

Abstract
The −1G mutant HBV is more prevalent in individuals co-infected with HIV/HBV than in individuals infected with HBV alone and in some cases is the dominant virus in circulation. This mutant is created by the deletion of a dGMP (−1G) from the guanine rich homopolymer sequence located at nts 2,085–2,090 (numbering from EcoRI site as position 1) in the HBV core gene. This deletion causes a frameshift generating a premature stop codon at 64Asn in the HBV core gene (codon 93 in the precore gene), that truncates the precore protein, precursor of the secreted hepatitis B “e” antigen (HBeAg), and the core protein which forms the viral nucleocapsid. However, the replication phenotype of the −1G mutant HBV is unknown. An in vitro cell culture model in which hepatoma cells were transiently transfected with infectious cDNAs was used to show that the −1G mutant HBV is incapable of autonomous replication and, as expected, replication was restored to wild-type (wt) levels by supplying HBV core protein in trans. Although the −1G mutation had no deleterious effect on intracellular HBV-DNA levels, high levels of −1G mutant HBV relative to wt HBV reduced virus secretion and HBeAg secretion relative to empty vector controls. Importantly, the −1G mutant HBV also caused intracellular retention of truncated precore protein in the endoplasmic reticulum (ER) and Golgi apparatus. Together, these effects may be contributing to the increased pathology observed in the setting of HIV/HBV co-infection.
Keywords: HBV, HIV, co-infection, precore, HBeAg, virus replication, ER stress
INTRODUCTION
Despite the availability of an effective vaccine, infection with the hepatitis B virus (HBV) remains a global health problem, with an estimated 400 million people living with chronic hepatitis B infection [Leemans et al., 2007]. HBV is the 10th leading cause of mortality [Lavanchy, 2004], resulting in at least 1.2 million deaths annually from liver cirrhosis and hepatocellular carcinoma [Leemans et al., 2007]. Co-infection of HBV with human immunodeficiency virus (HIV) is common due to shared modes of transmission and in the USA and Australia, up to 6% of those infected with HIV have active HBV co-infection [Rustgi et al., 1984]. Co-infection with HIV alters the natural history of chronic HBV infection [Thio et al., 2002], resulting in higher levels of HBV replication [Colin et al., 1999], lower rates of HBeAg seroconversion and rapid progression of liver disease [Gilson et al., 1997] for reasons that remain unclear. Studies using in vitro cell culture models show that co-infection with HIV increases the level of hepatitis B surface antigen (HBsAg) [Iser et al., 2010], although it is likely that multiple factors contribute to the increased pathogenicity observed in co-infected individuals.
A novel HBV precore/core mutant was identified previously that was significantly more prevalent in individuals from the USA and Australia that were co-infected with HIV [Revill et al., 2007]. This mutant was created by the deletion of a dGMP (−1G) from the guanine-rich homopolymer sequence located at nts 2,085–2,090 (numbering from EcoRI site as position 1) in the HBV core gene. This deletion causes a frameshift generating a premature stop codon at 64Asn in the HBV core gene (codon 93 in the precore gene), resulting in truncation of their predicted protein products [Revill et al., 2007]. The −1G mutant HBV has also been identified in a Japanese patient co-infected with HIV and HBV [Fukushima et al., 2008] and most recently an identical mutant has been identified in HBV mono-infected patients infected with subgenotype A1 who progressed rapidly to hepatocellular carcinoma. To date, this mutant has only been identified in HBV genotypes A and D [Revill et al., 2007; Fukushima et al., 2008; Skelton et al., 2012].
The wild-type (wt) core gene encodes a 21-kDa protein that forms the viral nucleocapsid and is a prerequisite for HBV replication, whereas the 25-kDa precore protein is the precursor of the secreted hepatitis B “e” antigen (HBeAg) which has no role in HBV replication but is required for the establishment of chronic infection [Hadziyannis, 1995] and is an important regulator of innate and adaptive immune responses [Milich et al., 1990; Visvanathan et al., 2007; Lang et al., 2011; Wilson et al., 2011] Interestingly, although the −1G mutation prematurely truncates the precore/HBeAg protein coding sequence, all patients in which the mutant has been identified to date were HBeAg positive. In some persons co-infected with HIV, the −1G mutant HBV forms up to 90% of the HBV quasispecies pool and has also been associated with high-serum HBV-DNA levels in vivo [Revill et al., 2007]. However, the replication efficiency and protein expression of this HBV mutant is unknown. Accordingly, this study investigated the in vitro replication phenotype of the −1G mutant HBV using Huh7 cells transiently transfected with −1G mutant and wt infectious cDNAs (genotype A2) and examined the subcellular localization of the −1G-encoded precore protein by confocal microscopy.
MATERIALS AND METHODS
Infectious cDNA Plasmids
Replication studies were performed using pBlue-script KS+ plasmids (Stratagene, La Jolla, CA) into which a 1.5-times genome-length copy of wt HBV genotype A, subtype adw2, was inserted [Bock et al., 1997; Chin et al., 2001]. In order to create a plasmid expressing −1G mutant HBV, deletion of nt G2090 from the C gene was accomplished by site-directed mutagenesis (SDM) using primer pairs −1G 1.5plus (2068GCAAGCCATTCTCTGCTGGGGGAATTGATGACTCTAGCTACC2110) and −1G 1.5minus (2110GGTAGCTAGAGTCATCAATTCCCCCAGCAGAGAATGGCTTGC2068) and a Quickchange SDM kit (Stratagene), as per manufacturer’s instructions.
