

Abstract
The relationship between the TCR repertoires of natural regulatory T (nTreg) and conventional T (Tconv) cells capable of responding to the same antigenic epitope is unknown. Here, we used TCRβ-chain transgenic mice to generate polyclonal nTreg and Tconv cell populations specific for a foreign antigen. CD4+ T cells from immunized 3.L2β+/− TCRα+/− Foxp3EGFP mice were re-stimulated in culture to yield nTreg cells (EGFP+) and Tconv cells (EGFP−) defined by their antigenic reactivity. Relative to Tconv cells, nTreg cell expansion was delayed, although a higher proportion of viable nTreg cells had divided after 72 hours. Spectratype analysis revealed that both the nTreg and Tconv cell responses were different and characterized by skewed distributions of CDR3 lengths. CDR3 sequences from nTreg cells displayed a divergent pattern of Jα usage, minimal CDR3 overlap (3.4%), and less diversity than CDR3 sequences derived from Tconv cells. These data indicate that foreign antigen-specific nTreg and Tconv cells are clonally distinct, and that foreign antigen-specific nTreg cells populations are constrained by a limited TCR repertoire.
INTRODUCTION
Natural regulatory T (nTreg)3 cells and conventional CD4+ T (Tconv) cells must both complete affinity-based selection in the thymus. During this process, interaction with a high affinity self-ligand (agonist) results in the expression of Foxp3 and the acquisition of regulatory function in cells committed to the nTreg cell lineage, while Tconv cells are eliminated by negative selection. In some studies, introduction of the cognate antigen into the thymus of TCR transgenic mice results in the development of a small population of Foxp3+ nTreg cells and elimination of most Tconv cells bearing high levels of the transgenic TCR (1, 2). However, other experiments have shown that exposure to the cognate antigen results in negative selection of nTreg cell precursors (3, 4). These findings were based on TCRs derived from Tconv cells. When transgenic TCRs are derived from Treg cell clones, nTreg cell development is a saturable process that requires a small precursor frequency to remain efficient (5, 6). These profound differences in thymic selection requirements strongly suggest that the TCR repertoire of nTreg and Tconv cells should be fundamentally distinct.
The question of how much the nTreg and Tconv cell TCR repertoires overlap was investigated initially using unselected T cell populations. In order to limit diversity, these studies used mice with “fixed” transgenic TCRβ chains in combination with restricted TCRα chain CDR3 analyses. In general, the findings from these models established that the nTreg and Tconv TCR repertoires were similarly diverse, while the reported degree of overlap between the two varied widely (7-11). These studies did not distinguish between nTreg cells and induced Treg (iTreg) cells, which could further complicate conclusions based on these experiments (12).
nTreg cells also possess TCRs with higher affinity for self-peptide/MHC ligands than CD4+ Tconv cells, consistent with the idea that the two T cell subsets recognize different sets of antigens (8). Indeed, the TCR repertoires of nTreg and antigen-experienced (CD44high) Tconv cells from mice with a transgenic TCRβ chain had minimal overlap, but had similar patterns of variability that were based on anatomical location (13). Collectively, these data point to a major role for self-antigens in shaping the peripheral Treg cell TCR repertoire. In contrast, foreign antigen exposure determines the repertoire and distribution of Tconv cells. While the aforementioned studies have broadly compared the TCR repertoires of nTreg and Tconv cells, the relationship between nTreg and Tconv cells that are capable of responding to the same antigen is unknown. This is a particularly important comparison, given the crucial role of Treg cells in controlling responses to both self and foreign antigens (10).
In order to compare the TCR repertoire of nTreg and Tconv cells activated by the same foreign antigenic epitope, we modified an approach first developed for the study of TCR allelic exclusion and fine specificity mapping (14, 15). We crossed 3.L2 TCRβ-chain transgenic mice with Foxp3EGFP and TCRα+/− mice to limit TCR diversity and allow discrimination between antigen-specific Tconv and nTreg cells. Following the immunization of progeny with Hb(64-76) peptide, popliteal and superficial inguinal lymph node cells were restimulated in culture, and the TCRα-chain repertoires of dividing antigen-specific nTreg and Tconv cells compared. We found the CDR3 length distribution in nTreg cells to be relatively narrow, and sequence analysis of TCRα-chain CDR3 regions showed almost no overlap between the two populations. TCR diversity calculations confirmed that the repertoire of nTreg cells was significantly less diverse than that of Tconv cells. Together, our findings demonstrate that Hb(64-76)-specific nTreg cell responses are limited and clonally distinct when compared to Tconv cells responding to the same foreign antigen.
MATERIALS AND METHODS
Mice
All mice utilized in this study were bred onto the B6.AKR background for >14 generations and analyzed between 6-12 weeks of age. The transgenic mice that express the beta chain of the 3.L2 TCR (3.L2β) were generated in the transgenic facility at the Medical College of Wisconsin with the same construct used to make 3.L2 αβ TCR transgenic mice (16). The creation and characterization of Foxp3EGFP mice are described elsewhere (17), and TCRα−/− mice were purchased from the Jackson laboratory. All mice were housed in the animal facility at the Medical College of Wisconsin and handled in accordance with institutional guidelines. The institutional review committee approved all animal studies.
Analytical flow cytometry and cell sorting
Single cell suspensions with up to 5 × 106 cells/sample were stained for cell surface markers as described (3). Data were acquired using an LSR-II flow cytometer (BD Biosciences), then analyzed using FlowJo software (Tree Star, Inc.). Cells were sorted using a FACSAria cell sorter (Becton Dickinson). Peripheral tissues were pooled before antibody staining and sorting. After culture, T cells were sorted to isolate nTreg and Tconv populations that were either unresponsive or reactive to Hb(64-76) peptide. The average purity of cell sorting was 98% for both the nTreg and Tconv populations.
Antibodies, peptides and cytokines
The antibodies used were obtained from BD Pharmingen or Caltag and included PE-Cy5-conjugated anti-mouse CD3 (145-2C11), Pacific Blue-conjugated anti-mouse CD4 (RM4-5), Pacific Orange-conjugated anti-mouse CD8 (53-6.7), PE-Cy7-conjugated anti-mouse CD25 (PC61), Allophycocyanin-conjugated anti-mouse CD44 (IM7), PE-TexasRed-conjugated anti-mouse CD62L (MEL14), Allophycocyanin-conjugated anti-mouse TCRβ (H57), and PE-conjugated anti-mouse Vβ8.3 (1B3.3). In cases where CD4 staining was done prior to sorting cells for culture, a different clone and fluorochrome-conjugate was used for CD4 staining in subsequent sorting procedures.
The peptides were synthesized, purified and analyzed, as previously described (18). The peptide sequences, in single-letter amino acid code, are: Hb(64-76) – GKKVITAFNEGLK (abbreviated “N72” for the Asn at position 72, the primary T cell contact residue); MCC(88-103) – ANERADLIAYLKQATK. Human IL-2 was obtained from Chiron.
