

Abstract
The poor prognosis of glioblastoma (GBM) routinely treated with ionizing radiation (IR) has been attributed to the relative radioresistance of glioma initiating cells (GIC). Other studies suggest that GIC are sensitive but the response is mediated by undefined factors in the microenvironment. GBM produce abundant transforming growth factor-β (TGFβ), a pleotropic cytokine that promotes effective DNA damage response. Consistent with this, radiation sensitivity, as measured by clonogenic assay, of cultured murine (GL261) and human (U251, U87MG) glioma cell lines, increased approximately 25% when treated with LY364947, a small molecule inhibitor of TGFβ type I receptor kinase, prior to irradiation. Mice bearing GL261 flank tumors treated with 1D11, a pan-isoform TGFβ neutralizing antibody, exhibited significantly increased tumor growth delay following IR. GL261 neurosphere cultures were used to evaluate GIC. LY364947 had no effect on primary or secondary neurosphere-forming capacity. IR decreased primary neurosphere formation by 28%, but did not reduce secondary neurosphere formation. In contrast, LY364947 prior to IR decreased primary neurosphere formation by 75% and secondary neurosphere formation by 68%. Notably, GL261 neurospheres produced 3.7-fold more TGFβ per cell compared to traditional culture, suggesting that TGFβ production by GIC promotes the DNA damage response and self-renewal and creates microenvironment mediated resistance. Consistent with this, LY364947 treatment in irradiated GL261 neurosphere-derived cells decreased DNA damage responses, H2AX and p53 phosphorylation, and induction of self-renewal signals, Notch1 and CXCR4. These data motivate the use of TGFβ inhibitors with radiation to improve therapeutic response in GBM patients.
Keywords: TGFβ, DNA damage response, radiosensitivity, glioblastoma
Introduction
Glioblastoma multiforme (GBM) remains a significant therapeutic challenge and an unmet medical need. Despite advances in radiation therapy and improvements in chemotherapeutics and targeted therapies, outcomes remain poor, with a median survival of 14.6 months when current standard of care, such as concurrent chemoradiation therapy and adjuvant chemotherapy, are used (1). While the exact origin of GBM (and other malignant brain tumors) is unknown, it is thought that a fraction of tumor cells have cancer stem cell characteristics (glioma initiating cells, GIC) and true tumorigenic potential (2, 3). Originally proposed twenty-five years ago (4), it is generally thought that the resistance of GIC contributes to the poor response to radiation and chemotherapy and inevitable tumor recurrence (reviewed in (5)). The exact mechanism of treatment resistance is unknown, but GIC intrinsic hyperactivation of the PI3K/Akt and PTEN pathways (6, 7) and increased activation of DNA damage checkpoint pathways (8) are thought to contribute.
Microenvironment can also contribute to radiation responses (reviewed in (9). Jamal et al. showed that GBM cells irradiated under orthotopic conditions have a greater capacity to repair DNA double strand breaks than GBM cells irradiated in vitro (10). A critical component of the GBM microenvironment is the pleotropic cytokine transforming growth factor-β (TGFβ). TGFβ has a range of effects on the glioma microenvironment, including extracellular matrix deposition, angiogenesis, and invasion (reviewed in (11)). Both TGFβ1 and TGFβ2 have been implicated in autocrine tumor growth regulation (12). TGFβ2 is overexpressed in gliomas (13). Higher levels of TGFβ1 have been found in anaplastic gliomas (WHO grade III) than in GBM (WHO grade IV), suggesting a potential role of TGFβ1 in the early stages of tumorigenesis (14).
The TGFβ family has been shown to play a role in both pluripotent stem cells (reviewed in (15)) and neural stem cells specifically (16). TGFβ has been implicated in GIC biology as well. Penuelas et al. showed that exposure of patient derived tumor neurospheres to TGFβ increased the number of neurospheres in a dose-dependent fashion and injection of these neurospheres into mice resulted in earlier appearance of more aggressive tumors (17). Ikushima et al. reported that autocrine TGFβ contributes to the tumorigenicity of the GIC population by activation of Sox4 and Sox2 (18). More recently, Anido et al. showed that TGFβ inhibitors affect a CD44high/Id1high GIC population via Id1 and Id3, which they propose controls the “master regulators” of the TGFβ-GIC gene program, including LIF, Sox2, Sox4 and CD44 (19).
Ionizing radiation (IR) induces TGFβ in vitro and in vivo in both normal and cancer cells (20-22). We have shown previously that reactive oxygen species are likely involved in the radiation-induced activation of TGFβ (23) and the process is mediated by a conformational change in latency-associated peptide (LAP)-TGFβ complex, allowing the release of active TGFβ1 (24). Our studies and others have directly linked TGFβ to DNA damage responses and radiosensitivity (25, 26). Inhibiting TGFβ decreases radiation-induced phosphorylation of p53, chk2, H2AX and rad17, all of which are substrates of ataxia telangectasia mutated (ATM), a protein kinase critical in the molecular response to IR-induced DNA double-strand breaks. ATM, a member of the phosphatidylinositol 3-kinase (PI3-kinase) family, is thought to be a master controller of cell cycle checkpoint signaling pathways that are required for cell response to DNA damage and for genome stability. Moreover, there is evidence using proteomic profiling that prolonged TGFβ treatment of cells can affect DNA damage repair such as Rad51 in a Smad-dependent manner (27). Notably, breast cancer cell lines treated with a small molecule TGFβ type I receptor kinase inhibitor showed increased radiosensitivity as measured by clonogenic assay and decreased DNA damage responses to radiation, including nuclear foci of the histone variant H2AX, regardless of sensitivity to TGFβ growth control. A syngeneic model of triple-negative breast cancer showed increased tumor growth delay in response to single or fractionated radiation treatment with the addition of TGFβ neutralizing antibodies during radiotherapy (28).
The present study is aimed at determining the effects of TGFβ inhibition on radiation sensitivity of the GIC population. To assess the therapeutic potential of TGFβ inhibition during radiotherapy, we determined the relationship between sensitivity to TGFβ mediated growth inhibition, GIC formation, molecular responses to radiation, and radiosensitivity in human and murine GBM in vitro and in vivo. We determined that neurosphere cultures compared to bulk populations produce more TGFβ, whose inhibition significantly compromises both DNA damage response and self-renewal of GIC.
Materials/Methods
Cell Culture
The murine glioma, GL261 (obtained from National Cancer Institute-Frederick Cancer Research Tumor Repository; Frederick, MD; authenticated in 2012 by Idexx Radil, West Sacremento, CA) and human glioma U251 (generous gift of Dr. Kevin Camphausen; authenticated in 2010 by Idexx Radil) cells were cultured in Dulbecco’s Modified Eagle Medium GlutaMAX™ (Gibco; Carlsbad, CA) supplemented with 10% fetal bovine serum (Sigma-Aldrich; St. Louis, MO) and 1% pyruvate (Gibco; Carlsbad, CA). Human glioma U87MG (obtained from ATCC in 2011) cells were cultured in Eagle’s Minimum Essential Medium (Gibco; Carlsbad, CA) with 10% fetal bovine serum, at 37°C with 5 % CO2. All cell lines were tested for Mycoplasma and were negative (Cellshipper Mycoplasma test; Bionique, Saranac Lake, NY).