Transfection of Huh7 Cells With HBV
Huh7 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM, Gibco BRL, New York, NY) supplemented with 10% fetal calf serum at 37°C in a humidified environment containing 5% CO2. Cells were seeded into 60 mm dishes or 12-well plates and each dish/well was transfected using Fugene6 transfection reagent (Roche Diagnostics, Mannheim, Germany), with 3.8 or 0.8 μg of plasmid DNA, respectively, as previously described [Chin et al., 2001]. Plasmid vectors encoding −1G mutant HBV and/or wt HBV were co-transfected with CMV-driven plasmids expressing the HBV core protein (pCIcore), precore protein (pCIprecore) [Wilson et al., 2011] or the empty vector (pCIev) at ratios of 1:1, 1:4, and 4:1 (Table I), as previously described [Gunther et al., 2000; Marschenz et al., 2006]. Briefly, the concentration of the wt or −1G DNA used in transfection varied, depending on the ratio used in the experiment. Therefore, if a sample was transfected with −1G: wt at 4:1, four times more −1G DNA was transfected than wt. However, the total concentration of DNA remained constant. Since transfection of a 60 mm dish required that 3.8 μg of DNA be added, a 4:1 −1G:wt ratio required 3.04 μg of −1G DNA and 0.76 μg of wt DNA, respectively. A 1:1 ratio indicated an equal amount of −1G and wt DNA (1.9 μg each) was transfected and a 1:4 ratio meant four times as much wt DNA was transfected, relative to −1G DNA. Transfection efficiency was monitored by measuring expression of secreted alkaline phosphatase (SEAP) after co-transfection with 0.1 μg of a SEAP-expressing plasmid, as previously described [Gunther et al., 2000].
TABLE I.
Ratios of Plasmid Constructs Used in Transfection Experiments
| Construct | Ratio | ||
|---|---|---|---|
| wt/pCIev | 1:1 | 4:1 | 1:4 |
| wt/pCIcore | 1:1 | 4:1 | 1:4 |
| −1G/pCIev | 1:1 | ||
| −1G/pCIcore | 1:1 | 4:1 | 1:4 |
| wt/−1G | 1:1 | 4:1 | 1:4 |
| −1G/pCIprecore | 1:1 | ||
Analysis of HBV DNA
HBV DNA isolated from intracellular HBV core particles harvested 5 days post-transfection was detected by Southern blotting and autoradiography as described previously [Chin et al., 2001]. Virions were immunoprecipitated from cell culture supernatant prior to Southern blotting to exclude unenveloped nucleocapsids, using previously described methods [Warner and Locarnini, 2008]. Image densities from at least five sets of independent experiments were quantified by densitometry and compared by one-way analysis of variance with multiple paired comparisons by Holm–Sidak tests, with the aid of SigmaStat (v2.0; SPSS, Chicago, IL). Differences giving P-values of ≤0.05 were considered significant.
Northern Hybridization Analysis of Core-Associated RNA
Virion-encapsidated pregenomic RNA (pgRNA) was isolated from intracellular core particles using QIAGEN Viral RNA Extraction Kits, which utilizes DNAse and RNAse treatment following cell lysis. This was followed by Proteinase K digestion, facilitating release of core-associated pgRNA. RNA purity and concentration were determined using a bioanalyzer (Waldbronn, Karlsruhe, Germany), or spectrophotometrically and 10 μg of RNA from each sample was electrophoresed through 1% agarose glyoxal gels before transfer to nylon membranes using standard protocols. Membranes were probed using a genomic length HBV-DNA probe, described previously [Chin et al., 2001] and pgRNA levels from three independent experiments were quantified by densitometry.
Western Blotting
Intracellular antigens in cell lysates were detected as previously described [Warner and Locarnini, 2008] using antibodies against the precore/HBeAg, HBcAg, or HBsAg. The anti-HBe-specific antibody (PRM-3) was prepared against the 10 unique amino acids at the HBeAg N-terminus (SKLCLGWLWG), which is highly conserved in all orthohepadnaviruses [Revill et al., 2010]. To generate the inoculum, a 10 mer peptide was synthesized by Mimotopes (Clayton, Victoria, Australia) and for stability purposes the cysteine at position 4 was modified to serine. Polyclonal rabbit antiserum was produced by IMVS (Adelaide, South Australia) following four intramuscular inoculations (1 mg each inoculation). Specificity of PRM-3 for the HBV precore protein and HBeAg was determined by immunoblotting and ELISA (not shown). A polyclonal anti-HBc antibody was purchased from Dako (Capinteria, CA; Cat. No. B0586) and anti-HBs IgM was kindly provided by Dr. Paul Coleman of Abbott Diagnostics (Chicago, IL). To examine viral antigen secretion, proteins were precipitated from cell culture supernatants using polyethylene glycol (PEG) or methanol using standard protocols.
Quantitative HBeAg Analysis
HBeAg concentrations in cell culture supernatants were measured by chemiluminescent microparticle immunoassay (CMIA) using the ARCHITECT HBeAg assay from Abbott Diagnostics. A 100 PE IU/ml HBeAg standard, obtained from the Paul Ehrlich Institute (Paul–Ehrlich Institute, Bundesamt fur Sera und Impstoffe, Paul–Ehrlich Strasse 51–59, D-63225 Langen, Germany) was used to calibrate a set of in-house HBeAg standards, prepared from a pool of high-titer HBeAg positive sera by diluting with ARCHITECT HBsAg diluent (which does not react with HBsAg, HIV-1 RNA or HIV-1 Ag, anti-HIV-1/HIV-2, anti-HCV, or anti-HBs). Standards were stored at −20°C. Replicate standards were run to validate the assay. Samples were analyzed by linear regression using the method of Fried et al. [2008] and when results were outside the linear range of the assay, the corresponding samples were serially diluted and re-assayed. HBeAg-negative human serum was used as a negative control.
