP-α-MSH | 5.86 ± 1.90 nM | 5 |
| 2 | 1.45 ± 0.44 nM | 6 |
| 4 | 3.62 ± 0.86 nM | 6 |
| MSH(4) | 1.15 ± 0.53 μM | 4 |
| 3 | 0.37 ± 0.12 μM | 6 |
| 5 | 0.42 ± 0.22 μM | 6 |
The IC50 value given is the average of n independent binding experiments, each done in quadruplicate.
Conclusion
This paper describes the synthesis of a branched flexible linker that terminates in amino and carboxylic acid functional groups to facilitate incorporation of the linker into multimeric peptide ligand constructs by solid-phase synthesis. The backbone of the linker is a hybrid poly(ethylene oxide)/ethylene imine that affords water solubility and biocompatibility to the linker. The branching of the linker permits incorporation of moieties for imaging and/or therapeutic use. In the present work, a fluorescent dansyl group was incorporated. The carboxyl terminus of the linker is based on a γ-alkoxybutanoic acid substructure rather than the common β-alkoxypropionic acid substructure. This prevents unwanted β-elimination reactions from interfering with synthetic manipulations. The utility of this linker was demonstrated by synthesis of compounds containing one or two copies of ligands targeted to the human melanocortin 4 receptor. In vitro binding studies established the noninterference of the linker with ligand binding to this receptor.
Experimental
tert-Butyl 1-Cyano-6,9,12-trioxa-3,15-diazahexadecanoate (7)
To a solution of 6 (4.34 g, 14.8 mmol) in MeOH (9 mL) was added acrylonitrile (1.56 mL, 1.26 g, 23.7 mmol) dropwise over 12 min. After 19 h, the mixture was concentrated in vacuo, and the residue was purified by flash column chromatography on silica gel (EtOAc/MeOH, 10:1 → 2:1) to give 7 (4.07 g, 11.8 mmol, 80%) as a colorless oil: analytical TLC on silica gel, 3:1 EtOAc/MeOH, Rf = 0.31 (ninhydrin); 1H NMR (CDCl3) δ 1.39 (9H, s), 2.49 (2H, t, J = 6.8 Hz), 2.78 (2H, t, J = 5.1 Hz), 2.90 (2H, t, J = 6.8 Hz), 3.26 (2H, m), 3.49 (2H, t, J = 5.1 Hz), 3.54–3.82 (10H, m), 5.22 (1H, br s); 13C NMR (CDCl3) δ 18.5, 28.4, 40.3, 45.0, 48.5, 70.1, 70.2, 70.3, 70.4, 79.0, 118.6, 156.0 (two signals could not be located because of their overlaps with other signals); HRMS (FAB+, NBA) calcd for C16H32N3O5 (M + H)+ 346.2342, found 346.2330.
12-(4-Methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)-3,6,9-trioxatridecan-1-ol (9)
To NaH (499 mg, 20.8 mmol) was added a solution of tri(ethylene glycol) (4.73 g, 4.20 mL, 31.5 mmol) in THF (20 mL) dropwise. After 30 min, a solution of 8 (3.10 g, 10.4 mmol) in THF (10 mL) was added dropwise. The mixture was heated to reflux, and after 2 h, the reaction was quenched with H2O (10 mL). The mixture was extracted with CH2Cl2 (20 mL × 5), and the combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc/Et3N, 29:67:4) to give 9 (2.18 g, 6.80 mmol, 65%) as a pale yellow oil: analytical TLC on silica gel, 12:1 EtOAc/MeOH, Rf = 0.42 (anisaldehyde); 1H NMR (CDCl3) δ 0.76 (3H, s), 1.69 (4H, m), 2.57 (1H, t, J = 6.8 Hz), 3.43 (2H, m), 3.51–3.68 (12H, m), 3.84 (6H, s); 13C NMR (CDCl3) δ 14.5, 23.4, 30.2, 33.2, 61.7, 69.8, 70.4, 70.6, 70.6, 70.9, 72.5, 72.5, 109.0; HRMS (FAB+, NBA) calcd for C15H29O7 (M + H)+ 321.1913, found 321.1907.