For cell proliferation experiments, cells were cultured in 10% serum replacement medium (SRM; Knockout SR, Life Technologies, Inc., Carlsbad, CA) containing either 2 ng/ml TGFβ1 (R&D Systems; Minneapolis, MN), 400 nM small molecule inhibitor of the TGFβ type I receptor kinase, LY364947 ([3-(Pyridin-2-yl)-4-(4-quinonyl)](4)-1H-pyrazole; Lilly designation HTS466284; Calbiochem; St. Louis, MO), 10 μg/ml of 1D11, a pan-isoform, neutralizing TGFβ monoclonal antibody, or 13C4, a murine monoclonal isotype control antibody (kindly provided by Genzyme Inc.; Framingham, MA). Cells were trypsinized and counted using a Coulter counter at 24 h and 48 h post-treatment.
Mink Lung Cell Luciferase Assay
To measure secreted active and latent TGFβ in conditioned media, luciferase induction by conditioned media was measured in the mink lung epithelial cells transfected with truncated plasminogen activator inhibitor-1 promoter fused to the firefly luciferase reporter gene as previously described (29). Active TGFβ was measured directly in untreated samples, whereas total TGFβ (active and latent) was measured following heat activation at 80°C for five minutes (30). Unconditioned media was used as a control in all experiments, which was subtracted, and each condition was repeated in the presence of pan-specific TGFβ neutralizing antibody to confirm specificity. All conditions were performed in triplicate, and results represent the mean measured value per 106 cultured cells.
Neurosphere Assay
Cells were diluted in serum-free growth medium (1000 cells/ml) and plated in 500μl in non-tissue culture coated 24-well plates (Corning-Costar; Lowell, MA). Cells were fed with 125 μl of serum-free growth medium every other day, for 14 days. The culture medium consisted of serum-free DMEM/F12 (Invitrogen, Grand Island, NY) supplemented with 10 U/ml heparin (Sigma-Aldrich; St. Louis, MO), 2% B27 (Invitrogen), human recombinant fibroblast growth factor 2 (FGF-2, 20 ng/ml, Sigma-Aldrich) and epidermal growth factor (EGF, 20 ng/ml, Sigma-Aldrich). After 14 days, spheres were measured and those > 100μm were counted as a neurosphere forming unit. To generate secondary neurospheres, primary neurospheres were gently centrifuged, mechanically dissociated, and then incubated with trypsin at 37°C for five minutes. After centrifugation and washing with PBS, cells were diluted using neurosphere media at 1000 cells/ml and plated into non-tissue culture coated 24-well plates, as above. Spheres were counted and measured from 6 different wells per experiment. The neurosphere number and size data shown is the average + S.E. of three independent experiments consisting of three replicates.
Clonogenic Assay
To assess clonogenic survival of cells in monolayer culture, human and murine glioma cell lines were grown for 48 h to 70% confluence upon which cells were incubated with serum replacement media containing 400 nM of LY364947 kinase inhibitor or 10 μg/ml pan-specific TGFβ neutralizing antibody 1D11 or control antibody 13C4 for 48 hours before- and 3 hour post-radiation exposure. Cells were irradiated with 1-8 Gy using a Varian Clinac 2300 C/D linear accelerator (Varian; Palo Alto, CA), trypsinized 3 h post-irradiation and plated in triplicates at three dilutions into six-well cell culture plates in serum containing media. Colonies were allowed to grow for 10-12 days followed by fixing and staining with crystal violet. Colonies containing >50 cells were counted to determine percent survival and the number of colonies obtained from three replicates was averaged for each treatment. These mean values were corrected according to plating efficiency of respective controls to calculate cell survival for each dose level.
To assess clonogenic survival of cells in neurosphere culture, neurospheres were cultured for 14 days under neurosphere conditions described above, then treated with 400 nM of LY364947 kinase inhibitor for 48 hours before- and 3 hours-post irradiation (2 Gy). Cells were centrifuged, media aspirated, and plated in triplicate at three dilutions into six-well cell culture plates in serum containing media. The remainder of the assay was performed as above.
Western Blotting
To examine the DNA damage response and TGFβ signaling, 5×105 cells were grown in complete media for 48 h, followed by LY364947 treatment (400 nM) in 10 % SRM for 24 h. The cells were irradiated with 5 Gy and lysed after 1 hour, or treated with 500 pg/ml TGFβ and lysed after 30 minutes. Then the extracts were subjected to immunoblot analysis with one of the primary antibodies: phospho-Smad2 on serine 465/467 at 1:500 (clone 138D4, CAT#3108, Cell Signaling; Beverly, MA), Smad2/3 at 1:500 (CAT#610842, BD Transduction Laboratories; Lexington, KY), phospho-p53 on serine 15 at 1:500 (CAT#92845, Cell Signaling), p53 at 1:500 (Clone Ab-8, CAT#MS-738-P, Neomarkers; Fremont, CA and CAT#554157, BD Biosciences), ATM serine1981 phosporylation at 1:500 (CAT#2152-1, Epitomics; Burlingame, CA) and ATM, clone 2C1 at 1:500 (CAT#GTX70103, GeneTex; San Antonio, TX). Protein estimation was carried out using the BCA protein assay kit (Pierce; Rockford, IL). One hundred μg of protein was electrophoresed on a 4-15 % gradient gel (BioRad; Hercules, CA) and transblotted on PVDF Immobilion™-FL membrane (Millipore Corporation; Ballerica, MA). The membrane was blocked in blocking buffer and probed with a primary antibody. The membrane was washed 3 times for 10 min with 0.1% TBST, followed by incubation with secondary antibodies (goat anti-mouse, CAT#926-32220 and goat anti-rabbit, CAT#926-32211, Odyssey; Lincoln, NE) for 1 h at room temperature. The membrane was washed 3 times for 10 min with TBST 0.1% and scanned on the Odyssey LICOR system. Using ImageJ 1.45s software (National Institute of Health, USA) the raw integrated density was measured for each band of the protein of interest in all 3 cells lines. After correction for loading using actin, the ratio of phosphorylated to total protein was determined, normalized to the control group and represented as fold change from the control group. Representative figures are displayed in grayscale.
Comet Assay
The persistence DNA damage following fractionated irradiation on neurosphere cultures was assessed by CometAssay®. Neurospheres were cultured and treated with 400 nM of LY364947 kinase inhibitor as described above and irradiated with 2 Gy for three consecutive days beginning on day 10 in culture. Neurospheres were dissociated and harvested 24hrs following the third fraction. Single cell gel electrophoresis at 19V (300mAMP, 40 min) was preformed by Alkaline CometAssay® (Trevigen) according to the manufacturer’s protocol. SYBR® Green stained DNA comets were imaged at 100× magnification and the extent of DNA breaks was quantified as tail moment using CometScore™ software.