Confocal Microscopy
Huh7 cells were seeded onto sterilized 25 mm diameter coverslips in six-well tissue culture plates. The next day, cells were transfected with wt HBV or −1G mutant HBV plasmids, with or without the rescue plasmid (pCIcore) or empty vector control. Cells were fixed in absolute methanol 2 days post-transfection and blocked with 5% skim milk/PBS + 0.2% Tween 20 for 1 hr at 37°C. After staining cell nuclei with 4′,6-diamidino-2-phenylindole (DAPI), cell organelles were tagged with specific antibodies: anti-calnexin for endoplasmic reticulum (ER) staining (AB2798) and anti-Golgi zone (AB14490; both from Abcam, Cambridge, MA) to identify the ER and Golgi apparatus, respectively, as well as anti-HBe (PRM-3). The secondary antibodies (also from Abcam) were anti-mouse conjugated to FITC (AB6785) used to visualize the cellular organelles and anti-rabbit conjugated to Texas Red (AB6719), to visualize the full-length and truncated HBeAg. All primary and secondary antibody-binding steps were performed at 37°C, with antibodies diluted in the blocking solution. After staining, the coverslips were washed in PBS + 0.2% Tween, then PBS, before mounting and examination using a Leica TCS SP2 Laser Scanning Confocal Microscope. To ensure that observed proteins were not artefacts of the staining procedures, a range of controls were used, including analysis of un-transfected cells using anti-HBc and ER/Golgi antibodies, as well as antibody controls (Supplementary Fig. 1).
ER Stress Assay
To determine if −1G mutant HBV altered GRP78 mRNA expression, we quantified GRP78 mRNA in Huh-7 cells by real-time PCR. RNA was extracted as described previously and its quality determined using an Agilent Bioanalyser. cDNA was synthesized using a High capacity RT kit (ABI) using RNA with an Agilent Bioanalyser RNA integrity number of ≥8, indicative of high-quality RNA. GRP78 mRNA expression was measured by real-time PCR using a Taqman HSPA5 Assays-On-Demand Gene Expression probe (Hs99999174.m1 probe; Applied Biosystems, Foster City, CA) and the ABI FAST 7500 Sequence Detection System (ABI). The relative quantities of mRNA were determined by combining data from three independent experiments, following normalization to an RPLP0 ribosomal cDNA internal control. To confirm that any observed reduction in GRP78 expression was not due to cell death, total protein in cells was measured using a Bio-Rad microtiter DC protein assay and total rRNA production was determined using the Agilent bioanalyser, according to the manufacturer’s instructions.
RESULTS
In Vitro Viral Replication Studies
To determine the replication phenotype of the genotype A −1G mutant HBV and its effect on wt HBV replication, permissive Huh7 cells were transfected with a replication competent infectious cDNA encoding the −1G mutation. HBV replication was assessed by Southern/Northern hybridization, measuring intracellular and secreted viral DNA and core-associated viral pre-genomic RNA.
HBV core-associated DNA
Densitometric analysis of viral core-associated DNA analyzed by Southern hybridization (following correction for transfection efficiency) showed that the −1G mutant HBV was incapable of autonomous replication as expected (Fig. 1, lane 8), whereas its replication was restored by co-transfection with a CMV-driven HBV core protein expressing plasmid (pCIcore; Fig. 1, lane 9). Following rescue, −1G mutant HBV (Fig. 1, lanes 9–11) displayed a replication capacity similar to that of wt HBV (Fig. 1, lanes 5–7). Co-transfection with mixtures of −1G mutant HBV and wt HBV at different ratios, showed that transfection of mutant:wt at a 1:1 ratio (Fig. 1, lane 12), or 1:4 ratio (Fig. 1, lane 13) resulted in levels of DNA replication similar to transfection with high levels of wt HBV alone (Fig. 1, lane 4). However, increasing the mutant:wt ratio to 4:1 (Fig. 1, lane 14) markedly reduced the level of intracellular core-associated DNA, reflecting the inability of −1G mutant HBV to replicate autonomously in the absence of sufficient core protein. In summary, the −1G mutant HBV did not have a high replication phenotype and high levels of mutant reduced overall HBV replication.
Fig. 1.
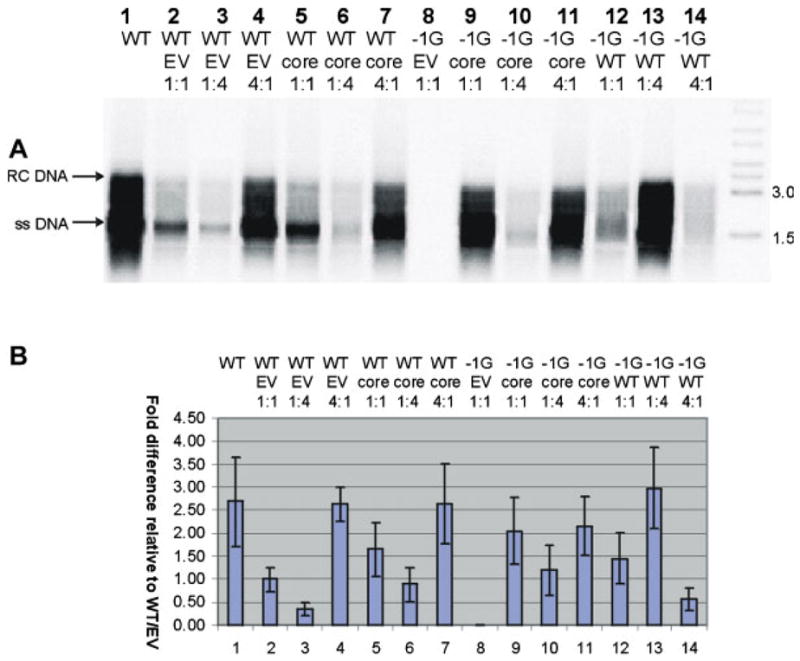
A: Representative autoradiograph of a Southern blot of intracellular core-associated DNA, following transfection of Huh7 cells with different ratios of −1G mutant and/or wt HBV (WT), with or without core expressing plasmid (pCIcore = core) or empty vector (EV). The initials RC and SS on the left hand side of (A) indicate relaxed circular and single-stranded HBV DNA, respectively. B. Differences in amounts of intracellular HBV DNA extracted from cells transfected with different ratios of −1G mutant and or wt HBV, following correction for transfection efficiency and quantification of Southern autoradigraphs by densitometry. Columns represent means of results from five independent experiments; error bars indicate standard errors of the means. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jmv]
Secreted virion DNA
We then asked whether the −1G mutation altered HBV virion secretion. Southern hybridization of extracellular virion DNA showed that following rescue with the HBV capsid protein, significantly less virion DNA was secreted following co-transfection with −1G mutant HBV compared with wt HBV (Fig. 2, lanes 5 vs. 9). Additionally, following co-transfection of the −1G mutant HBV with wt HBV at a ratio of 1:4, significantly less extracellular DNA was detected than in the supernatant of cells transfected with wt HBV and an equivalent amount of empty vector (Fig. 2, lanes 4 vs. 13). This suggests that the co-presence of −1G mutant HBV had a direct effect on viral secretion.