Immunization
To create a source of activated APCs for cell culture, B6.AKR mice were immunized subcutaneously with CFA emulsified 1:1 with PBS (100μl per mouse). Trigenic 3.L2β Foxp3EGFP TCRα+/− mice were similarly immunized subcutaneously, but one day later and with CFA emulsified 1:1 with PBS containing 400μM Hb(64-76) peptide for a final peptide concentration of 200μM (20nmoles, 100μl per mouse). One week after each respective immunization, single-cell suspensions were derived from the draining lymph nodes and spleen, and stained for cell sorting.
PKH26 and CellTracker™ staining
The PKH26 mixture was freshly prepared per the manufacturer’s recommendations and used at a final concentration of 1 μM. The diluted PKH26 dye solution was combined 1:1 with sorted CD3+CD4+ T cells that were resuspended at a concentration of 2-4 million cells/ml. Cells were stained for three minutes at room temperature before stopping the reaction with calf serum. Violet CellTracker™ (Invitrogen) was used following the manufacturer’s recommendations.
Cell culture
All cell cultures were carried out at 37°C and 5% CO2 in a final volume of 200μl per well in a 96-well plate. CD3−CD4− APC populations were initially plated in 100μl one day preceding the addition of PKH26-stained CD3+CD4+ T cells in another 100μl of media. APCs and T cells were cultured for three more days before analysis. The number of APCs and T cells cultured per well was 4.5 × 105 and 5 × 104, respectively. All peptide/cytokine treatments were kept constant throughout the entire length of the culture. All samples were treated with 50U/ml of hIL-2 and 1 μg/ml of anti-CD28. Experimental samples were supplemented with 10μM of exogenous Hb(64-76) peptide, while control samples included those cultured in media with or without 10μM of exogenously added moth cytochrome C peptide.
RNA and cDNA isolation
For sorted cell yields over 2.5 × 104 cells, total RNA was isolated using Qiagen’s RNeasy Mini kit following the manufacturer’s protocol. An 8μl aliquot of RNA was reverse-transcribed using Invitrogen’s Superscript™ First-strand Synthesis kit, also following the manufactuer’s protocol. For lower sorted cell yields, cDNA was isolated directly using Invitrogen’s Superscript™ III CellsDirect cDNA Synthesis System following the manufacturer’s protocol and upscaling the volumes as needed. All cDNA samples were either used immediately or stored at -20°C.
TCRα spectratyping
Murine TCRα variable (Vα) gene primers were developed as listed in Unit 10.28 of the Current Protocols in Immunology (19). Each Vα region primer was used with a FAM-labeled or unlabeled constant (Cα) region primer to generate PCR products for subsequent TCRα spectratyping and cloning. FAM-labeled PCR products were analyzed using GeneMapper software to determine the distribution and proportion of CDR3 lengths. Individual bands were separated via polyacrylamide gel electrophoresis (PAGE) using a BIORAD sequencing gel apparatus. The resultant bands were then visualized using a Typhoon scanner and purified using Qiagen’s QIAEX II DNA Extraction kit following the manufacturer’s protocol.
The various spectratypes were compared by generating an overall skew value for each experimental population relative to CD4+ peripheral T cell control data. Spectratype data collected as fluorescent peak areas from GeneMapper® software and were converted to relative frequencies (RF) by dividing each fluorescent peak area by the total peak fluorescence. This compensates for small changes in the dataset due to unequal sample size. For generating the control distribution, splenocytes from two or three untreated mice were analyzed, and when possible an average normal distribution assigned. The skew was calculated for each CDR3 length as the difference between the control value and the experimental value. The overall skew represents the sum of the absolute value of the differences for each length. This can be envisioned as an analog equivalent to a Hamming distance calculation in which 100 intensity markers are distributed among N bins and the skew counts how many changes have to be made in the experimental distribution of markers in order to arrive at the control distribution. The minimum skew would be 0 and the maximum is 2. Some Vα families were poorly represented in the 3.L2 TCRβ mice and a control distribution could not be calculated. Nevertheless, stimulation resulted in expansion of T cells that used these families (e.g. Vα17). For a few Vα families, the length distribution differed between the experimental mice, in which case the skews were calculated on a mouse-by-mouse basis (e.g. Vα3, 6, and 13).
TA-TOPO cloning and sequencing
To improve the cloning efficiency, PCR products were reamplified for an additional 5 cycles using unlabeled primers to dilute out the FAM label. Each reamplified sample was combined with the pCR®4-TOPO® vector, incubated at room temperature for 30 minutes, then added to One Shot® TOP10 chemically competent E. coli. The bacteria were plated on LB agar plates containing ampicillin and colonies were randomly picked, grown for 16 hours, and shipped to Agencourt® Bioscience Corporation (500 Cummings Center, Suite 2450, Beverly, MA 01915) for further processing and CDR3 sequencing.
Statistics
Two-tailed Student’s t-tests were used to determine significance between two given groups. For analyses containing more than two groups, one-way analysis of variance and Tukey’s test were applied. The abundance coverage estimator and Morisita-Horn Index were calculated using the EstimateS 8.0.0 software package (http://viceroy.eeb.uconn.edu/EstimateS/).
RESULTS
Characterization of 3.L2β+/- x TCRα+/- mice
We first compared B6.AKR TCRα+/− Foxp3EGFP mice (WT) with 3.L2β+/− TCRα+/− Foxp3EGFP mice that expressed the 3.L2 TCRβ chain (3.L2β). In peripheral lymph nodes, there was no significant difference in the percent and number of CD4+ Tconv or nTreg cells between the two strains (Figure 1A). In 3.L2β transgenic mice, nearly all (98%) CD4+ T cells expressed Vβ8.3. Many nTreg cells from these mice also had high levels of CD25 and CD44, while CD62L expression was relatively low (Figure 1B), consistent with the nTreg cell surface phenotype (20). Tconv cells from 3.L2β mice had low levels of CD25 and CD44, with high levels of CD62L, consistent with naïve Tconv cell phenotype. When we stimulated unprimed splenocytes from 3.L2β mice with Hb(64-76) peptide (N72) in cell culture, we detected a dose-dependent proliferative response. This indicated a higher frequency of N72-reactive clones than found in B6.AKR controls (Figure 1C), making 3.L2β mice a useful model from which to derive an amplitude of commonly specific T cell populations after immunization with N72.
Figure 1. 3.L2β+/- × TCRα+/- mice generate Tconv cells and nTreg cells that are reactive to Hb(64-76) peptide.