1-tert-Butyl 27-Methyl 14-(2-Cyanoethyl)-5,8,11,17,20,23-hexaoxa-2,14-diazaheptacosanedioate (12)
To a solution of oxalyl chloride (856 μL, 9.81 mmol) in CH2Cl2 (28 mL) at −78 °C was added a solution of DMSO (1.39 mL, 19.6 mmol) in CH2Cl2 (7 mL) over 5 min. After 3 min, a solution of 9 (2.86 g, 8.92 mmol) in CH2Cl2 (10 mL) was added through a cannula over 7 min. After 15 min, Et3N (6.22 mL, 44.6 mmol) was added over 3 min, and after 75 min, the cooling bath was removed. After 10 min, H2O (35 mL) and CH2Cl2 (100 mL) were added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (150 mL × 2). The combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo. The residue was mixed with 7 (3.35 g, 9.70 mmol), and the mixture was dissolved in 1,2-dichloroethane (100 mL). To the solution at 0 °C was added NaBH(OAc)3 (3.03 g, 14.3 mmol) in one portion. After 3 h, additional NaBH(OAc)3 (377 mg, 1.78 mmol) was added. After 40 min, H2O (35 mL) was added, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (150 mL × 3), and the organic layers were combined, dried (MgSO4), filtered, and concentrated in vacuo. The residue was dissolved in MeOH (80 mL)–H2O (1 mL). To the solution was added p-TsOH·H2O (899 mg, 4.46 mmol), and after 4.5 h, additional p-TsOH·H2O (170 mg, 0.892 mmol) was added. After 19 h, saturated aqueous NaHCO3 (5 mL) was added, and the mixture was concentrated in vacuo to ca. 15 mL. EtOAc (300 mL) and brine (20 mL) were added, and the layers were separated. The aqueous layer was extracted with EtOAc (300 mL), and the combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo. The residue was dissolved in distilled MeOH (100 mL), and K2CO3 (2.10 g, 15.2 mmol) was added to the solution at rt in one portion. After 1.5 h, the mixture was cooled to 0 °C, and EtOAc (140 mL) and saturated aqueous NH4Cl (140 mL) were added. The layers were separated, and the organic layer was washed with brine (20 mL). The combined aqueous layers were extracted with EtOAc (200 mL × 2), and the organic layers were combined, dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/MeOH, 30:1 → 7:1) to give 12 (2.95 g, 5.11 mmol, 57% from 9) as a pale yellow oil: analytical TLC on silica gel, 5:1 EtOAc/MeOH, Rf) = 0.47 (ninhydrin); 1H NMR (CDCl3) δ 1.41 (9H, s), 1.89 (2H, m), 2.37 (2H, d, J = 7.3 Hz), 2.49 (2H, br m), 2.76 (4H, br, m), 2.95 (2H, br m), 3.28 (2H, m), 3.44–3.88 (27H, m), 5.05 (1H, br s); 13C NMR (CDCl3) δ 16.3, 24.9, 28.4, 30.7, 40.3, 50.9, 51.5, 54.0, 54.0, 68.7, 70.1, 70.2, 70.4, 70.4, 70.5, 70.5, 79.1, 119.3, 156.0, 173.9 (five signals could not be located because of their overlaps with other signals); HRMS (FAB+, NBA) calcd for C27H52N3O10 (M + H)+ 578.3653, found 578.3660.
1-tert-Butyl 27-Methyl 14-[3-(N-Ethyl-N-dansyl)aminopropyl]-5,8,11,17,20,23-hexaoxa-2,14-diazaheptacosanedioate (14)