Fig. 2.
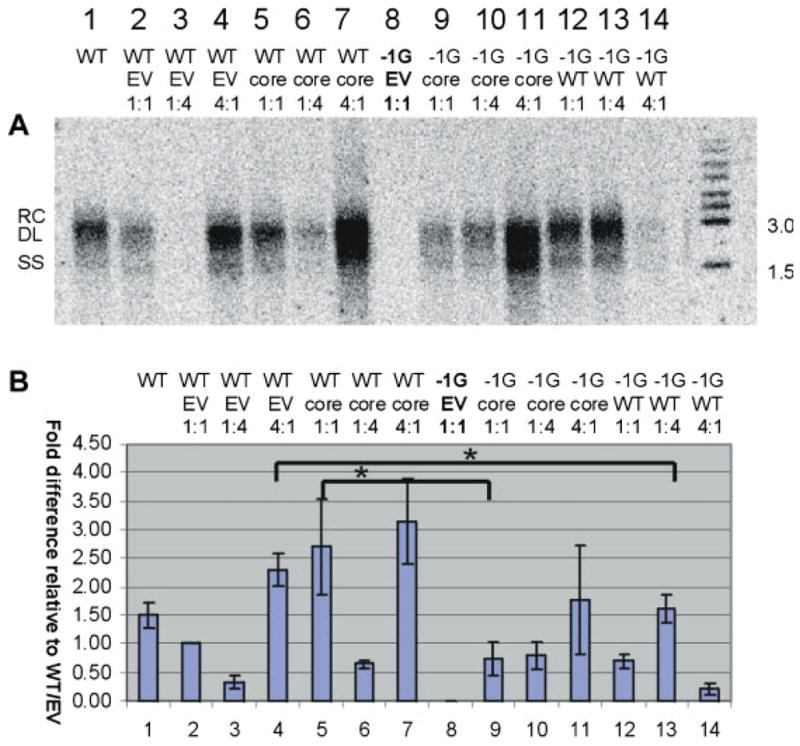
A: Representative autoradiograph of a Southern blot of extracellular virion DNA following immunoprecipitation of cell culture supernatant with anti-HBs. The initials RC, DL, and SS on the left hand side of (A) indicate relaxed circular, duplex linear and single-stranded HBV DNA, respectively. B: Differences in amounts of extracellular virion DNA extracted from cell culture supernatant, following correction for transfection efficiency and quantification by densitometry. Columns represent means of results of five independent experiments; error bars represent the SE. Statistically different treatments of interest are indicated by *P < 0.05. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jmv]
HBV pregenomic RNA
Northern hybridization analysis of core-associated HBV RNA showed that the relative amounts of 3.5 kb pregenomic RNA (pgRNA, Fig. 3) in each experiment were generally proportional to the amount of core-associated HBV DNA present (Fig. 1). pgRNA was not detected in core preparations extracted from cells transfected with −1G mutant HBV alone (Fig. 3, lane 7), reflecting the failure of this mutant to produce nucleocapsids. Although there was a trend towards increased pgRNA in capsids isolated from cells transfected with high levels of −1G mutant HBV following rescue with core plasmid (Fig. 3A and B, lane 10) relative to wt HBV (Fig. 3, lane 6), this failed to reach statistical significance following analysis of multiple experiments.
Fig. 3.
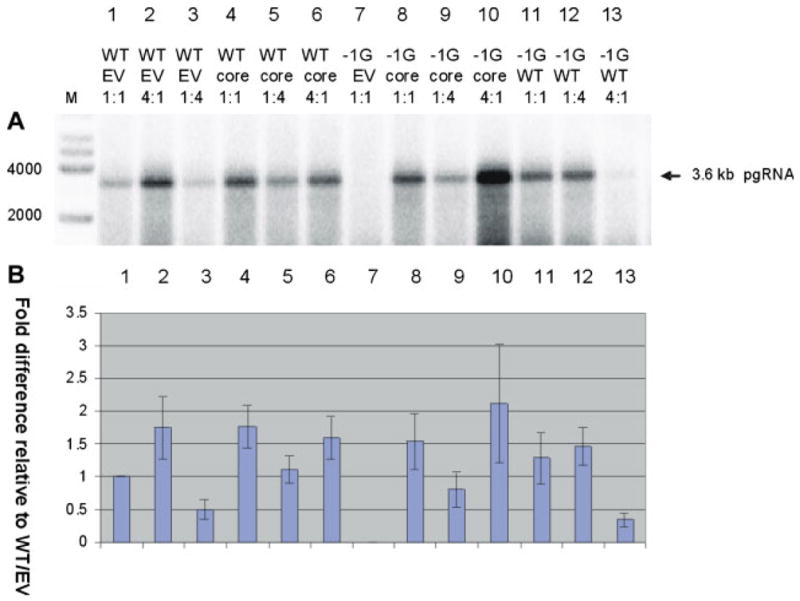
A: Representative autoradiograph of a Northern blot showing intracellular core-associated HBV RNA extracted from different Huh-7 transfectants. The sizes of MW markers are indicated. B. Densitometry plot showing relative differences in amounts of core-associated HBV RNA, following correction for transfection efficiency and quantification of northern autoradiographs by densitometry. Columns represent means of results from three independent experiments; error bars indicate standard errors of the means. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jmv]
Viral Protein Synthesis
To determine the effect of the −1G mutation on pre-core and core protein expression, cellular and secreted protein expression was analyzed by Western blotting, using (i) an in house antibody specific for the HBV precore protein (PRM-3), and (ii) a commercial (Dako) core antibody, which shows a small degree of cross-reactivity for the precore protein. The level of secreted HBeAg in cell culture supernatant and patient serum was quantified using a sensitive ARCHITECT HBeAg assay (Abbott Diagnostics) and the localization of truncated proteins produced by the −1G mutant HBV was determined by confocal microscopy.