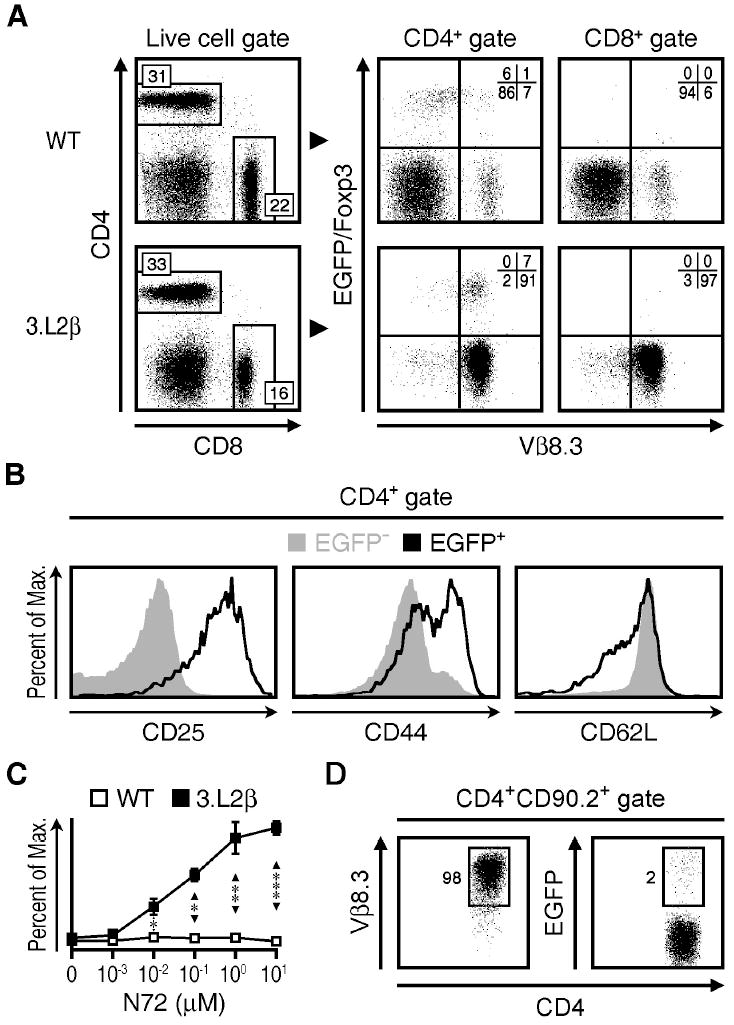
A) CD4 versus CD8 staining (left) of live lymphocytes and Foxp3 versus Vβ8.3 expression (right) of gated CD4+ or CD8+ lymphocytes from TCRα+/- mice (WT) or 3.L2β+/- × TCRα+/- mice (3.L2β). B) Histograms of CD25 (left), CD44 (middle) and CD62L (right) staining, for CD4+EGFP− (solid gray) and CD4+EGFP+ (open black) cell populations from 3.L2β mice. C) Proliferation assay showing percent of maximum (2.5 × 103) counts versus Hb(64-76) peptide (N72) concentration used to activate WT (open) and 3.L2β (closed) splenocytes in culture for 72 hours. D) Vβ8.3 staining (left) and EGFP expression (right) versus CD4 staining of EGFP−CD4+CD90.2+ lymphocytes that were transferred into CD90.1+ mice subsequently immunized with N72 peptide and examined 5 days later. These data are representative of three to five independent experiments, with three to nine mice per group. All quadrant values are means and all error bars represent S.E.M. * p<0.05, ** p<0.005, *** p<0.0005
We were particularly interested in comparing the respective N72-specific TCR repertoires of Tconv cells and nTreg cells without including induced regulatory T (iTreg) cells that express Foxp3 more transiently than their “natural” counterpart (21). To this end, we determined the extent to which Tconv cells converted to Foxp3+ iTreg cells after immunization by transferring 5.0 × 106 EGFP− Tconv cells from CD90.2+ 3.L2β mice into CD90.1+ B6.AKR mice, followed by immunization with 20 nmoles of N72 peptide in CFA. Results showed that 2% of the transferred CD90.2+ cells upregulated their expression of EGFP after 5 days (Figure 1D). These data were consistent with our previous studies that demonstrated a 2-3% iTreg cell conversion rate in TCRαβ transgenic T cells following immunization (17). Overall, this approach of “fixing” the TCRβ chain increased the frequency of N72-reactive cells in both unmanipulated and primed mice, but did not significantly alter the frequency of nTreg or Tconv cells within the CD4+ T cell compartment. Following immunization, conversion of antigen-specific EGFP− cells to EGFP+ cells occurred rarely.
In vitro proliferation of N72-specific nTreg and Tconv cells
In order to isolate larger quantities of N72-specific Treg cells and Tconv cells for repertoire analysis, we immunized 3.L2β Foxp3EGFP Hbβs/s mice with N72 peptide in CFA and harvested CD4+ T cells from the draining lymph nodes at the peak of the T cell response (day 7). 3.L2β Foxp3EGFP Hbβd/s mice, which naturally express the Hbβd minor (N72 epitope) and negatively select CD4+ T cells with N72-reactive TCRs served as controls. We then labeled these cells with PKH26 or Violet CellTracker™ and co-cultured them with N72 peptide-treated B6.AKR splenocytes as APCs. Flow cytometric analysis of EGFP versus PKH26 fluorescence at 12-hour intervals tracked the degree of N72-reactivity and rate of proliferation in the nTreg and Tconv cell populations. After 72 hours, 60.1% of the surviving nTreg cells and 25.7% of the Tconv cells had undergone cell division in response to stimulation with N72 peptide (Figure 2A). Co-cultures with no peptide, with the unrelated MCC(88-103) peptide that like N72 also binds to I-Ek (22) with PPD, or with CD4+ T cells from immunized 3.L2β Hbβd/s mice resulted in little to no proliferation and demonstrated the specificity of the response for the N72 (Figure 2A, Supplemental Figure 1). A more in-depth view of this response revealed that the proportion of divided Tconv cells showed a significant increase after 48 hours in culture, while a similarly significant increase did not occur in the nTreg cells until after 72 hours (Figure 2B). As the frequency of antigen-specific precursors in the larger Tconv cell subset could impact the proportional representation of dividing nTreg cells, we plotted the number of divided cells within each population as a percent of their respective 72-hour end points (Figure 2C, dotted lines). The lag in nTreg cell division after 48 hours was also evident in this analysis, consistent with the delay in nTreg cell proliferation observed in the draining lymph nodes of immunized mice and with the requirement for high levels of exogenous IL-2 for nTreg cell division (17, 23). Despite these differences, the proportion of divided nTreg cells (PKH26lowEGFP+) after 72 hours was significantly greater than that found among the Tconv cells at this time point (Figure 2C, bars).
Figure 2. In vitro proliferation of N72-specific nTreg and Tconv cells from immunized mice.