Immunofluorescence
GL261, U251 and U87MG cells (2×104) were grown in chamber slides in complete media for 48 h, followed by LY364947 treatment (400 nM) in 10% SRM for 24 h prior to 2 Gy radiation. GL261-derived neurospheres, tumor cryosections or cells prepared as above were fixed using 2% paraformaldehyde for 20 min at room temperature followed by permeabilization with 100 % methanol for 20 min at -20°C. Then specimens were blocked with the supernatant of 0.5% casein/PBS, stirred for 1 h, incubated with mouse detective antigen (Biocare Medical; Concord, CA) for an additional 3 hrs for murine glioma tumors, and incubated with a mouse monoclonal γH2AX antibody (clone JBW301, Upstate Biotechnology; Charlottesville, VA) at 1:500, rabbit monoclonal phospho-serine 465/467 Smad2 antibody at 1:100 (clone 138D4, CAT#3108, Cell Signaling), goat polyclonal CXCR4antibody (CAT# ab1670, Abcam; Cambridge, MA) at 1:300, or rabbit monoclonal Notch1antibody (CAT# ab8925, Abcam) at 1:100, overnight at 4°C followed by washes and incubation with Alexa-488 or Alexa-594 labeled anti-mouse/anti-rabbit/anti-goat secondary antibodies (Molecular Probes; Eugene, OR) for 1 h at room temperature. Specimens were counter-stained with 4′,6-diamidino-2-phenylindole (DAPI), and washed in PBS-Tween20 0.1% before mounting with Vectashield mounting medium (Vector Labs; Burlingame, CA). Specimens were imaged using a 40X objective with 0.95 numerical aperture Zeiss Plan-Apochromat objective on a Zeiss Axiovert (Zeiss; Wetzlar, Germany) equipped with epifluorescence. All images were acquired with a CCD Hamamatsu Photonics (Herrsching am Ammersee, Germany) monochrome camera at 1392×1040 pixel size, 12 bits per pixel (bpp) depth and assembled as false color images using the Metamorph imaging platform (Molecular Devices, Inc.; Sunnyvale, CA). Foci were enumerated as previously described (31).
In Vivo Tumor Studies
Animal studies were conducted using protocols that had undergone institutional review and approval. Female C57/BL6 mice age 6-8 weeks obtained from Taconic (Hudson, NY) were used for animal experiments. Animals were housed in a temperature-controlled animal care facility with a 12-h light-dark cycle and allowed chow and water ad libitum. GL261 cells (106) were injected into the right flank of mice and allowed to grow. Once tumors reached average 140mm3, animals (n=10 in each group) were randomized to receive 1D11 TGFβ neutralizing antibody or 13C4 control antibody (10mg/kg, intraperitoneal injection). Twenty-four hours later, tumors were irradiated with a dose of 6 Gy using a Varian Clinac 2300 C/D linear accelerator fitted with a 25-mm radiosurgery conical collimator (BrainLAB AG; Feldkirchen, Germany). Superflab bolus (1.5 cm tissue equivalent material) was placed over the tumor, and a source-to-skin distance of 100 cm was set. Radiation was delivered at 600 cGy/min with 6 MV X-rays. Mice were monitored thrice weekly for signs of toxicity and tumor volumes were measured with a caliper. Tumor volumes were calculated as length × width2 × 0.52 with all measurements in mm. Animals were sacrificed when tumors reached 10mm x 10mm in two dimensions. Two hours before sacrifice, animals were injected with pimonidazole (60 mg/kg intraperitoneal, HPI; Burlington, MA). Tumors were harvested and portions were formalin fixed and frozen in O.C.T. (Sakura Tissue-Tek; Torrance, CA). All animal experiments were performed in accordance with New York University’s institutional animal care and use committee. For analysis, each tumor was normalized to its pre-treatment volume.
Statistical Analysis
The significance of the difference between mean values was calculated by performing a two-way Students t-test. The significance of the difference between the mean values for graded doses of radiation in clonogenic assays, neurosphere formation, and γH2AX foci quantification was calculated by performing a one-way ANOVA test with Tukey post-test. ANOVA with the Student’s Newman-Keuls Multiple Comparison post-test was used to determine significance between in vivo tumor growth delay measured by time-to-reach three times pre-treatment volume. A p value of < 0.05 was considered significant.
Results
Inhibition of TGFβ radiosensitizes glioma cells
In serum-free conditions, glioma murine GL261 and human U251 and U87MG cell lines produced comparable amounts of active and latent TGFβ; GL261 162.3+11.7 pg/ml and 477.6+67.8 pg/ml; U251 123.1+15.0 pg/ml and 420.5+97.9 pg/ml; U87MG 82.5+18.6 pg/ml and 350.5+144.2 pg/ml, respectively, and demonstrated intact TGFβ signaling through Smad phosphorylation (Figure 1A). Murine GL261 and human U251MG cells were not responsive to growth modulation by exogenous TGFβ or blockade of endogenous TGFβ signaling by the addition of the TGFβ small molecule inhibitor LY364947. U87MG cells were growth inhibited by addition of TGFβ by 19.7+8.8% at 24hrs and 55.3+9.3% at 48hrs, an effect that was reversed by the addition of the TGFβ small molecule inhibitor LY364947 (data not shown).
Figure 1. TGFβ inhibition radiosensitizes glioma cells independent of effects on proliferation.
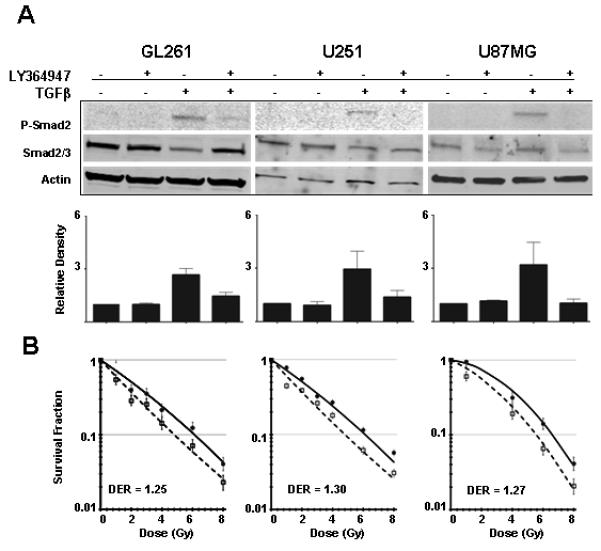
(A) Human (U251 and U87MG) and mouse (GL261) glioma cell lines respond to exogenous TGFβ1 (2 ng/mL), as measured by Smad phosphorylation, an effect that was blocked with the addition of the TGFβ type 1 receptor kinase inhibitor, LY364947. Representative immunoblots of three replicates and bar plots of densitometry quantitation of mean + S.E. are shown. (B) Clonogenic assay of GL261, U251, and U87MG glioma cells with (open symbols) and without (closed symbols) pre-treatment with LY364947 24 hours before radiation exposure shows that TGFβ inhibition significantly radiosensitizes all three glioma cell lines. The DER at 10% survival is between 1.25-1.30. Mean ± SD values of triplicate determinations are shown. GL261, P = 0.04; U251, P = 0.03; U87MG, P = 0.03, ANOVA with Tukey post-test.