Western blotting
Western blotting of cell lysates showed that although the −1G mutant HBV expressed similar levels of HBsAg compared with wt HBV following rescue (data not shown), the −1G mutant HBV did not express full-length precore protein, HBeAg, or core/capsid protein (Fig. 4A–D, lanes 7–10). That the −1G mutant HBV produced HBsAg in the absence of core protein (Fig. 4C, anti-HBs, lane 7) confirmed the plasmid itself was transcriptionally active. Core protein visible in lanes 8–10 was produced by the CMV-driven expression plasmid used to rescue replication of the −1G mutant HBV.
Fig. 4.
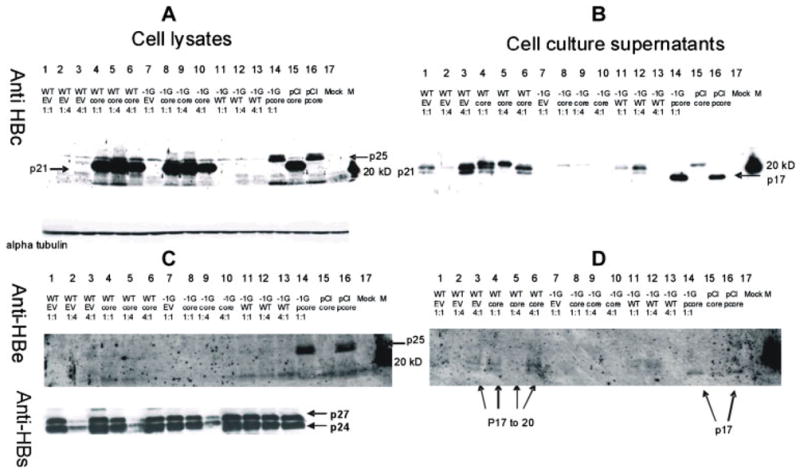
Representative Western blots of viral proteins, following transfection of Huh7 cells with different ratios of −1G mutant and/or wt HBV (WT), with or without core-expressing plasmid (pCIcore) or empty vector (EV). Western blots were probed with antibodies directed against the HBV core (panels A and B), precore (panels C and D) or HBsAg (panel C) proteins. Panels A and C show intracellular proteins and panels B and D show extracellular proteins extracted from cell culture supernatant following methanol precipitation. To control for protein loading, blots A and C were stripped and reprobed with an antibody directed against endogenous cellular alpha-tubulin (shown below blot A).
Western blotting analysis of lysates from transfected Huh7 cells using Dako anti-HBc antibody identified low concentrations of core protein in cells transfected at a high wt: empty vector ratio (Fig. 4A, lane 3) but core protein was not detected in cells co-transfected with −1G mutant HBV and empty vector (Fig. 4A, lane 7). As expected, large amounts of core protein were detected in cells that had been co-transfected with pCIcore (Fig. 4A, lanes 4–6, 8–10, 15).
Analysis of intracellular HBV precore protein expression using an in-house polyclonal antibody to the precore N-terminus (PRM-3) identified the p25 precore protein in cells transfected with the CMV-driven pre-core expression plasmid (pCIprecore, Fig. 4C, lanes 14 and 16) and in cells transfected with high levels of wt HBV, albeit faintly (Fig. 4C, lane 3). The precore protein was not detected in cells transfected with −1G mutant HBV (Fig. 4C, lanes 7–10). The specificity of the PRM-3 antibody for the precore protein was confirmed by its inability to detect the HBV core protein produced by pCIcore (Fig. 4C, lanes 4–6, 8–10, 15) or detect core protein produced by wt HBV.
Western blot analysis of cell culture supernatants using the PRM-3 HBeAg-specific antibody detected HBeAg (p17) when transfected with pCIprecore, reflecting the correct processing and secretion of the P25 protein (Fig. 4D, lanes 14 and 16). In supernatants of cells transfected with wt HBV, two precore-specific bands were detected, of approximately 14–17 kDa (Fig. 4D, lanes 3–6). The intensities of the corresponding bands were decreased in supernatants from cells following co-transfection with both wt and −1G mutant HBV, reflecting the inability of the −1G mutant HBV to produce the precore protein (Fig. 4D, lanes 11–13). Similarly, no HBeAg was detected in cell culture supernatants following transfection with the −1G mutant HBV, either in isolation or following co-transfection with pCIcore (Fig. 4D, lanes 7–10).
Quantification of HBeAg Expression
In vitro studies
The serological phenotype of patients infected with −1G mutant HBV was HBeAg-negative. Quantitative analysis of HBeAg in cell culture supernatants by CMIA (Abbott ARCHITECT) from cells transfected with −1G mutant HBV alone failed to detect HBeAg (Fig. 5, lane 8), and in co-transfection experiments, the concentration of secreted HBeAg was inversely proportional to the −1G mutant:wt HBV ratio (Fig. 5, lanes 12–14). The amount of HBeAg produced in cells co-transfected with high levels of −1G mutant HBV relative to wt HBV (Fig. 5, lane 14) was significantly lower than cells co-transfected with high levels of empty vector relative to wt HBV (Fig. 5, lane 3).
Fig. 5.

Relative concentrations (y-axis) of HBeAg (PE IU/ml) in post-transfection cell culture supernatants as measured by enzyme immunoassay after correction for transfection efficiency. Columns represent the mean results from three experiments; error bars represent SE. Note that HBeAg values for cells transfected with −1G/pCI precore plasmid (1:1) are not presented, as they were off the scale (8.3-fold increase in HBeAg with a standard error of 3.2). [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jmv]
Confocal microscopy
The −1G mutation introduced a frameshift that truncated the deduced precore and core genes. Using confocal microscopy of transfected Huh7 cells probed with the HBeAg-specific PRM-3 (Fig. 6A and B, lanes 3 and 4, −1G mutant) we showed that following transfection of the −1G mutant HBV DNA, large aggregates of the −1G precore protein were formed and localized to both the ER (Fig. 6, panel A) and Golgi (Fig. 6, panel B). These large aggregates were observed in approximately 20% of cells examined (data not shown) and were not observed in negative controls (Supplementary Fig. 1). The large aggregates were not identified in cells transfected with plasmids encoding wt HBV alone (Fig. 6, lane 4, WT) in which the HBeAg showed a more diffuse pattern, as seen previously in the livers of patients infected with HBeAg positive chronic hepatitis B infection [Lindh et al., 1999].