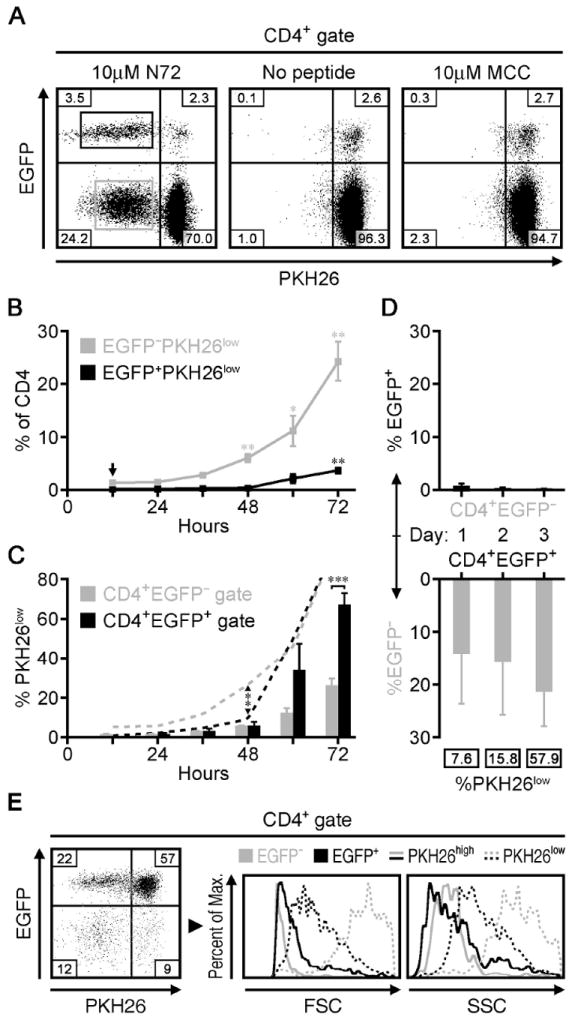
A) EGFP expression versus residual PKH26 staining of CD4+ T cells cultured for 72 hours after treatment with 10μM N72 (left), no peptide (middle), or 10μM MCC peptide (right). Quadrant numbers are means (n=4) and the boxed populations depict the gates used to sort N72-reactive (PKH26low) nTreg cells (EGFP+) versus Tconv cells (EGFP−). B) nTreg cells and Tconv cells shown as a percent of CD4+ cells at the indicated time points after treatment with 10μM N72 in culture. Significance for changes in percentage is based on comparison to the 12 hour time point (arrow). C) The proportion of nTreg cells and Tconv cells that are PKH26low within their respective populations (bars) at the indicated time points as in B, along with the percent of cells that have already divided at each time point (dotted lines) relative to the end point at 72 hours. Arrows and brackets show significant differences between the two cell populations at the given time points. D) The proportions of sorted EGFP− (upper) and EGFP+ (lower) cells that upregulated or lost EGFP expression, respectively, after treatment with 10μM N72 peptide in vitro for the indicated time periods. Boxed numbers represent PKH26low cells as a percent those cells that lost EGFP expression while in culture. E) EGFP expression versus PKH26 staining (left) of sorted CD4+EGFP+ T cells cultured for 72 hours after treatment with 10μM N72. Right panels show histograms of the forward scatter (FSC) and side scatter (SSC) profiles of the PKH26high (solid lines) and PKH26low (dotted lines) cells within both the nTreg cell and Tconv cell populations. These data are representative of two to four independent experiments, with a total of three to four mice per group. EGFP+ and EGFP− cell populations are represented using black and gray colors, respectively. All quadrant values are means and all error bars represent S.E.M. * p<0.05, ** p<0.005, *** p<0.0005
Next, we gauged the capacity of Tconv cells to express Foxp3 and the stability of Foxp3 expression among nTreg cells using similar in vitro experiments that started with sorted EGFP− T cells and EGFP+ T cells, respectively. With an initial EGFP− T cell input, 0.3% of the CD4+ T cells expressed EGFP after 72 hours (Figure 2D, upper panel), indicating that iTreg cells were not produced under these culture conditions. In the reciprocal experiments starting with EGFP+ cells, we noted an early loss of EGFP expression. After 24 hours, 14.1% of the surviving CD4+ T cells lost EGFP expression, although only 7.6% of these were PKH26low (Figure 2D, lower panel). This indicated that a fraction of the EGFP+ T cells had unstable Foxp3 expression that resulted in its loss before cell division. After 72 hours, 21.3% of the surviving CD4+ T cells were EGFP− and 57.9% of these had divided (Figure 2D, E). These data indicated that the ‘unstable’ fraction of cells was capable of cell division upon restimulation and that the 79.2% of CD4+ T cells that remained EGFP+ were enriched for the ‘stable’ nTreg phenotype. Cell size and granularity analysis of the stable versus unstable fractions showed phenotypic differences between these two populations. Although all non-divided T cells showed similar profiles (Figure 2E, solid lines), the proliferating EGFP− T cells were noticeably larger and more granular than those divided nTreg cells that retained EGFP expression (Figure 2E, dotted lines). This difference may be related to their presumably different function.
Vα TCR repertoires of N72-specific nTreg and Tconv cells
In these experiments, TCR variability is constrained by rearrangements at a single TCRα chain locus in the context of a transgenic TCRβ chain. This allowed us to evaluate TCR repertoires using a series of previously designed Vα primers to amplify the complementarity determining region 3 (CDR3) and flanking segments of the TCRα sequences for each sorted N72-specific T cell population (19). For the purposes of this study, we enumerated Vα and Jα regions based on the nomenclature in the Current Protocols of Immunology (19), the former of which differs from the TRAV designations in the IMGT® database (24). Resolving the PCR products by length indicated that there were a number of Vα families for which either the Tconv or nTreg were skewed away from the normal length distribution associated with peripheral CD4+ T cells (Table 1). The average peripheral length was established by examining the splenocytes from two or three control animals. The fluorescent peak areas corresponding to the different CDR3 lengths were converted to relative frequencies for each length and peaks compared (Supplemental Figure 2A). Downstream analyses focused on select Vα samples, (Vα2, Vα3, Vα5, Vα6, Vα13, Vα15, Vα17, Vα18), that deviated from the typical Gaussian distribution of CDR3 lengths. Within each cell type, CDR3 spectra for most Vα products were similar between mice (Figure 3), which allowed us to pool the data accordingly. These data show that both nTreg and Tconv populations are highly skewed relative to controls (p<0.0005 for both nTreg and Tconv cells), which indicates that they are highly selected. When compared to each other, the overall skews for the nTreg and Tconv cell populations were not significantly different (p= 0.35), suggesting a similar level of skewing in each repertoire.
Table 1. CDR3 length skewing after stimulation.
The peripheral control skew was calculated by comparing the spectratypes from the individual animals used to generate the control data. The relatively high value for Vα6 most likely reflects the effect of the transgenic β-chain on the selection process. Multiple values are shown for individual mice in those cases where the spectratypes were different for each animal. The maximum skew possible is 2.
| Tconv | nTreg | CD4+ T cells | |
|---|---|---|---|
| Vα 2 | 1.20 | 1.08 | 0.03 |
| Vα 3 | 0.30 | 1.09, 1.15, 0.49 | 0.03 |
| Vα 5 | 0.38 | 1.10 | 0.17 |
| Vα 6 | 1.15, 1.36, 1.17 | 1.76, 1.23 | 0.30 |
| Vα 13 | 0.79 | 1.12, 0.59, 1.58 | 0.04 |
| Vα 15 | 1.10 | 1.31 | 0.08 |
| Vα 18 | 1.22 | 0.54 | 0.04 |
Figure 3. The Vα TCR repertoires of N72-specific nTreg and Tconv cells.