Regardless of TGFβ-mediated growth modulation, addition of the TGFβ inhibitor LY364947 significantly increased the radiosensitivity of all three cell lines as measured in clonogenic assay (Figure 1B; GL261 p=0.04, U251 p=0.03, U87MG p=0.03, ANOVA with Tukey post-test). The dose enhancement ratio (DER) at 10% cell survival was 1.25-1.30, which indicates that 25% less dose was necessary to kill 90% of the cells by radiation when TGFβ signaling was blocked. LY364947 also significantly decreased radiation-induced phosphorylation of ATM Ser1981 and P53 Ser15 (Figure 2A). Consistent with prior studies in human and mouse epithelial cells (25, 28), radiosensitization was correlated with a significantly fewer γH2AX foci, a marker of DNA damage response (Figure 2B). These data indicate that TGFβ inhibition in glioma cells abrogates the response to DNA damage and increases radiosensitivity.
Figure 2. Radiosensitization of glioma cells by TGFβ inhibition correlates with abrogation of the DNA damage response.
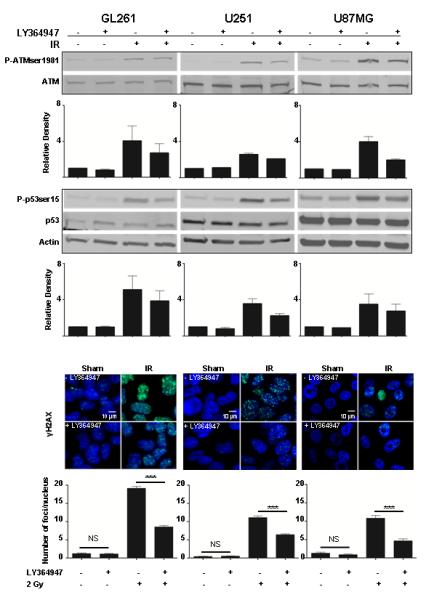
(A) Aspects of the DNA damage response of the murine GL261 and human U251 and U87 MG glioma cell lines assessed by immunoblotting cell lysates obtained 30 minutes after 2 Gy of ionizing radiation. Pre-treatment with TGFβ inhibitor LY364947 decreased IR-induced ATM phosphorylation at serine 1981 and p53 phosphorylation at serine 15 in all 3 cell lines. Representative immunoblots of 2-3 replicates and bar plots of densitometry quantitation of mean + S.E. are shown. (B) TGFβ inhibition with LY364947 significantly decreased γH2AX foci (green) in murine GL261 and human U251 and U87 MG glioma cells. Nuclei are counterstained with DAPI (blue). Quantification of γH2AX foci shown below each panel revealed a significant reduction in the number of radiation-induced γH2AX foci with LY354947 in GL261 cells (19.2+0.5 vs. 8.6+0.4) U251 cells (11.1+0.4 vs 6.4+0.2) and U87MG cells (10.8+0.7 vs 4.7+0.6). *** p<0.0001, ANOVA.
There are three pharmacological routes to blocking TGFβ: neutralizing the ligand, inhibiting expression, and truncating the signaling cascade (32). The pharmacokinetic properties of antibody and small molecule kinase inhibitors result in considerable differences in the duration of TGFβ signal modulation. Several TGFβ neutralizing antibodies that are in clinical development have demonstrated safety and efficacy in fibrotic disorders (32, 33). We compared in vitro efficacy of TGFβ ligand captured using 1D11 pan-TGFβ neutralizing antibodies to that of LY364947 in monolayer GL261 cells. Pre-treatment with pan-specific TGFβ neutralizing antibody, 1D11, produced the same level of radiosensitization as LY364947 (Figure 3A; p=0.04, ANOVA with Tukey post-test).
Figure 3. Treatment with 1D11 pan-specific TGFβ monoclonal antibody radiosensitizes glioma cells in vitro and in vivo.
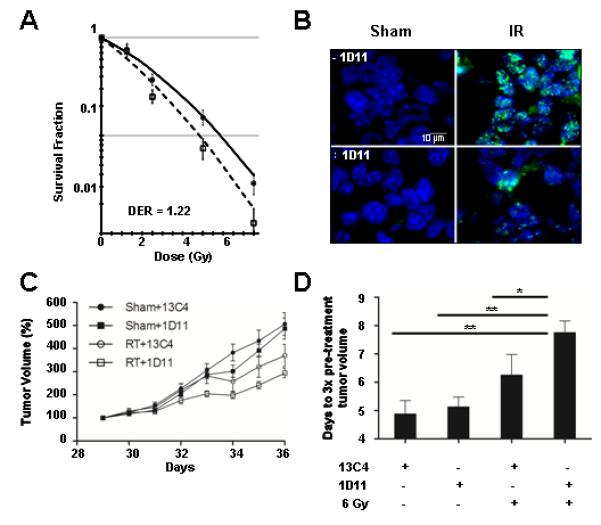
(A) Inhibition of TGFβ using a pan-specific monoclonal antibody, 1D11, resulted in similar level of radiosensitization (1.22 at 10% cell survival) as seen with the use of the small molecule inhibitor LY364947. P = 0.04, ANOVA with Tukey post-test. (B) Treatment with 1D11 neutralizing antibody resulted in decreased γH2AX immunofluorescence (green) 1 hr after 2 Gy compared to irradiated mice receiving control antibody 13C4. Nuclei are counterstained with DAPI (blue). (C) A single intraperitoneal injection of 1D11 antibody 24 hours prior to irradiation (6 Gy) of GL261 flank tumors resulted in greater tumor growth delay compared to mice receiving 13C4 control antibody. The y-axis represents tumor volume normalized to pre-treatment volume. RT/13C4 vs RT/1D11, p<0.05; RT/1D11 vs Sham/1D11, p<0.01; ANOVA with Newman-Keuls multiple comparison post-test (D) The time-to-reach 3 times pre-treatment volume was significantly increased by 1D11 treatment compared to antibody control 13C4. * p<0.05 and ** p<0.01, ANOVA with Newman-Keuls multiple comparison post-test.
To test whether increased radiosensitivity conferred therapeutic benefit, we established flank tumors of GL261 cells in C57bl mice. A single intraperitoneal injection of 1D11 antibody (10 mg/kg) did not affect tumor growth rate of established tumors (~150 mm3) compared to control antibody treated mice. Immunohistochemical detection of γH2AX in tumors harvested 1 hr after radiation was reduced in mice treated with 1D11 (Figure 3B). Consistent with inhibition of DNA damage recognition, the tumor growth delay of mice treated with 1D11 injected 24 hr before a single 6 Gy fraction was significantly increased compared to IR alone (Figure 3C-D).
Radiosensitivity of GL261 neurosphere formation
Tumor regrowth is thought to be, in large part, due to the relative response of GIC (5), hence we next examined the effect of TGFβ on GL261 neurosphere forming capacity as a surrogate of GIC. Addition of LY364947 to GL261 neurosphere cultures did not affect either primary or secondary neurosphere-forming capacity. Irradiation (2 Gy) of monolayer GL261 cells significantly decreased primary neurosphere-forming capacity by 28% (p<0.001; ANOVA). Addition of TGFβ inhibitor LY364947 decreased primary neurosphere formation by another 47% for a total reduction of 75% (Figure 4A; p<0.001; ANOVA). Surprisingly, irradiation of primary neurospheres did not affect secondary neurosphere formation. However, LY364947 treated, irradiated primary neurosphere cultures showed significantly decreased secondary neurosphere-forming capacity, leading to a 68% reduction (Figure 4B; p<0.001; ANOVA).