To a solution of 12 (338 mg, 0.585 mmol) in MeOH (1.8 mL) was added 5% Rh/C (N. E. Chemcat, Tokyo, Japan, 101 mg), and the system was sealed with a septum. EtNH2 solution (2 M) in MeOH (2.93 mL, 5.85 mmol) was added, and after 30 min, the air inside was replaced with hydrogen (balloon) by three vacuum/H2 cycles and additional 2 M EtNH2 solution in MeOH (1.47 mL, 2.93 mmol) was added. After 21 h, the mixture was passed through a Celite pad, and the pad was washed with MeOH (100 mL). The combined filtrates were concentrated in vacuo to give a 6:4 mixture (331 mg) of monoethylated amine and unethylated amine. The amines mixture was dissolved in MeOH (3.2 mL), and MeCN (143 μL, 2.73 mmol) and 5% Rh/C (97.8 mg) were added. The air inside the flask was replaced with H2 (balloon) as described above and stirred at rt. After 21 h, the mixture was passed through a Celite pad, and the pad was washed with MeOH (100 mL). The combined filtrates were concentrated in vacuo to give a mixture (328 mg) of monoethylated amine, diethylated amine, and unethylated amine in a ratio of 87:4:9, respectively. The amines mixture was dissolved in MeOH (3.0 mL), and MeCN (134 μL, 2.57 mmol) and 5% Rh/C (93.9 mg) were added. The air inside the flask was replaced with H2 (balloon) as described above and the mixture stirred at rt. After 22 h, the mixture was passed through a Celite pad, and the pad was washed with MeOH (100 mL). The combined filtrates were concentrated in vacuo to give a mixture of monoethylated amine, diethylated amine, and unethylated amine (306 mg) in a ratio of 87:9:4, respectively. Proton NMR data for the monoethylated amine 13: 1H NMR (CDCl3) δ 1.08 (3H, t, J = 6.8 Hz), 1.38 (9H, s), 1.62 (2H, quin, J = 6.8 Hz), 1.84 (2H, quin, J = 6.8 Hz), 2.34 (2H, t, J = 6.8 Hz), 2.54 (2H, t, J = 6.8 Hz), 2.60–2.67 (8H, m), 3.25 (4H, m), 3.41–3.61 (23H, m), 5.14 (1H, br s). The mixture of amines was dissolved in a mixture of CH2Cl2 (15 mL) and THF (1 mL), and dansyl chloride (170 mg, 0.629 mmol) was added. After 1 h, Et3N (33.8 μL, 0.242 mmol) was added, and after 14.5 h, additional Et3N (33.8 μL, 0.242 mmol) was added. After 1 h, EtOAc (150 mL) and H2O (50 mL) were added, and the layers were separated. The organic layer was washed with brine (10 mL), and the combined aqueous layers were extracted with EtOAc (150 mL). The organic layers were combined, dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (CH2Cl2/MeOH, 30:1 → 12:1) to give 14 (212 mg, 0.251 mmol, 47% from 12) as a light green oil. Data for 14: analytical TLC on silica gel, 10:1 CH2Cl2/MeOH, Rf = 0.30 (longwave UV); 1H NMR (CDCl3) δ 1.04 (3H, t, J = 6.8 Hz), 1.40 (9H, s), 1.60 (2H, br quin, J = 6.8 Hz), 1.86 (2H, quin, J = 6.8 Hz), 2.36 (4H, m), 2.57 (4H, m), 2.84 (6H, s), 3.24–3.36 (6H, m), 3.40–3.57 (24H, m), 3.63 (3H, s), 5.02 (1H, br s), 7.13 (1H, d, J = 7.3 Hz), 7.44–7.52 (2H, m), 8.13 (1H, d, J = 7.3 Hz), 8.24 (1H, d, J = 8.5 Hz), 8.48 (1H, d, J = 8.5 Hz); 13C NMR (CDCl3) δ 13.6, 24.8, 26.1, 28.4, 30.7, 40.3, 41.5, 44.5, 45.4, 51.5, 52.5, 53.7, 69.6, 70.0, 70.1, 70.2, 70.3, 70.5, 70.5, 79.1, 115.0, 119.6, 123.0, 127.8, 129.4, 130.0, 130.0, 135.4, 151.6, 155.9, 173.9 (seven signals could not be located because of their overlaps with other signals); HRMS (FAB+, MIX) calcd for C41H71N4O12S (M + H)+ 843.4789, found 843.4777.