Fig. 6.

Confocal fluoromicrographs of Huh7 cells following probing with antibodies specific for the HBV precore protein, showing intracellular accumulation of aggregated −1G-encoded truncated pre-core protein (arrowed) in the (A) ER and (B) Golgi. Nuclei were stained with DAPI (lane 1). Fluorescent probes were specific for ER (lane 2A) or Golgi (lane 2B). Images from cells probed for HBeAg are shown in lane 3. The merged images showing co-localization of the wt or −1G-encoded precore proteins in the ER/Golgi are presented in lane 4. Cells were transfected with: (1) empty vector (EV); (2) WT/EV (WT); (3) −1G/EV; or (4) −1G/WT. All co-transfections were performed at a 1:1 ratio. Scale bar = 20 μM.
Endoplasmic Reticulum Stress Assays
Since expression of truncated HBV proteins has previously been associated with ER stress and disease progression [Wang et al., 2003; Chua et al., 2005], we were interested to determine whether the truncated precore protein encoded by the −1G mutant HBV caused ER stress using an in vitro cell culture model. ER stress is often manifested as up-regulation of heat shock proteins such as glucose regulatory protein 78 (GRP78), which bind to unfolded proteins and facilitate their removal from the ER lumen to the cytoplasm [Bertolotti et al., 2000]. To determine if −1G-encoded precore protein altered GRP-78 mRNA transcription, we measured GRP78 mRNA in transfected cells by real-time PCR. Transfection of Huh7 cells with plasmids encoding −1G mutant HBV or wt HBV had no effect on GRP78 levels, relative to cells transfected with empty vector (Fig. 7). This contrasted with an eightfold increase in GRP78 mRNA levels in cells treated with the ER stress inducer brefeldin A.
Fig. 7.

Fold difference relative to untransfected cells (lane 7) in GRP78 mRNA expression following co-transfection of Huh7 cells with the following plasmids: (1) Empty vector (EV), (2) −1G, (3) −1G/pCIcore1:1, (4) −1G/pCIcore1:4, (5) −1G/pCIcore 4:1 (6) wt HBV, (7) Mock (untransfected), (8) EV plus brefeldin A. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jmv]
DISCUSSION
In this study, we have characterized an HBV mutant that occurs mostly in patients with chronic HBV infection that are co-infected with HIV [Revill et al., 2007]. Although incapable of autonomous replication because it lacks a functional core protein, this mutant replicated to near wt levels when the core protein was supplied in trans. Similar findings have been reported for other HBV core deletion mutants that emerge during chronic hepatitis B mono-infection [Gunther et al., 2000; Marschenz et al., 2006], but some of the latter mutants replicated more efficiently than the corresponding wt. By contrast although the presence of −1G mutant HBV was associated with high HBV concentrations in vivo [Revill et al., 2007], the “rescued” mutant did not replicate more efficiently than wt, even when an unlimited supply of core protein was available. Indeed, transfection with mixtures containing high levels of −1G mutant HBV relative to wt HBV decreased overall viral replication, most likely due to the limited availability of viral nucleocapsid. However, secreted HBV-DNA levels were lower in cells transfected with plasmids expressing −1G mutant HBV and core protein at a 1:1 ratio and in cells co-transfected with −1G mutant and high levels of wt HBV, relative to cells transfected with wt HBV alone. This suggested the −1G mutant HBV had a secretion defect or altered secretion of wt HBV.
The finding that the −1G mutant HBV does not have a high-replication phenotype in vitro appears to contradict previous findings that this mutant was associated with high HBV-DNA levels in persons co-infected with HIV [Revill et al., 2007]. The reason(s) for the apparent incongruity between previous in vivo and current in vitro findings are unclear, but may reflect differences in HBV replication in the setting of chronic infection, compared with short-term cell culture studies. Differences in HBV replicative capacity for the same mutant in vitro and in vivo are not unknown, with the most common precore HBV mutant (G1896A precore stop codon mutant) having a high-replication phenotype in vitro [Scaglioni et al., 1997; Chen et al., 2003], yet is associated with reduced HBV-DNA levels in patients [Lok and McMahon, 2009]. However, the differences in HBV replication in vitro and in vivo may also reflect (i) the fact that in vitro studies were performed in the setting of HBV mono-infection, not HIV/HBV co-infection and (ii) the absence of a humoral immune response in cell culture.
The −1G mutation produces a frameshift that disrupts HBV core and precore reading frames, truncating the deduced core and precore proteins, respectively. The precore protein is the precursor of the HBeAg, a potent immune suppressor that regulates innate immune responses to HBV infection [Visvanathan et al., 2007; Lang et al., 2011; Wilson et al., 2011] and the core protein forms the viral nucleocapsid, critical for packaging and replication. This truncation decreases the deduced molecular weights of the precore and core proteins to 10.5 and 7.5 kDa, respectively [Revill et al., 2007]. Although we did not detect these proteins by immunoblotting, this may reflect their small size and the insensitivity of the assay used rather than the absence of any protein, as evidenced by the low levels of wt precore proteins detected using these antibodies. Nonetheless, confocal microscopy showed that although low amounts of anti-HBe-reactive proteins were produced, they accumulated in organelles of cells transfected with −1G mutant HBV.