Figure 4. TGFβ inhibition in conjunction with radiation decreases GL261 neurosphere-forming capacity and radiosensitizes neurosphere-derived cells.
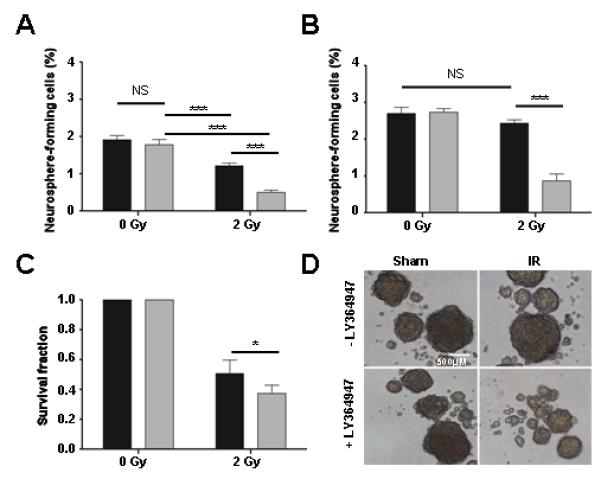
GL261 murine glioma cells were cultured under neurosphere conditions. (A) Treatment with LY364947 (24hrs, 400nM) alone had no effect on primary neurosphere formation. Irradiation (2Gy) decreased primary neurosphere-forming capacity by 28%, and LY364947 treatment for 24 hours prior to irradiation decreased neurosphere formation by an additional 47%, resulting in 75% fewer neurospheres. (B) Treatment with LY364947 (24hrs, 400nM) alone had no effect on secondary neurosphere formation. Irradiation of primary neurospheres had no effect on secondary neurosphere-forming capacity, yet LY364947 treatment of primary neurospheres before irradiation decreased secondary neurosphere formation by 68%. (C) Pre-treatment with LY364947 for 24 hr decreased neurosphere-derived clonogenic cell survival after irradiation (2Gy). After 2 Gy irradiation the survival fraction of untreated neurospheres was reduced by 43% while the survival fraction of neurospheres treated with 2Gy and LY354947 was further reduced by an additional 20%. (D) Representative images of neurospheres from sham, control-treated, sham, LY364947 -treated, 2Gy, control-treated, and 2Gy, LY364947 -treated. Data are means+S.D. of triplicate determinations and representative three experiments. NS, not significant, * p<0.05 and *** p<0.0001, ANOVA.
Given that secondary neurosphere formation, a measure of GIC self-renewal, was resistant to radiation but the majority of cells in a neurosphere are not GIC, we asked whether both populations were afforded the same degree of resistance by measuring clonogenic survival of GL261 cells dissociated from treated neurospheres. Consistent with monolayer cultures, radiation decreased colony forming efficiency and addition of TGFβ inhibitor LY364947 further increased the radiosensitivity of these cells by a similar magnitude to that effect seen in GL261 bulk culture. Irradiation (2 Gy) reduced the surviving fraction by 43% and the addition of LY354947 further reduced clonogenic survival by an additional 20% (Figure 4C). Thus, non-GICs were not afforded protection from IR in neurosphere culture. These data support the contention that GIC are specifically protected from radiation (8), but that this is conditional, i.e. specific to neurosphere culture.
TGFβ inhibition reduces DNA damage response in GL261 neurospheres and radiosensitizes the GIC population
We speculated that lack of radiation effect on secondary neurosphere formation, which is indicative of self-renewal, would be reflected in the DNA damage response. To test this, γH2AX foci induced by 2 Gy were measured at 30 minutes post-radiation in GL261 neurospheres (Figure 5A). Surprisingly, we observed that the number of foci per cell was 6 in neurosphere-derived cells versus 19 in monolayer culture at the same time after the same dose. The difference between monolayer and neurosphere culture could be either a failure to recognize damage, or a more effective DNA damage response, or both (34). Nonetheless, addition of TGFβ inhibitor LY364947 in neurosphere culture reduced radiation induced γH2AX foci by 5.2 fold (6.2 foci/nucleus vs 1.2 foci/nucleus) (Figure 5B), indicating that TGFβ inhibition compromised the molecular recognition of DNA damage in this population, as well as GL261 bulk culture.
Figure 5. TGFβ inhibition affects DNA damage response and self-renewal pathways up-regulated by ionizing radiation.
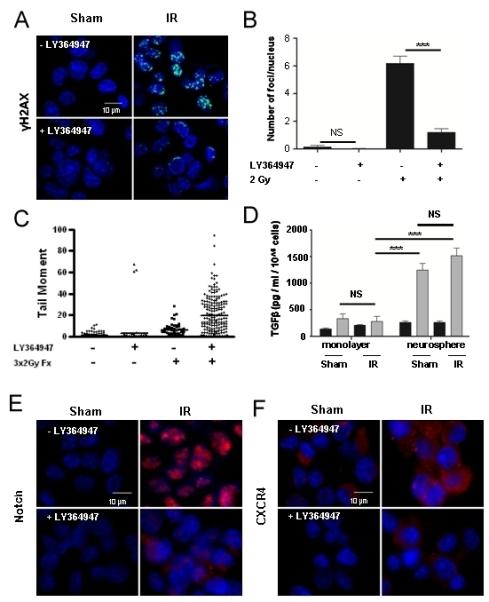
(A) LY364947 pre-treatment for 24 hours prior to 2 Gy decreased radiation-induced γH2AX foci in GL261 cells derived from neurosphere. (B) Quantification revealed an 81% decrease in the number of radiation-induced γH2AX foci with LY364947 treatment (6.2 +0.5 vs 1.2+0.3). (C) Comet assay was used to determine that LY364947 inhibited DNA repair following fractionated (3×2 Gy) daily radiation exposure of neuropsphere cultures. Data shown are representative of three experiments. Comets measured for control, N= 317; LY364947 treated, N=74; fractionated 3×2 Gy, N=52; LY364947 treated and fractionated 3×2, N=197. (D) TGFβ production by GL261 cells was measured in conditioned media obtained following 48 hours in monolayer or neurosphere growth conditions. GL261 cells grown under neurosphere conditions produced 3.7 fold more total and 1.9 fold more active TGFβ per cell than cells grown in monolayer culture. *** indicates p<0.0001, single-tailed unpaired t-test. (E) Inhibition of TGFβ with LY364947 also blocked radiation-induced Notch1 immunofluorescence (red), and (F) CXCR4 immunofluorescence (red), in GL261 cells dissociated from neurospheres three hours after irradiation with 2 Gy. Nuclei are counterstained with DAPI (blue).
These data suggest that DNA damage recognition is compromised and that fractionation, which is standard in radiotherapy, would amplify this effect and compromise DNA repair. To test this, we exposed GL261 neurospheres to 3 daily fractions of 2 Gy with or without LY364947 and performed comet assays to evaluate unresolved DNA damage at 24 hr after the final dose (Figure 5C). Radiation significantly increased the mean tail moment of cells isolated from neurospheres. The mean of those treated with radiation and LY364947 was 19.8+17 S.D. compared to 6+6 S.D. for fractionated radiation alone. Less than 5% of non-irradiated cells had tail moments above 10, compared to 17% for those treated with fractionated radiation and 64% of those from cells treated with the small molecule TGFβ signaling inhibition and fractionated radiation. These data suggest that TGFβ inhibition not only compromises DNA damage recognition but prevents DNA repair.