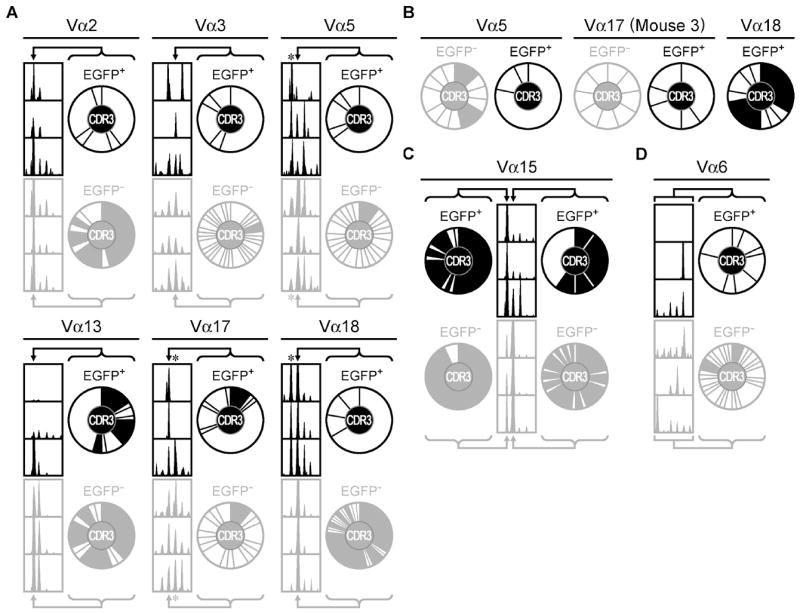
A) Fragment analyses of FAM-labeled PCR products showing the distribution of CDR3 lengths (left). CDR3 pie charts (right) depict the distribution of unique CDR3 amino acid sequences at the indicated CDR3 lengths (arrows), which are equivalent to 12 (Vα2, Vα13), 13 (Vα3, Vα18), or 14 (Vα5, Vα17) amino acids, respectively. The size of the slice is proportional to the clonotype frequency. Shaded slices indicate public amino acid sequences that appear in more than one mouse. B) Pie charts of the unique CDR3 amino acid sequences at the lengths indicated by the asterisks in (A), which are equivalent to 13 (Vα5), 15 (Vα17), or 12 (Vα18) amino acids, respectively. C) Similar analyses as in (A) for clones expressing Vα15, with indicated lengths equivalent to 12-13 amino acids. D) Similar analyses as in (A) for clones expressing Vα6, with indicated lengths equivalent 12-19 amino acids. These data are representative of three independent experiments, with a total of three mice per group. EGFP+ and EGFP− cell populations are represented using black and gray colors, respectively.
For TCR repertoire analysis, we cloned and sequenced the amplified nTreg and Tconv TCRα CDR3 regions from each mouse. From the 8 Vα families analyzed, we recovered 819 in-frame nucleotide sequences corresponding to 326 unique amino acid sequences (222 Tconv, 92 nTreg, 11 overlapping sequences). Ln-rank versus ln-rank frequency plots for the Tconv and nTreg amino acid repertoires revealed an excellent fit for a power law-like relationship. This indicates similar results at all scales of measurement and is consistent with adequate sampling of the repertoires (Supplemental Figure 2B). An overview of the sequencing data is shown in Figure 3. Within a Vα family, each unique CDR3 amino acid sequence was considered as its own distinct ‘species’ and is proportionally represented by slices in the pie charts. For those samples that shared spectratype peaks at common CDR3 lengths (Vα2, Vα3, Vα5, Vα13, Vα17, Vα18; Figure 3A), we analyzed sequences from the same sized products from the respective Tconv and nTreg cell TCR repertoires. Two of these samples (Vα5 and Vα18) also contained a notable nTreg peak that was one codon shorter than the dominant Tconv peak, which we included in the analysis (Figure 3B). Analysis of the Vα18+ Tconv population at this shorter length was not informative due to a lack of CDR3 sequences (data not shown). For Vα15-containing nTreg and Tconv clones, a distinguishing feature of their spectratypes is that each was largely restricted to a dominant CDR3 length, with the nTreg pool expressing CDR3 regions that were predominantly one amino acid shorter than the Tconv cells (Figure 3C). The Vα6+ samples demonstrated little consistency between mice, although each spectratype contained a single dominant CDR3 length. We therefore considered the entire range of CDR3 lengths for Vα6+ clones in a pooled analysis (Figure 3D). Consistent with the skewed spectratypes and fewer CDR3 species, the nTreg pool employed fewer unique Jα regions and exhibited a different pattern of Jα usage than the corresponding Tconv population for each of these Vα analyses (Supplemental Figure 3). Furthermore, Jα segments in common between the two populations showed a bias in Jα usage toward one of the given T cell subsets. The similar spectratype skewing with a reduced number CDR3 species indicates that the N72-specific nTreg TCR repertoire is less diverse than the corresponding N72-specific Tconv TCR repertoire.
Next, we examined the ‘public’ and ‘private’ composition of the repertoires. We considered any CDR3 amino acid sequence to be public if it was recovered from two or more mice. Most Tconv and nTreg sequences at the dominant CDR3 length were private (Figure 3A, filled slices, 85% and 89% private, respectively). Thus antigen-specific nTreg and Tconv TCR repertoires are largely private. In apparent contrast, the Vα15+ Tconv and nTreg populations were mostly public (Figure 3C, filled slices). These data indicate that the public versus private nature of the repertoires tracks with use of certain V segments and is not closely associated with T cell function.
TCR repertoire comparisons of antigen-specific nTreg and Tconv cells
In order to estimate the total size of the two repertoires and the clonal relationships between them, we compared the nTreg cell TCR repertoire with that of the Tconv cells using statistical estimation of species richness (number of different CDR3 sequences) and shared species (overlap between CDR3 repertoires). Previous studies have applied these methods (8, 25), which include the abundance coverage estimator (ACE) and the Morisita-Horn index (MHI). The ACE is a non-parametric statistical tool that uses the number of rare CDR3 sequences (<10 occurrences) and the number of singletons to estimate the number of “unseen” CDR3 sequences and therefore the total number of CDR3 sequences present in the population. The MHI is an abundance–based similarity index that calculates the overlap between two populations, with values ranging from 0 (no overlap) to 1 (complete overlap). The MHI is relatively resistant to undersampling, since the most abundant CDR3 sequences dominate the probability of overlap and are always present in the sample.