HBeAg was not detectable in the culture medium from cells transfected with −1G mutant HBV, regardless of whether the HBV core protein was supplied in trans. Furthermore, co-transfection with higher −1G mutant: wt ratios resulted in proportional reductions in the concentration of HBeAg detectable in cell culture supernatants, confirming that the −1G mutation confers an HBeAg-negative phenotype in vitro. Indeed, a small yet significant reduction in HBeAg was observed in the supernatant of cells transfected with wt HBV in the presence of −1G mutant compared with cells transfected with wt HBV and an equivalent amount of empty vector, suggesting that the −1G mutant HBV may be altering secretion of wt HBeAg. It is possible that the aggregation of truncated precore protein in the ER and Golgi observed in confocal microscopy studies affected the secretion of wt HBeAg, although this requires further investigation. Although the −1G mutation abrogates production of the HBeAg in vitro, all patients infected with the −1G mutant identified to date were HBeAg positive, even when −1G mutant formed 90% of the quasispecies pool [Revill et al., 2007]. This is most probably due to the high viral loads in these patients, such that even if only 10% of the circulating virus was wild-type, it was sufficient to confer HBeAg-positive status.
Intracellular retention of truncated viral proteins may result in ER stress and associated up-regulation of chaperone proteins such as GRP78 mRNA (reviewed by Zhang et al. [2005]). Whilst aggregation of −1G-encoded protein was observed in the ER and Golgi, we observed no difference in GRP78 mRNA or protein expression in cells transfected with rescued −1G mutant, or wt HBV, compared with mock cells or cells transfected with empty vector. These findings using genomic length DNA may be analogous to previous studies of HBV preS1/preS2 mutants associated with hepatocellular carcinoma and serious liver disease. These studies showed that over-expression of HBV proteins resulted in ER stress [Wang et al., 2003], yet transfection with genomic-length HBV encoding the same mutations [Chua et al., 2005] had no such effect. Future studies will focus on the generation of CMV-driven plasmids expressing the truncated −1G precore and core proteins. Since we only observed −1G mutant HBV precore protein aggregates in approximately 20% of cells transfected with genomic-length −1G mutant HBV, it is also possible that the low level of aberrant protein was insufficient to trigger a measurable response in vitro.
HBV-associated liver disease progresses more rapidly in the setting of HIV co-infection than HBV mono-infection. We now demonstrate that an HBV mutant usually associated with HIV co-infection [Revill et al., 2007] alters HBV viral secretion and protein expression in this setting. Yet, the reasons this mutant is predominantly associated with HIV/HBV co-infection are unclear. Perhaps the truncated precore/core proteins encoded by the −1G mutant HBV elicit a strong immune response, which is ameliorated in the setting of immunosuppression, enabling this mutant to proliferate. Since the wt precore protein is an important regulator of innate and adaptive immune responses, future studies will also focus on how the truncated proteins encoded by the −1G mutant HBV alter immune responses, which may also contribute to the more severe disease pathogenesis observed in persons co-infected with HBV and HIV.
Supplementary Material
Acknowledgments
We thank Lucy Selleck for maintenance of the cell cultures and Alan Breschkin, Stav Corby, Julia Darmanin, and Adam Enriquez for their assistance with the HBsAg ELISA assays. We also thank Matt Burton (MCRI, Melbourne, Australia) and Elizabeth Vincan for assistance with confocal microscopy.
Grant sponsor: NIH; Grant number: AI060449.
Footnotes
Additional supporting information may be found in the online version of this article.
References
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bock CT, Tillmann HL, Maschek HJ, Manns MP, Trautwein C. A preS mutation isolated from a patient with chronic hepatitis B infection leads to virus retention and misassembly. Gastroenterology. 1997;113:1976–1982. doi: 10.1016/s0016-5085(97)70018-0. [DOI] [PubMed] [Google Scholar]
- Chen RY, Edwards R, Shaw T, Colledge D, Delaney WET, Isom H, Bowden S, Desmond P, Locarnini SA. Effect of the G1896A precore mutation on drug sensitivity and replication yield of lamivudine-resistant HBV in vitro. Hepatology. 2003;37:27–35. doi: 10.1053/jhep.2003.50012. [DOI] [PubMed] [Google Scholar]
- Chin R, Shaw T, Torresi J, Sozzi V, Trautwein C, Bock T, Manns M, Isom H, Furman P, Locarnini S. In vitro susceptibilities of wild-type or drug-resistant hepatitis B virus to (−)-beta-D-2,6-diaminopurine dioxolane and 2′-fluoro-5-methyl-beta-L-arabinofuranosyluracil. Antimicrob Agents Chemother. 2001;45:2495–2501. doi: 10.1128/AAC.45.9.2495-2501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua PK, Wang RY, Lin MH, Masuda T, Suk FM, Shih C. Reduced secretion of virions and hepatitis B virus (HBV) surface antigen of a naturally occurring HBV variant correlates with the accumulation of the small S envelope protein in the endoplasmic reticulum and Golgi apparatus. J Virol. 2005;79:13483–13496. doi: 10.1128/JVI.79.21.13483-13496.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin JF, Cazals-Hatem D, Loriot MA, Martinot-Peignoux M, Pham BN, Auperin A, Degott C, Benhamou JP, Erlinger S, Valla D, Marcellin P. Influence of human immunodeficiency virus infection on chronic hepatitis B in homosexual men. Hepatology. 1999;29:1306–1310. doi: 10.1002/hep.510290447. [DOI] [PubMed] [Google Scholar]