Our prior studies showed that Tgfb1 null cells fail to mount the full DNA damage response and were radiosensitive (25), as was observed by inhibiting TGFβ signaling in monolayer GBM cultures. Since GIC appeared to be radioresistant, but were sensitized by TGFβ blockade, we speculated that increased TGFβ production and/or activity could underlie their phenotype. Active and total TGFβ levels in media conditioned for seventy-two hours from GL261 monolayers or neurospheres were measured using the mink lung epithelial cell luciferase assay. As expected, the TGFβ2 isoform accounts for 85% of total TGFβ produced by cells grown under either condition, determined by isoform-specific neutralizing antibodies (data not shown). GL261 cells in neurosphere culture produced significantly more (3.7+1.4 fold, p<0.001, ANOVA) total TGFβ and a trend toward more active TGFβ (1.9+1.5 fold, p>0.05) than GL261 cells in monolayer culture (Figure 5D). Irradiation before conditioning media under either of the culture conditions did not significantly affect TGFβ levels. Thus, increased production of TGFβ by GL261 neurospheres could be protective as evidenced by a more effective molecular response to radiation induced DNA damage leading to radiation resistance.
To test the idea that TGFβ inhibition affects GIC self-renewal following IR, we examined CXCR4 and Notch1, which have been implicated in GIC self-renewal pathways (35, 36). Irradiation (2 Gy) of primary neurospheres significantly induced both markers, measured three hours following radiation treatment, which was blocked by TGFβ inhibition with LY364947 (Figure 5 E, F). Thus, we concluded that GIC are protected from radiation-induced cell kill by increased TGFβ production under conditions approximating the niche (i.e. neurosphere culture), which promotes effective DNA damage response and self-renewal via CXCR4 and Notch 1. Inhibition of TGFβ signaling compromises both mechanisms.
Discussion
GBM is a cancer characterized by a high degree of radioresistance, evidenced by inevitable local and/or disseminated recurrence. Our study suggests that high TGFβ levels confer resistance for both GIC and more differentiated tumor cells to DNA damage. Tumors have been described as “wounds that do not heal”, whose considerable similarities with the process of wound healing include TGFβ activity. Ionizing radiation is certainly another source of injury to tumor cells and tumor microenvironment, as well as surrounding normal tissue (20-22, 37). Pharmaceutical TGFβ inhibition circumvents this microenvironment mediated protection by compromising DNA damage recognition and therefore repair, as evidenced by increased clonogenic cell death and unrepaired DNA measured by comet assay. The increase in therapeutic index is likely even higher in situ, as there are multiple sources of TGFβ and multiple modes of action of TGFβ besides the DNA damage response on both cell behavior and tumor microenvironment. Indeed, several studies have demonstrated potential therapeutic benefit for TGFβ inhibition in preclinical glioma models, including anti-angiogenesis (38) and anti-invasion (39, 40).
Our previous work showed that genetic depletion of TGFβ1 compromises the DNA damage response in vivo and in vitro (25, 26) and we have now shown in vitro and in vivo radiosensitization with TGFβ inhibitors in breast cancer models (28). The negative impact of TGFβ on response to radiation therapy has recently been demonstrated in multiple tumor types, further illustrating the well-established pleotropic effects of this cytokine in the tumor microenvironment. Addition of TGFβ inhibitors improves radiation response in preclinical models of GBM (41, 42). Zhang et al. specifically reported that the addition of the small molecule inhibitor of TGFβ receptor type I and II kinase, LY2109761, to the current standard of care treatment, radiation and the oral alkylating agent temozolomide provided benefit. In addition to radiosensitization and tumor growth delay, TGFβ signaling blockade had anti-angiogenic and anti-migration effects as well. Mengxian et al. similarly reported radiosensitization, tumor growth delay, and improved survival with the addition of the same small molecule inhibitor of TGFβ, LY2109761, without combining with temozolomide. They further demonstrated that either TGFβ inhibition or radiation decreased self-renewal of glioma stem-like cells in a neurosphere assay, and a greater decrease when these were combined. Our study provides the explanation of these findings in that autocrine TGFβ potentiates an effective molecular DNA damage response as well as self-renewal.
TGFβ inhibitors are already in phase II/III clinical trials for fibrosis (32, 33). Phase I/II clinical studies using the antisense oligonucleotide AP-12009 (Antisense Pharma, Regensberg, Germany) to target TGFβ2 in recurrent or refractory WHO grade III or IV glioma demonstrated prolonged survival when compared to historical controls (43). More recently, a randomized phase IIb study of AP-12009 in patients with recurrent or refractory GBM or anaplastic astrocytoma showed tumor control rate superiority of lower dose AP-12009 over standard chemotherapy in anaplastic astrocytoma and comparable survival in GBM, with lower rates of toxicity observed with AP-12009 compared to chemotherapy (44).
Our study includes several aspects that add significantly to the growing body of evidence that TGFβ is a therapeutic target in GBM. First, we use both a small molecule inhibitor of TGFβ type I receptor kinase, LY364947, as well as a pan-specific TGFβ neutralizing antibody, 1D11, to target the TGFβ pathway in combination with IR. Potential advantages seen with clinical use of neutralizing antibodies to TGFβ or its receptor include their longer half-life and potentially more consistent inhibition than small molecule inhibitors, as well as targeting of all three TGFβ isoforms that could be beneficial by affecting not only tumor cells but also the tumor microenvironment. Although the blood brain barrier does prevent antibody entry into normal brain tissue, the situation is much more complex in the setting of GBM, in which the tumor itself can modulate the blood brain barrier’s permeability (45). Indeed, the pan-specific TGFβ neutralizing antibody 1D11 has been shown in preclinical orthotopic models to concentrate intratumorally (46). Further, the recent success with bevacizumab (Avastin®, an anti-VEGF monoclonal antibody) in both recurrent and newly-diagnosed GBM highlights the feasibility of therapeutic antibodies in CNS tumors (reviewed in (47)). Although outcomes have improved, concern over the altered pattern of relapse in bevacizumab-treated GBM patients, characterized by distant infiltration of the brain by tumors that demonstrate increased invasiveness, has emerged (48). Combination therapy with strategies to inhibit invasion have been proposed, and the TGFβ pathway is a logical approach given the documented role TGFβ plays in glioma migration and invasion (11).