In every case, the nTreg cell ACE values were lower than those for the Tconv cells (Figure 4). This result indicated less diversity among the nTreg TCRs, in agreement with the spectratype data. Of the seven Vα groups that allowed comparison of equivalent CDR3 lengths between the two T cell populations, four of them showed no overlap (Figure 4A). Of the remaining three Vα groups, the percent overlap appeared highest in the Vα13 sample (Figure 4B) with 17% of unique CDR3 sequences or 34% of the total number of Vα13+ samples. However, taking into consideration the biodiversity of each Vα13+ population yielded an MHI of only 0.103 (Figure 4B). This was in fact lower than the maximal MHI value of 0.119 that we observed in the Vα2+ sample, which also showed comparatively less overlap at 6% of unique CDR3 sequences or 18% of the total number of samples. Nevertheless, while some overlap did exist between the Tconv cell and nTreg cell TCR repertoires, it remained minimal and amounted to only 3.4% of all the unique sequences that we considered in our study.
Figure 4. Overlap between the nTreg cell N72-specific TCR repertoire and its Tconv cell counterpart.
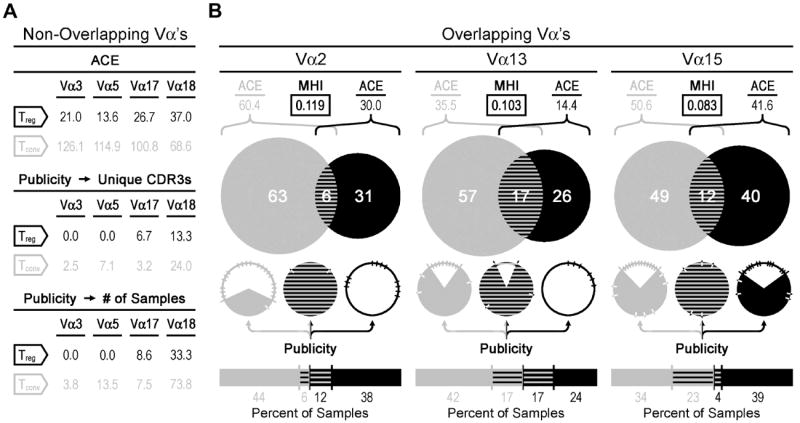
A) Statistical data are shown for those select Vα samples (Vα3, Vα5, Vα17, Vα18) that had no apparent overlap (MHI = 0.00) between the nTreg cell and Tconv cell repertoires. B) For those Vα repertoires that exhibited some degree of overlap (Vα2, Vα13, Vα15), the Venn diagrams show the proportion and number of unique CDR3 sequences that were found either solely in the Tconv cell (gray) population, nTreg cell (black) population, or in both (striped) populations. The pie charts and bar graphs below the Venn diagrams represent the publicity data. In the pie charts, filled slices represent public sequences and notches show the number and proportion of unique CDR3 amino acid sequences. The underlying bar graphs and numbers represent the percent distribution of public sequences recovered from the Tconv cell (left) and nTreg cell (right) populations. For each given Vα analysis, CDR3 amino acid sequences are considered public if retrieved from two or more mice. All publicity data is presented in the pie charts and bar graphs (B) or in tabular format (A) as a percentage based either on giving equal weight to each unique CDR3 amino acid sequence or on the number of samples in each group as indicated. Biodiversity estimations include the Morisita-Horn index (MHI, boxed) and the abundance-based coverage estimator (ACE). These data are representative of three independent experiments, with a total of three mice per group. Sample sizes are as follows: Vα2=89, Vα3=84, Vα5=89, Vα13=109, Vα15=161, Vα17=98, Vα18=110.
Interestingly, the overlapping sequences that we observed were consistently more public than those sequences that were unique to either the Tconv cell or nTreg cell populations (Figure 4B). Publicity ranged from 75% to 100% of overlapping TCR amino acid sequences, whereas 0% to 38% of the non-overlapping TCRs within these Vα groups were public (Figure 4B). These data demonstrate that among those Tconv and nTreg cells that respond to the same epitope, shared TCRs are more likely be public.
TCRα CDR3 amino acid sequences
Our data thus far support the notion that when considering foreign antigen-specific responses, the TCR repertoire of nTreg cells is relatively limited and distinct from that of Tconv cells. The bulk of each CDR3α sequence is made up of the Jα region. A closer look at the primary sequences within the Vα groups (excluding Vα6) that showed no CDR3 overlap revealed that most involved N- and/or P-nucleotide additions (Figure 5A-C). Each of these groups used certain Jα regions multiple times, in which case variability within CDR3s that used a common Jα region was limited to the intersection between the variable and junctional regions (Figure 5A-C, boxed amino acids). CDR3 variability in most of these Tconv subsets (Vα3, Vα5, Vα17) was high, as demonstrated by each of these Vα groups being most frequently populated by singletons (44-69%). The notable exception was the Vα18 group, in which the most frequently recovered sequence was that of the original 3.L2 TCRα (line #5). The Vα6 samples also showed no overlap between the Tconv cell and nTreg cell populations (Figure 5B), although the dominant CDR3 lengths were inconsistent between mice.
Figure 5. TCRα CDR3 amino acid sequences from N72-specific nTreg and Tconv cell clones.

A-C) CDR3 sequences found two or more times are shown using single-letter amino acid code. Singleton sequences are pooled. The frequency of each CDR3 amino acid sequence is given in the “Treg” and “Tconv” columns. Asterisks in the “V” column indicate use of two or more V-region subfamilies, and the number of mice containing the sequence is listed in the “M” column if the sequence was recovered from more than one mouse. The “Nuc” column indicates the use of N- (N), P- (P), or both N- and P-nucleotides (NP) in one or more samples within each of these respective sequences. Lines with sequences found in both nTreg cell and Tconv cell populations are outlined in gray. At given CDR3 lengths, common Jα usages and the corresponding amino acid positions that show variability in sequence are bolded and outlined in black. Sample sizes are listed in the header of each respective Vα analysis. Conserved amino acids that have more than one underlying codon sequence are also bolded and colored light gray, dark gray or black for 2, 3, or 4+ unique codons, respectively. Line numbers (“#” column, far left) are provided for organizational purposes. A) Analysis of non-overlapping Vα groups based on CDR3 length-selected clones. B) Analysis of the non-overlapping Vα6 group, where the CDR3 regions were not preselected for sequencing based on their length. C) Analysis of CDR3 size-selected Vα groups that show some overlap between the nTreg and Tconv cell populations. D) Venn diagrams show the number of unique clones (n=76) that were found either solely in the Tconv cell (gray) population (n=49), nTreg cell (black) population (n=23), or both (striped, n=4). To be considered clonal, PCR products needed to share the same sequence and come from the same mouse. The respective underlying bar graphs and numbers are similarly colored and represent the percent distribution of samples recovered from the Tconv cell (left) and nTreg cell (right) populations. These data are representative of three independent experiments, with a total of three mice per group.