- Fried MW, Piratvisuth T, Lau GK, Marcellin P, Chow WC, Cooksley G, Luo KX, Paik SW, Liaw YF, Button P, Popescu M. HBeAg and hepatitis B virus DNA as outcome predictors during therapy with peginterferon alfa-2a for HBeAg-positive chronic hepatitis B. Hepatology. 2008;47:428–434. doi: 10.1002/hep.22065. [DOI] [PubMed] [Google Scholar]
- Fukushima K, Ueno Y, Inoue J, Wakui Y, Obara N, Kimura O, Kido O, Nakagome Y, Kakazu E, Matsuda Y, Kogure T, Kondo Y, Nagasaki F, Yamagiwa Y, Ashino Y, Shimosegawa T. A case of HIV co-infected with hepatitis B virus precore/core deletion mutant treated by entecavir. Hepatol Res. 2008;38:842–846. doi: 10.1111/j.1872-034X.2008.00332.x. [DOI] [PubMed] [Google Scholar]
- Gilson RJ, Hawkins AE, Beecham MR, Ross E, Waite J, Briggs M, McNally T, Kelly GE, Tedder RS, Weller IV. Interactions between HIV and hepatitis B virus in homosexual men: Effects on the natural history of infection. AIDS. 1997;11:597–606. doi: 10.1097/00002030-199705000-00007. [DOI] [PubMed] [Google Scholar]
- Gunther S, Piwon N, Jung A, Iwanska A, Schmitz H, Will H. Enhanced replication contributes to enrichment of hepatitis B virus with a deletion in the core gene. Virology. 2000;273:286–299. doi: 10.1006/viro.2000.0432. [DOI] [PubMed] [Google Scholar]
- Hadziyannis SJ. Hepatitis B e antigen negative chronic hepatitis B: From clinical recognition to pathogenesis and treatment. Viral Hepat Rev. 1995;101:7–36. [Google Scholar]
- Iser DM, Warner N, Revill PA, Solomon A, Wightman F, Saleh S, Crane M, Cameron PU, Bowden S, Nguyen T, Pereira CF, Desmond PV, Locarnini SA, Lewin SR. Coinfection of hepatic cell lines with human immunodeficiency virus and hepatitis B virus leads to an increase in intracellular hepatitis B surface antigen. J Virol. 2010;84:5860–5867. doi: 10.1128/JVI.02594-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J Hepatol. 2011;55:762–769. doi: 10.1016/j.jhep.2010.12.042. [DOI] [PubMed] [Google Scholar]
- Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat. 2004;11:97–107. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- Leemans WF, Janssen HL, de Man RA. Future prospectives for the management of chronic hepatitis B. World J Gastroenterol. 2007;13:2554–2567. doi: 10.3748/wjg.v13.i18.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindh M, Savage K, Rees J, Garwood L, Horal P, Norkrans G, Dhillon AP. HBeAg immunostaining of liver tissue in various stages of chronic hepatitis B. Liver. 1999;19:294–298. doi: 10.1111/j.1478-3231.1999.tb00052.x. [DOI] [PubMed] [Google Scholar]
- Lok AS, McMahon BJ. Chronic hepatitis B: Update 2009. Hepatology. 2009;50:661–662. doi: 10.1002/hep.23190. [DOI] [PubMed] [Google Scholar]
- Marschenz S, Endres AS, Brinckmann A, Heise T, Kristiansen G, Nurnberg P, Kruger DH, Gunther S, Meisel H. Functional analysis of complex hepatitis B virus variants associated with development of liver cirrhosis. Gastroenterology. 2006;131:765–780. doi: 10.1053/j.gastro.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Milich DR, Jones JE, Hughes JL, Price J, Raney AK, McLachlan A. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc Natl Acad Sci USA. 1990;87:6599–6603. doi: 10.1073/pnas.87.17.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revill PA, Littlejohn M, Ayres A, Yuen L, Colledge D, Bartholomeusz A, Sasaduesz J, Lewin SR, Dore GJ, Matthews GV, Thio CL, Locarnini SA. Identification of a novel hepatitis B virus precore/core deletion mutant in HIV/hepatitis B virus co-infected individuals. AIDS. 2007;21:1701–1710. doi: 10.1097/QAD.0b013e32826fb305. [DOI] [PubMed] [Google Scholar]
- Revill P, Yuen L, Walsh R, Perrault M, Locarnini S, Kramvis A. Bioinformatic analysis of the hepadnavirus e-antigen and its precursor identifies remarkable sequence conservation in all orthohepadnaviruses. J Med Virol. 2010;82:104–115. doi: 10.1002/jmv.21645. [DOI] [PubMed] [Google Scholar]
- Rustgi VK, Hoofnagle JH, Gerin JL, Gelmann EP, Reichert CM, Cooper JN, Macher AM. Hepatitis B virus infection in the acquired immunodeficiency syndrome. Ann Intern Med. 1984;101:795–797. doi: 10.7326/0003-4819-101-6-795. [DOI] [PubMed] [Google Scholar]
- Scaglioni PP, Melegari M, Wands JR. Posttranscriptional regulation of hepatitis B virus replication by the precore protein. J Virol. 1997;71:345–353. doi: 10.1128/jvi.71.1.345-353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton M, Kew MC, Kramvis A. Distinct mutant hepatitis B virus genomes, with alterations in all four open reading frames, in a single South African hepatocellular carcinoma patient. Virus Res. 2012;163:59–65. doi: 10.1016/j.virusres.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Thio CL, Seaberg EC, Skolasky R, Jr, Phair J, Visscher B, Munoz A, Thomas DL. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS) Lancet. 2002;360:1921–1926. doi: 10.1016/s0140-6736(02)11913-1. [DOI] [PubMed] [Google Scholar]
- Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, Rodgers S, Kurtovic J, Chang J, Lewin S, Desmond P, Locarnini S. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology. 2007;45:102–110. doi: 10.1002/hep.21482. [DOI] [PubMed] [Google Scholar]
- Wang HC, Wu HC, Chen CF, Fausto N, Lei HY, Su IJ. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am J Pathol. 2003;163:2441–2449. doi: 10.1016/S0002-9440(10)63599-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner N, Locarnini S. The antiviral drug selected hepatitis B virus rtA181T/sW172* mutant has a dominant negative secretion defect and alters the typical profile of viral rebound. Hepatology. 2008;48:88–98. doi: 10.1002/hep.22295. [DOI] [PubMed] [Google Scholar]
- Wilson R, Warner N, Ryan K, Selleck L, Colledge D, Rodgers S, Li K, Revill P, Locarnini S. The hepatitis B e antigen suppresses IL-1beta-mediated NF-kappaB activation in hepatocytes. J Viral Hepat. 2011;18:499–507. doi: 10.1111/j.1365-2893.2011.01484.x. [DOI] [PubMed] [Google Scholar]
- Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.