14-[3-(N-Ethyl-N-dansyl)aminopropyl]-25-trifluoroacetylamino-5,8,11,17,20,23-hexaoxa-14-azapentacosanoic Acid (17)
To a solution of 14 (212 mg, 0.251 mmol) in EtOAc (6 mL)–MeOH (2 mL) at rt was added 2 M HCl in Et2O solution (5.00 mL, 10.0 mmol). After 2 h, additional 2 M HCl in Et2O solution (7.50 mL, 15.0 mmol) was added, and after an additional 2 h, the mixture was concentrated in vacuo. The residue was coevaporated with MeOH (5 mL × 3). Analytical TLC on silica gel, 10:1 CH2Cl2/MeOH, Rf = 0.11 or 5:1 CH2Cl2/MeOH, Rf = 0.32 (longwave UV). To the residue were added MeOH (4 mL), THF (2 mL), and 2 M NaOH (2.51 mL, 5.02 mmol) successively. After 4 h, additional 2 M NaOH (1.26 mL, 2.51 mmol) was added. After 3 h, the mixture was acidified to pH 4 with 2 M HCl, n-BuOH (80 mL) and brine (5 mL) were added, and the layers were separated. The organic layer was washed with brine (5 mL), and the combined aqueous layers were extracted with n-BuOH (80 mL × 3). The organic layers were combined, dried (Na2SO4), filtered, and concentrated in vacuo. Analytical TLC on silica gel, 5:1 CH2Cl2/MeOH, Rf = 0.07 (longwave UV). The residue was dissolved in MeOH (15 mL), and Et3N (176 μL, 1.26 mmol) and CF3CO2Et (150 μL, 1.26 mmol) were added. After 18 h, the mixture was concentrated in vacuo. EtOAc (100 mL) and brine (10 mL) were added, and the layers were separated. The aqueous layer was extracted with EtOAc (100 mL × 3), and the combined organic layers were dried (Na2SO4), filtered, and concentrated in vacuo. The residue was separated by PLC (CHCl3/MeOH, 6:1, developed five times, longwave UV), and the silica gel containing product was collected in a flash column and eluted with CHCl3/MeOH (4:1 → 3:1). The eluent was concentrated in vacuo, EtOAc (100 mL) and 2 M HCl (30 mL) were added to the residue, and the layers were separated. The organic layer was discarded, and the aqueous layer was adjusted to pH 6 with 2 M NaOH and extracted with EtOAc (100 mL). The organic layer was washed with brine (10 mL), and the combined aqueous layer was extracted with EtOAc (300 mL × 2). The organic layers were dried (Na2SO4) and concentrated in vacuo. CHCl3 (10 mL) was added to the residue, passed through a cotton-packed pasteur pipet, and washed with CHCl3 (20 mL). The filtrate was concentrated in vacuo to give 17 (192 mg, 0.233 mmol, 93%) as a light green oil: analytical TLC on silica gel, 5:1 CH2Cl2/MeOH, Rf = 0.35 (longwave UV); λmax 343 nm (ε = 4500, DMSO); 1H NMR (CDCl3) δ 0.99 (3H, t, J = 6.8 Hz), 1.85 (2H, quin, J = 6.4 Hz), 2.03 (2H, br m), 2.38 (2H, t, J = 6.4 Hz), 2.85 (6H, s), 3.04 (2H, br m), 3.15 (4H, br m), 3.29–3.38 (4H, m), 3.48–3.61 (22H, m), 3.81 (4H, m), 7.15 (1H, d, J = 7.3 Hz), 7.46–7.55 (2H, m), 7.57 (1H, br s), 8.11 (1H, d, J = 7.3 Hz), 8.21 (1H, d, J = 8.5 Hz), 8.50 (1H, d, J = 8.5 Hz); 13C NMR (CDCl3) δ 13.4, 23.3, 24.9, 31.2, 39.6, 41.8, 44.0, 45.4, 52.3, 52.8, 53.2, 66.2, 66.4, 68.6, 70.1, 70.2, 70.2, 70.3, 70.4, 70.5, 70.7, 115.2, 115.9 (q, J = 288 Hz), 119.3, 123.2, 128.1, 129.4, 130.0, 130.3, 135.0, 151.8, 157.3 (q, J) = 37 Hz), 176.2 (three signals could not be located because of their overlaps with other signals); 19F NMR (CDCl3) δ −76.6; HRMS (FAB+, NBA) calcd for C37H60F3N4O11S (M + H)+ 825.3931, found 825.3943.
Solid-Phase Synthesis of Compounds 2–5
The Rink resin was washed with DMF, and the Nα-Fmoc protecting group was removed with 1:4 piperidine in DMF (1 × 2 min and 1 × 20 min). The resin was washed successively with DMF, CH2Cl2, and DMF. The next Nα-Fmoc amino acid was coupled by using the HCTU/i-Pr2EtN procedure (3 equiv of Nα-Fmoc amino acid, 3 equiv of HCTU, and 3 equiv of i-Pr2EtN; 1 min preactivation) for 1 h. The coupling procedure was repeated if the Kaiser test showed a purple color. If the second coupling did not result in a negative Kaiser test, the resin was washed with DMF and the free amino groups were capped with 50% Ac2O in pyridine for 10 min.