The Vα samples that did show some overlap between the Tconv and nTreg populations also consisted largely of CDR3s that included N- and/or P-nucleotides (Figure 5C). Additionally, most of the overlapping sequences showed variability in upstream sequences, which suggested that these TCRs were more likely to employ different Vα subfamily members. A number of amino acids were each encoded by more than one codon, and this occurred more often among the overlapping sequences when compared to those that showed no overlap. All overlapping Vα2+ and Vα13+ CDR3s used the Jα52 and Jα27 regions, respectively, while the overlapping Vα15+ CDR3s used a wider variety of Jα regions. Next, we considered the possibility that each shared CDR3 sequence may be derived from a unique clone. If this were true, we would expect the respective Tconv and nTreg nucleotide sequences from any given mouse to be identical in their entirety. Instead, we found that 65% (49/76) and 30% (23/76) were specific to the Tconv and nTreg populations, respectively (Figure 5D). Only ~5% of the unique nucleotide sequences that made up the pool of overlapping CDR3 amino acid sequences were contained within both T cell populations. This indicated that within an individual, overlapping sequences were not clonally related. In fact, there was a bias toward use of certain Jα regions, which resulted in separate Vα groups sharing a common CDR3 amino acid sequence (Supplemental Figure 4). Similarities in the Jα27 versus Jα52 primary sequence also allowed one of the overlapping sequences (CAAGANTGKLTF) to have incidences of both of these Jα segments.
DISCUSSION
Our study compared a foreign antigen-specific TCR repertoire of nTreg cells to the corresponding TCRs of a Tconv cell population that responded to the same epitope. This type of analysis is particularly significant, since good protection from pathogens involves participation of both the Tconv cell and nTreg cell subsets to keep the response adequate yet well controlled. In this context, we found the nTreg TCR repertoire to be distinct, restricted and less diverse than that of the Tconv cells.
A restriction in nTreg cell foreign antigen-specific responses may originate during thymic selection. In the thymus, nTreg precursors have different affinity requirements for their maturation compared to developing Tconv thymocytes (1, 2). Induction of Foxp3 requires interaction with an agonist self-peptide, and the frequency of Foxp3+ cells is directly related to the strength of the agonist (3). Consequently, the differences in affinity-based selection should result in populations of Tconv and nTreg cells with discrete TCR repertoires. In combination with the autoreactivity of the nTreg population (7, 26), these data support the idea that the recognition of foreign antigens by nTreg cells relies on polyspecificity (27). However, some studies have shown that nTreg precursors are subject to the process of negative selection (26, 28, 29), which functions to eliminate cells with crossreactive TCRs (30). It follows that the autospecific nTreg cell population should be relatively deficient in foreign antigen recognition. Our data are in agreement with this interpretation. For each Vα analysis, the nTreg population was consistently less diverse as determined by the lower ACE value. Taken together, these data support a model in which negative selection acts to shape the nTreg TCR repertoire. This conclusion leads us to propose that if foreign antigen recognition by nTreg cells depends on polyspecificity, it may be important to limit the extent of this crossreactivity within the nTreg cell compartment in order to allow for the generation of effective immune responses to foreign antigens. This hypothesis is not directly addressed herein.
Our findings stand in an apparent contrast to previous analyses of naïve polyclonal nTreg populations, which show that nTreg and Tconv TCR repertoires are at least equally diverse (7, 9). Such unselected repertoire analyses estimate the total diversity potential contained within an individual. In this report, we examined a TCR repertoire that was defined by its functional capacity to respond to a specific foreign antigen. Thus, the relatively restricted nTreg TCR repertoire described herein is a reflection of an antigen-driven selection process in the periphery rather than an estimate of the theoretical potential for nTreg cell diversity generated by developmental processes in the thymus. The smaller size of the resultant nTreg TCR repertoire was also not due to less cell division after antigenic stimulation. While the nTreg cells showed a lag in the kinetics of their response, the extent to which they proliferated was greater than that of Tconv cells. Alterations in the proximal TCR signaling of nTreg cells could have contributed to their delay in proliferation (31, 32). Additionally, nTreg cells have relatively reduced levels of CD4, which may in fact require them to have higher TCR-ligand affinity for activation compared to their Tconv cell counterparts. Consistent with this hypothesis, elimination of CD4 in Tconv cells increases the affinity threshold for T cell selection in the thymus (33). For mature nTreg cells, such an increase in the activation threshold would tend to limit the repertoire of clones capable of responding to any given antigen. Thus, while the respective TCR diversity potentials of the two populations are equivalent, the number of nTreg TCRs responding to a specific antigen may also be constrained by cell-intrinsic criteria unrelated to the origin (self versus foreign) of that antigen.
The antigen-specific nTreg cell responses in our system generally did not share CDR3α amino acid sequences with the corresponding Tconv cell pool (11 of 326 clones, or 3.4%). This observation was more pronounced when considering these sequences at the nucleic acid level. Only 7.1% of the DNA sequences that composed these overlapping amino acid sequences were identical between the nTreg cell and Tconv cell populations isolated from the same mouse. In other words, overlapping CDR3 amino acid sequences were rare and were derived from different clones. These data are consistent with our previous studies that found a limited overlap between nTreg cells and iTreg cells, the latter of which arises from the Tconv population and can comprise up to 10-15% of the peripheral Treg cell pool (12). In the present study, we also found that 14% of EGFP+ T cells lost Foxp3 expression within the first 24 hours following restimulation in culture. Taken together, these data suggest that the loss of Foxp3 expression described herein might be attributable to the downregulation of Foxp3 in iTreg cells. Indeed, if iTreg cells were maintained during in vitro restimulation, they would be scored as nTreg cells and would increase the overlap between nTreg and Tconv cells. As an alternative explanation, it has been shown that nTreg cells may also lose Foxp3 expression and become “ex-Foxp3” effector/memory cells (34). Misidentified “ex-Foxp3” cells would be scored as Tconv cells, thereby creating the appearance of a more extensive overlap, which is not what we observed. Thus the unstable fraction of Foxp3+ cells deserves special attention, and it will be interesting to further explore the TCR repertoire and functional capacity of these proliferating “dropout” cells in future studies.
In summary, when we compared antigen-specific Tconv cells to an enriched, stable nTreg cell pool responding to the same foreign epitope, we found virtually no overlap in their respective TCR repertoires and a comparatively restricted and less diverse nTreg cell response. We propose that foreign antigen recognition by nTreg cells, by definition, relies on the polyspecificity of nTreg cell TCRs, which is central to this limitation. When the nTreg cell repertoire is insufficient to control the Tconv response to any foreign antigen, the resulting increase in inflammation transiently supports the production of iTreg cells proportional to the needs of the host. Our data are therefore consistent with the more fundamental hypothesis that iTreg cells broaden the diversity of regulatory responses to foreign antigens (12).
Supplementary Material
Acknowledgments
We thank Jane Ebert for technical assistance, as well as Talal Chatila, Christopher Mayne and James Verbsky for critical reading of the manuscript.