The magnitude of radiosensitization seen in the current study (DER ~1.25 by clonogenic assay) must be taken into context of a disease as difficult to treat as GBM. The dose enhancement ratios currently reported are well within the level of radiosensitization seen in models of glioma. Zheng et al. used siRNA silencing of TNF receptor-associated Factor 2 (TRAF2) to radiosensitize U251 glioma cells in vitro with a DER of 1.2-1.39 (49). Golding et al. reported radiosensization with DERs of 1.6-2.1 in vitro using ATM kinase inhibitors KU-55933 in U87MG glioma cells (50) and KU-60019 in U1242 glioma cells (51). Our study showed that ATM kinase activity is reduced with TGFβ inhibition. More importantly, Kil et al. demonstrated a DER of 1.32 using U251 glioma cells treated with temozolomide (52). Considering that the addition of temozolomide to radiation therapy in the treatment of GBM was one of the largest breakthroughs in this disease in decades and is now considered standard of care, radiosensitization of this magnitude reported here must be considered significant, particularly since the radiation sensitivity of GIC increased nearly 3-fold.
Debate exists about whether GIC are more (8) or less (53) radioresistant than the tumor cell population as a whole, but it is thought that the GIC population contributes to the inevitable recurrence of GBM (54). Several recent studies have shown that the TGFβ pathway is important GIC biology (17-19). While we did not observe inhibition of neurosphere self-renewal with TGFβ inhibition alone (seen in several of the above mentioned studies), we did find that TGFβ inhibition in combination with IR prevents self-renewal mediated by CXCR4 and Notch1. These data resolve the paradoxical literature as to GIC radiation sensitivity. We postulate that TGFβ production in the GIC niche is evidence of microenvironment mediated resistance, and as such represents a very promising target to improve GBM radiotherapy and provide multi-faceted benefits that could prevent GBM recurrence.
Acknowledgments
1D11 and 13C4 antibody was provided by Genzyme Inc. Funding for this study was provided by NYU Department of Radiation Oncology, NCI Integrative Cancer Biology Program, U54-CA149233 and the NCI Training Program in Molecular Oncology and Immunology, 5 T32 CA009161-36 (CMMR).
Abbreviations
- TGFβ
transforming growth factor β
- GBM
glioblastoma
- GIC
glioma-initiating cell
- IR
ionizing radiation
- DER
dose enhancement ratio
- LAP
latency-associated peptide
- ATM
ataxia telangiectasia mutated
- PI3-kinase
phosphatidylinositol 3-kinase
- DAPI
4′,6-diamidino-2-phenylindole
Footnotes
Conflict of Interest: SML is employed by Genzyme, Inc.
References
- 1.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 4.Rosenblum ML, Gerosa M, Dougherty DV, Reese C, Barger GR, Davis RL, et al. Age-related chemosensitivity of stem cells from human malignant brain tumours. Lancet. 1982;1:885–7. doi: 10.1016/s0140-6736(82)92154-7. [DOI] [PubMed] [Google Scholar]
- 5.Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res. 2009;7:989–99. doi: 10.1158/1541-7786.MCR-09-0030. [DOI] [PubMed] [Google Scholar]
- 6.Castellino RC, Durden DL. Mechanisms of disease: the PI3K-Akt-PTEN signaling node--an intercept point for the control of angiogenesis in brain tumors. Nat Clin Pract Neurol. 2007;3:682–93. doi: 10.1038/ncpneuro0661. [DOI] [PubMed] [Google Scholar]
- 7.Hambardzumyan D, Squatrito M, Carbajal E, Holland EC. Glioma formation, cancer stem cells, and akt signaling. Stem Cell Rev. 2008;4:203–10. doi: 10.1007/s12015-008-9021-5. [DOI] [PubMed] [Google Scholar]
- 8.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 9.Mannino M, Chalmers AJ. Radioresistance of glioma stem cells: Intrinsic characteristic or property of the ‘microenvironment-stem cell unit’? Molecular Oncology. 2011;5:374–86. doi: 10.1016/j.molonc.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamal M, Rath BH, Williams ES, Camphausen K, Tofilon PJ. Microenvironmental regulation of glioblastoma radioresponse. Clin Cancer Res. 2010;16:6049–59. doi: 10.1158/1078-0432.CCR-10-2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barcellos-Hoff MH, Newcomb EW, Zagzag D, Narayana A. Therapeutic targets in malignant glioblastoma microenvironment. Semin Radiat Oncol. 2009;19:163–70. doi: 10.1016/j.semradonc.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jachimczak P, Hessdorfer B, Fabel-Schulte K, Wismeth C, B W, Schlingensiepen KH, et al. Transforming growth factor-beta-mediated autocrine growth regulation of gliomas as detected with phosphorothioate antisense oligonucleotides. Int J Cancer. 1996;65:332. doi: 10.1002/(SICI)1097-0215(19960126)65:3<332::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 13.Hau P, Jachimczak P, Schlaier J, Bogdahn U. TGF-β2 Signaling in High-Grade Gliomas. Curr Pharm Biotechnol. 2011 doi: 10.2174/138920111798808347. [DOI] [PubMed] [Google Scholar]
- 14.Baritaki S, Chatzinikola AM, Vakis AF, Soulitzis N, Karabetsos DA, Neonakis I, et al. YY1 Over-expression in human brain gliomas and meningiomas correlates with TGF-beta1, IGF-1 and FGF-2 mRNA levels. Cancer Invest. 2009;27:184–92. doi: 10.1080/07357900802210760. [DOI] [PubMed] [Google Scholar]
- 15.Pera MF, Tam PP. Extrinsic regulation of pluripotent stem cells. Nature. 2010;465:713–20. doi: 10.1038/nature09228. [DOI] [PubMed] [Google Scholar]
- 16.Aigner L, Bogdahn U. TGF-beta in neural stem cells and in tumors of the central nervous system. Cell Tissue Res. 2008;331:225–41. doi: 10.1007/s00441-007-0466-7. [DOI] [PubMed] [Google Scholar]
- 17.Peñuelas S, Anido J, Prieto-Sánchez RM, Folch G, Barba I, Cuartas I, et al. TGF-[beta] Increases Glioma-Initiating Cell Self-Renewal through the Induction of LIF in Human Glioblastoma. Cancer Cell. 2009;15:315–27. doi: 10.1016/j.ccr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5:504–14. doi: 10.1016/j.stem.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 19.Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell. 2010;18:655–68. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 20.Barcellos-Hoff MH. Radiation-induced transforming growth factor β and subsequent extracellular matrix reorganization in murine mammary gland. Cancer Res. 1993;53:3880–6. [PubMed] [Google Scholar]
- 21.Ehrhart EJ, Carroll A, Segarini P, Tsang ML-S, Barcellos-Hoff MH. Latent transforming growth factor-β activation in situ: Quantitative and functional evidence following low dose irradiation. FASEB J. 1997;11:991–1002. doi: 10.1096/fasebj.11.12.9337152. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Zheng H, Sung C-C, Richter KK, Hauer-Jensen M. Cellular Sources of Transforming Growth Factor-ß Isoforms in Early and Chronic Radiation Enteropathy. Am J Pathol. 1998;153:1531–40. doi: 10.1016/s0002-9440(10)65741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-β1. Molec Endocrin. 1996;10:1077–83. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 24.Jobling MF, Mott JD, Finnegan M, Erickson AC, Taylor SE, Ledbetter S, et al. Isoform specificity of redox-mediated TGF-β activation. Radiat Res. 2006;166:839–48. doi: 10.1667/RR0695.1. [DOI] [PubMed] [Google Scholar]