After coupling Val, Pro, Lys(Boc), Gly, Trp(Boc), Arg(Pbf), d-Phe, His(Trt), Glu(O-t-Bu), Nle , Ser(t-Bu), Tyr(t-Bu), and Ser(t-Bu) sequentially to the Rink amide resin, the dansylated PEG linker 17 was coupled as above. The trifluoroacetamide group was removed using a solution of hydrazine in MeOH/THF (15/15/70 v/v/v) for 6 h. The resulting resin 21 was divided into two portions, one of which was acetylated to give 24. Assembly of the second NDP-α-MSH ligand was carried out on the second portion of resin 21 by the procedure described above to give 22. N-Terminal acetylation of 22 produced 23. Cleavage of the peptides 2 and 4 from the resins 23 and 24, respectively, with concomitant side-chain deprotection was effected using a cleavage cocktail (10 mL per 1 g of the resin) consisting of TFA (91%), H2O (3%), HSCH2-CH2SH (3%), and PhSMe (3%) at rt for 3 h. The solution was filtered off, the resin was washed with TFA (2 × 3 min), the liquid phases were concentrated under a stream of nitrogen, and the product was precipitated using cold Et2O. The product was washed three times with cold Et2O, lyophilized, purified, and characterized (see the Supporting Information for methods and Table 1 for compound characterization data). Peptides 3 and 5 were synthesized, purified, and characterized similarly.
Supplementary Material
Acknowledgment
We sincerely thank Prof. Hironao Sajiki at the Gifu Pharmaceutical University for a gift of 5% Rh/C. This work was supported by Grants R33 CA 95944, RO1 CA 97360, and P30 CA 23074 from the National Cancer Institute.
Footnotes
Supporting Information Available: General experimental methods for linker synthesis, solid-phase synthesis, competitive binding experiments, and copies of the 1H NMR and 13C NMR spectra of compounds 7, 9, 12, 14, and 17. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Gillies RJ, Hoffman JM, Lam KS, Menkens AE, Piwnica-Worms DR, Sullivan DC, Weissleder R. Molecular Imaging. 2005;4:98–103. doi: 10.1162/15353500200505115. [DOI] [PubMed] [Google Scholar]; (b) Gillies RJ, Hruby VJ. Expert Opin. Ther. Targets. 2003;7:137–139. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]
- 2.Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Expert Opin. Ther. Targets. 2004;8:565–586. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 3.(a) Mammen M, Chio SK, Whitesides GM. Angew. Chem., Int. Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; (b) Kiessling LL, Gestwicki JE, Strong LE. Curr. Opin. Chem. Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 4.Tron L, Szollosi J, Szabo G, Jr., Matyus L, Damjanovich S. Symp. Biol. Hung. 1984;26:307–328. Small molecule dimensions are on the order of ten angstroms, while the distance between receptors is on the order of tens to hundreds of angstroms. See: [Google Scholar]
- 5.(a) Maison W, Frangioni JV, Pannier N. Org. Lett. 2004;6:4567–4569. doi: 10.1021/ol048055j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Monguchi Y, Vagner J, Handl HL, Jana U, Begay LJ, Hruby VJ, Gillies RJ, Mash EA. Tetrahedron Lett. 2005;46:7589–7592. [Google Scholar]; (c) Semetey V, Moustakas D, Whitesides GM. Angew. Chem., Int. Ed. 2006;45:588–591. doi: 10.1002/anie.200502991. [DOI] [PubMed] [Google Scholar]; (d) Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chem. 2006;17:1545–1550. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Vagner J, Handl HL, Gillies RJ, Hruby VJ. Bioorg. Med. Chem. Lett. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]; (b) Raju N, Ranganathan RS, Tweedle MF, Swenson RE. Tetrahedron Lett. 2005;46:1463–1465. [Google Scholar]
- 7.(a) Hashimoto M, Yang J, Holman GD. ChemBioChem. 2001;2:52–59. doi: 10.1002/1439-7633(20010105)2:1<52::AID-CBIC52>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]; (b) Warnecke A, Kratz F. Bioconjugate Chem. 2003;14:377–387. doi: 10.1021/bc0256289. [DOI] [PubMed] [Google Scholar]; (c) Herforth C, Heidler P, Franke S, Link A. Bioorg. Med. Chem. 2004;12:2895–2902. doi: 10.1016/j.bmc.2004.03.038. [DOI] [PubMed] [Google Scholar]; (d) Jensen TW, Hu B-H, Delatore SM, Garcia AS, Messersmith PB, Miller WH. J. Am. Chem. Soc. 2004;126:15223–15230. doi: 10.1021/ja048684o. [DOI] [PubMed] [Google Scholar]; (e) Warneke A, Fichtner I, Garmann D, Jaehde U, Kratz F. Bioconjugate Chem. 2004;15:1349–1359. doi: 10.1021/bc049829j. [DOI] [PubMed] [Google Scholar]
- 8. Available from Quanta BioDesign, Ltd.