Footnotes
This work was supported by National Institute of Health grants R01 AI073731, R01 AI085090, N01 50032, and the D.B. and Marjorie Reinhart Family Foundation
Abbreviations used in this paper: Hb, hemoglobin; MCC, moth cytochrome C; nTreg, natural regulatory T; Tconv, conventional T; iTreg, induced regulatory T
References
- 1.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol. 2002;3:756–763. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- 2.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 3.Relland LM, Mishra MK, Haribhai D, Edwards B, Ziegelbauer J, Williams CB. Affinity-based selection of regulatory T cells occurs independent of agonist-mediated induction of Foxp3 expression. J Immunol. 2009;182:1341–1350. doi: 10.4049/jimmunol.182.3.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aschenbrenner K, D’Cruz LM, Vollmann EH, Hinterberger M, Emmerich J, Swee LK, Rolink A, Klein L. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol. 2007;8:351–358. doi: 10.1038/ni1444. [DOI] [PubMed] [Google Scholar]
- 5.Bautista JL, Lio CW, Lathrop SK, Forbush K, Liang Y, Luo J, Rudensky AY, Hsieh CS. Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat Immunol. 2009;10:610–617. doi: 10.1038/ni.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leung MW, Shen S, Lafaille JJ. TCR-dependent differentiation of thymic Foxp3+ cells is limited to small clonal sizes. J Exp Med. 2009;206:2121–2130. doi: 10.1084/jem.20091033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 9.Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006;25:249–259. doi: 10.1016/j.immuni.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Pacholczyk R, Kern J, Singh N, Iwashima M, Kraj P, Ignatowicz L. Nonself-antigens are the cognate specificities of Foxp3+ regulatory T cells. Immunity. 2007;27:493–504. doi: 10.1016/j.immuni.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C. Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J Immunol. 2007;178:7032–7041. doi: 10.4049/jimmunol.178.11.7032. [DOI] [PubMed] [Google Scholar]
- 12.Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, Ziegelbauer J, Yassai M, Li SH, Relland LM, Wise PM, Chen A, Zheng YQ, Simpson PM, Gorski J, Salzman NH, Hessner MJ, Chatila TA, Williams CB. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35:109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lathrop SK, Santacruz NA, Pham D, Luo J, Hsieh CS. Antigen-specific peripheral shaping of the natural regulatory T cell population. J Exp Med. 2008;205:3105–3117. doi: 10.1084/jem.20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fenton RG, Marrack P, Kappler JW, Kanagawa O, Seidman JG. Isotypic exclusion of gamma delta T cell receptors in transgenic mice bearing a rearranged beta-chain gene. Science. 1988;241:1089–1092. doi: 10.1126/science.2970670. [DOI] [PubMed] [Google Scholar]
- 15.Jorgensen JL, Esser U, Fazekas de St Groth B, Reay PA, Davis MM. Mapping T-cell receptor-peptide contacts by variant peptide immunization of single-chain transgenics. Nature. 1992;355:224–230. doi: 10.1038/355224a0. [DOI] [PubMed] [Google Scholar]
- 16.Kersh GJ, Donermeyer DL, Frederick KE, White JM, Hsu BL, Allen PM. TCR transgenic mice in which usage of transgenic alpha- and beta-chains is highly dependent on the level of selecting ligand. J Immunol. 1998;161:585–593. [PubMed] [Google Scholar]
- 17.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol. 2007;178:2961–2972. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- 18.Kersh GJ, Allen PM. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J Exp Med. 1996;184:1259–1268. doi: 10.1084/jem.184.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Currier JR, Robinson MA. Spectratype/immunoscope analysis of the expressed TCR repertoire. Curr Protoc Immunol. 2001;Chapter 10(Unit 10):28. doi: 10.1002/0471142735.im1028s38. [DOI] [PubMed] [Google Scholar]
- 20.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 21.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsui K, Boniface JJ, Steffner P, Reay PA, Davis MM. Kinetics of T-cell receptor binding to peptide/I-Ek complexes: correlation of the dissociation rate with T-cell responsiveness. Proc Natl Acad Sci U S A. 1994;91:12862–12866. doi: 10.1073/pnas.91.26.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng G, Yu A, Malek TR. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol Rev. 2011;241:63–76. doi: 10.1111/j.1600-065X.2011.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lefranc MP, Pommie C, Ruiz M, Giudicelli V, Foulquier E, Truong L, Thouvenin-Contet V, Lefranc G. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol. 2003;27:55–77. doi: 10.1016/s0145-305x(02)00039-3. [DOI] [PubMed] [Google Scholar]
- 25.Adeegbe D, Matsutani T, Yang J, Altman NH, Malek TR. CD4(+) CD25(+) Foxp3(+) T regulatory cells with limited TCR diversity in control of autoimmunity. J Immunol. 2010;184:56–66. doi: 10.4049/jimmunol.0902379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romagnoli P, Hudrisier D, van Meerwijk JP. Preferential recognition of self antigens despite normal thymic deletion of CD4(+)CD25(+) regulatory T cells. J Immunol. 2002;168:1644–1648. doi: 10.4049/jimmunol.168.4.1644. [DOI] [PubMed] [Google Scholar]
- 27.Felix NJ, Donermeyer DL, Horvath S, Walters JJ, Gross ML, Suri A, Allen PM. Alloreactive T cells respond specifically to multiple distinct peptide-MHC complexes. Nat Immunol. 2007;8:388–397. doi: 10.1038/ni1446. [DOI] [PubMed] [Google Scholar]
- 28.van Santen HM, Benoist C, Mathis D. Number of T reg cells that differentiate does not increase upon encounter of agonist ligand on thymic epithelial cells. J Exp Med. 2004;200:1221–1230. doi: 10.1084/jem.20041022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wirnsberger G, Hinterberger M, Klein L. Regulatory T-cell differentiation versus clonal deletion of autoreactive thymocytes. Immunol Cell Biol. 2011;89:45–53. doi: 10.1038/icb.2010.123. [DOI] [PubMed] [Google Scholar]
- 30.Huseby ES, White J, Crawford F, Vass T, Becker D, Pinilla C, Marrack P, Kappler JW. How the T cell repertoire becomes peptide and MHC specific. Cell. 2005;122:247–260. doi: 10.1016/j.cell.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Zanin-Zhorov A, Ding Y, Kumari S, Attur M, Hippen KL, Brown M, Blazar BR, Abramson SB, Lafaille JJ, Dustin ML. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science. 2010;328:372–376. doi: 10.1126/science.1186068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hickman SP, Yang J, Thomas RM, Wells AD, Turka LA. Defective activation of protein kinase C and Ras-ERK pathways limits IL-2 production and proliferation by CD4+CD25+ regulatory T cells. J Immunol. 2006;177:2186–2194. doi: 10.4049/jimmunol.177.4.2186. [DOI] [PubMed] [Google Scholar]
- 33.Kao H, Allen PM. An antagonist peptide mediates positive selection and CD4 lineage commitment of MHC class II-restricted T cells in the absence of CD4. J Exp Med. 2005;201:149–158. doi: 10.1084/jem.20041574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.