- 25.Kirshner J, Jobling MF, Pajares MJ, Ravani SA, Glick A, Lavin M, et al. Inhibition of TGFβ1 signaling attenuates ATM activity in response to genotoxic stress. Cancer Res. 2006;66:10861–68. doi: 10.1158/0008-5472.CAN-06-2565. [DOI] [PubMed] [Google Scholar]
- 26.Wiegman EM, Blaese MA, Loeffler H, Coppes RP, Rodemann HP. TGFbeta-1 dependent fast stimulation of ATM and p53 phosphorylation following exposure to ionizing radiation does not involve TGFbeta-receptor I signalling. Radiother Oncol. 2007;83:289–95. doi: 10.1016/j.radonc.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 27.Kanamoto T, Hellman U, Heldin CH, Souchelnytskyi S. Functional proteomics of transforming growth factor-beta1-stimulated Mv1Lu epithelial cells: Rad51 as a target of TGFbeta1-dependent regulation of DNA repair. EMBO J. 2002;21:1219–30. doi: 10.1093/emboj/21.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouquet SF, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, et al. Transforming growth factor ®1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res. 2011;17:6754–65. doi: 10.1158/1078-0432.CCR-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Analyt Biochem. 1994;216:276–84. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 30.Brown PD, Wakefield LM, Levinson AD, Sporn MB. Physicochemical activation of recombinant latent transforming growth factor-beta’s 1, 2, and 3. Growth Factors. 1990;3:35–43. doi: 10.3109/08977199009037500. [DOI] [PubMed] [Google Scholar]
- 31.Costes SV, Ponomarev A, Chen JL, Nguyen D, Cucinotta FA, Barcellos-Hoff MH. Image-Based Modeling Reveals Dynamic Redistribution of DNA Damage into Nuclear Sub-Domains. PLoS Comput Biol. 2007;3:e155. doi: 10.1371/journal.pcbi.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3:1011–22. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- 33.Akhurst RJ. Large- and small-molecule inhibitors of transforming growth factor-beta signaling. Curr Opin Investig Drugs. 2006;7:513–21. [PubMed] [Google Scholar]
- 34.Neumaier T, Swenson J, Pham C, Polyzos A, Lo AT, Yang P, et al. Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells. Proceedings of the National Academy of Sciences. 2012;109:443–8. doi: 10.1073/pnas.1117849108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schulte A, Gunther HS, Phillips HS, Kemming D, Martens T, Kharbanda S, et al. A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia. 2011;59:590–602. doi: 10.1002/glia.21127. [DOI] [PubMed] [Google Scholar]
- 36.Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hauer-Jensen M, Richter KK, Wang J, Abe E, Sung CC, Hardin JW. Changes in transforming growth factor beta1 gene expression and immunoreactivity levels during development of chronic radiation enteropathy. Radiat Res. 1998;150:673–80. [PubMed] [Google Scholar]
- 38.Pen A, Moreno MJ, Durocher Y, Deb-Rinker P, Stanimirovic DB. Glioblastoma-secreted factors induce IGFBP7 and angiogenesis by modulating Smad-2-dependent TGF-beta signaling. Oncogene. 2008;27:6834–44. doi: 10.1038/onc.2008.287. [DOI] [PubMed] [Google Scholar]
- 39.Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, et al. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion--an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene. 2008;27:918–30. doi: 10.1038/sj.onc.1210683. [DOI] [PubMed] [Google Scholar]
- 40.Baumann F, Leukel P, Doerfelt A, Beier CP, Dettmer K, Oefner PJ, et al. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009;11:368–80. doi: 10.1215/15228517-2008-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M, Herion TW, Timke C, Han N, Hauser K, Weber KJ, et al. Trimodal Glioblastoma Treatment Consisting of Concurrent Radiotherapy, Temozolomide, and the Novel TGF-β Receptor I Kinase Inhibitor LY2109761. Neoplasia. 2011;13:537–49. doi: 10.1593/neo.11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mengxian Z, Kleber S, Roehrich M, Timke C, Han N, Tuettenberg J, et al. Blockade of TGF-beta signaling by the TGF{beta}R-I kinase Inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-11-1212. [DOI] [PubMed] [Google Scholar]
- 43.Hau P, Jachimczak P, Schlingensiepen R, Schulmeyer F, Jauch T, Steinbrecher A, et al. Inhibition of TGF-β2 with AP 12009 in Recurrent Malignant Gliomas: From Preclinical to Phase I/II Studies. Oligonucleotides. 2007;17:201–12. doi: 10.1089/oli.2006.0053. [DOI] [PubMed] [Google Scholar]
- 44.Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro-Oncology. 2011;13:132–42. doi: 10.1093/neuonc/noq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lampson LA. Monoclonal antibodies in neuro-oncology: Getting past the blood-brain barrier. MAbs. 2011;3:153–60. doi: 10.4161/mabs.3.2.14239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hulper P, Schulz-Schaeffer W, Dullin C, Hoffmann P, Harper J, Kurtzberg L, et al. Tumor localization of an anti-TGF-beta antibody and its effects on gliomas. Int J Oncol. 2011;38:51–9. [PubMed] [Google Scholar]
- 47.Beal K, Abrey LE, Gutin PH. Antiangiogenic agents in the treatment of recurrent or newly diagnosed glioblastoma: analysis of single-agent and combined modality approaches. Radiat Oncol. 2011;6:2. doi: 10.1186/1748-717X-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Narayana A, Kunnakkat SD, Medabalmi P, Golfinos J, Parker E, Knopp E, et al. Change in Pattern of Relapse After Antiangiogenic Therapy in High-Grade Glioma. Int J Radiat Oncol Biol Phys. 2010 doi: 10.1016/j.ijrobp.2010.10.038. [DOI] [PubMed] [Google Scholar]
- 49.Zheng M, Morgan-Lappe SE, Yang J, Bockbrader KM, Pamarthy D, Thomas D, et al. Growth inhibition and radiosensitization of glioblastoma and lung cancer cells by small interfering RNA silencing of tumor necrosis factor receptor-associated factor 2. Cancer Res. 2008;68:7570–8. doi: 10.1158/0008-5472.CAN-08-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009;8:2894–902. doi: 10.1158/1535-7163.MCT-09-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Golding SE, Rosenberg E, Adams BR, Wignarajah S, Beckta JM, O’Connor MJ, et al. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle. 2012;11:1167–73. doi: 10.4161/cc.11.6.19576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kil WJ, Cerna D, Burgan WE, Beam K, Carter D, Steeg PS, et al. In vitro and in vivo radiosensitization induced by the DNA methylating agent temozolomide. Clin Cancer Res. 2008;14:931–8. doi: 10.1158/1078-0432.CCR-07-1856. [DOI] [PubMed] [Google Scholar]
- 53.McCord AM, Jamal M, Williams ES, Camphausen K, Tofilon PJ. CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin Cancer Res. 2009;15:5145–53. doi: 10.1158/1078-0432.CCR-09-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krause M, Yaromina A, Eicheler W, Koch U, Baumann M. Cancer Stem Cells: Targets and Potential Biomarkers for Radiotherapy. Clinical Cancer Research. 2011;17:7224–9. doi: 10.1158/1078-0432.CCR-10-2639. [DOI] [PubMed] [Google Scholar]