- 9.(a) Thumshirn G, Hersel U, Goodman SL, Kessler H. Chem. Eur. J. 2003;9:2717–2725. doi: 10.1002/chem.200204304. The use of coupled lysine/poly(ethylene glycol) linkers has been reported; see. [DOI] [PubMed] [Google Scholar]; (b) Song A, Wang X, Zhang J, Marík J, Lebrilla CB, Lam KS. Bioorg. Med. Chem. Lett. 2004;14:161–165. doi: 10.1016/j.bmcl.2003.09.067. [DOI] [PubMed] [Google Scholar]
- 10.(a) Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. Proc. Natl. Acad. Sci. U.S.A. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. The “high-affinity” ligand employed here was based on NDP-α-MSH (Ser-Tyr-Ser-Nle-Glu-His-d-Phe-Arg-Trp-Gly-Lys-Pro-Val); see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hadley ME, Anderson B, Heward CB, Sawyer TK, Hruby VJ. Science. 1981;213:1025–1027. doi: 10.1126/science.6973820. The “low-affinity” ligand employed was based on the minimal active sequence for full agonist activity of α-MSH (His-d-Phe-Arg-Trp) [DOI] [PubMed] [Google Scholar]; (c) Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, DeVaux AE, Dym O, de L. Castrucci AM, Hintz MF, Riehm JP, Rao KR. J. Med. Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. see: [DOI] [PubMed] [Google Scholar]; (d) de Lauro Castrucci AM, Hadley ME, Sawyer TK, Wilkes BC, Al-Obiedi F, Staples DJ, DeVaux AE, Dym O, Hintz MF, Riehm JP, Rao KR, Hruby VJ. Gen. Comp. Endocrinol. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]; (e) Haskell-Luevano C, Hendrata S, North C, Sawyer TK, Hadley ME, Hruby VJ, Dickinson C, Gantz I. J. Med. Chem. 1997;40:2133–2139. doi: 10.1021/jm960840h. [DOI] [PubMed] [Google Scholar]
- 11.(a) Bendavid A, Burns CJ, Field LD, Hashimoto K, Ridley DD, Sandanayake KRAS, Wieczorek L. J. Org. Chem. 2001;66:3709–3716. doi: 10.1021/jo0057147. [DOI] [PubMed] [Google Scholar]; (b) Zhang W, Nowlan DT, III, Thomson LM, Lackowski WM, Simanek EE. J. Am. Chem. Soc. 2001;123:8914–8922. doi: 10.1021/ja0041369. [DOI] [PubMed] [Google Scholar]; (c) McReynolds KD, Bhat A, Conboy JC, Saavedra SS, Gervay-Hague J. Bioorg. Med. Chem. 2002;10:625–637. doi: 10.1016/s0968-0896(01)00325-x. [DOI] [PubMed] [Google Scholar]
- 12.(a) Atkins MP, Golding BT, Howes DA, Sellars PJ. J. Chem. Soc., Chem. Commun. 1980:207–208. [Google Scholar]; (b) Corey EJ, Kyler K, Raju N. Tetrahedron Lett. 1984;25:5115–5118. [Google Scholar]; (c) Baldwin JE, Adlington RM, Robertson J. Tetrahedron. 1991;47:6795–6812. [Google Scholar]
- 13.Sajiki H, Ikawa T, Hirota K. Org. Lett. 2004;6:4977–4980. doi: 10.1021/ol047871o. [DOI] [PubMed] [Google Scholar]
- 14.Aavula BR, Ali MA, Mash EA, Bednarczyk D, Wright SH. Synth. Commun. 2006;36:701–705. [Google Scholar]
- 15.(a) Merrifield RB. J. Am. Chem. Soc. 1963;85:2149–2154. [Google Scholar]; (b) Hruby VJ, Meyer J-P. In: Bioorganic Chemistry: Peptides and Proteins. Hecht SM, editor. Oxford University Press; New York: 1998. pp. 27–64. [Google Scholar]
- 16.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Anal. Biochem. 2004;330:242–250. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 17.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. J. Biol. Chem. 1993;268:15174–15179. The hMC4R vector was originally received from Dr. Ira Gantz; see: [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.