

Abstract
Bispecific antibodies (bscAbs), particularly those of the bispecific T-cell engager (BiTE) subclass, have been shown to effectively redirect T cells against cancer. Previous efforts to target antigens expressed in both tumors and normal tissues have produced significant toxicity, however. Moreover, like other large molecules, bscAbs may be restricted from entry into the “immunologically privileged” CNS. A tumor-specific mutation of the epidermal growth factor receptor, EGFRvIII, is a constitutively activated tyrosine kinase not found in normal tissues but frequently expressed in glioblastomas and many other neoplasms. Because it is localized solely to tumor tissue, EGFRvIII presents an ideal target for immunotherapy. Here we report the preclinical evaluation of an EGFRvIII-targeted BiTE, bscEGFRvIIIxCD3. Our results show that bscEGFRvIIIxCD3 activates T cells to mediate potent and antigen-specific lysis of EGFRvIII-expressing gliomas in vitro (P < 0.001) at exceedingly low concentrations (10 ng/mL) and effector-to-target ratios (2.5:1). Treatment with i.v. bscEGFRvIIIxCD3 yielded extended survival in mice with well-established intracerebral tumors (P < 0.05) and achieved durable complete cure at rates up to 75%. Antitumor efficacy was significantly abrogated on blockade of EGFRvIII binding, demonstrating the need for target antigen specificity both in vitro and in vivo. These results demonstrate that BiTEs can be used to elicit functional antitumor immunity in the CNS, and that peptide blockade of BiTE-mediated activity may greatly enhance the safety profile for antibody-redirected T-cell therapies. Finally, bscEGFRvIIIxCD3 represents a unique advancement in BiTE technology given its exquisite tumor specificity, which enables precise elimination of cancer without the risk of autoimmune toxicity.
Keywords: central nervous system, blood–brain barrier, immunomodulation, therapeutics
Over the past 3 decades, only modest progress has been made in the management of patients with glioblastoma (GBM). Despite image-guided surgical resection (1), maximal radiation therapy, and effective chemotherapy, GBM remains universally fatal, with a median survival of only 15 mo (2). These conventional therapies also lack specificity and are limited by incapacitating damage to surrounding normal brain and systemic tissues (3). A promising alternative is the use of immunotherapy, which has the capacity to target cancer cells specifically (4).
Substantial evidence suggests that T cells are predominant effectors in the immune-mediated eradication of cancer (5); however, attempts to mount and sustain antigen-specific T cells endogenously through vaccines have been largely disappointing (6). Other efforts to foster effective antitumor immune responses rely on the adoptive transfer of ex vivo expanded or genetically manipulated T cells. Although promising, these approaches are laborious, inconsistent, and further complicated by the need for viral transduction (7, 8). To avoid the complex preparation required for cell-based therapy, T cells instead can be activated in vivo by agonistic antibodies, which obviates the need for clonal T-cell survival and expansion; however, this approach indiscriminately activates circulating T cells throughout the body, producing disastrous autoimmune side effects (9).
An alternative approach is the use of bispecific antibodies (bscAbs) that elicit cytotoxic activity from circulating T cells but do so only in the proximity of their cognate tumor antigen (10). One bscAb class that has demonstrated remarkable efficacy is the bispecific T-cell engager (BiTE), consisting of a tumor-targeting single-chain variable antibody fragment (scFv) translated in tandem with another scFv directed against the T-cell activation ligand CD3 (11). Compared with previously described bscAbs, BiTEs have several distinguishing characteristics, including the ability to induce immunologic synapses as well as trigger serial rounds of killing from even unresponsive tumor-infiltrating T-cell populations without the need for classic costimulatory signals, conventional peptide-MHC recognition, or clonal T-cell persistence and expansion (12). Various mouse models and clinical trials have demonstrated the promising potential of antibody-redirected T-cell therapies either through chimeric antigen receptors or, more recently, via BiTEs against tumor-associated antigens, including ErbB-2, carcinoembryonic antigen, and CD19 (13-16). However, because these targets are not strictly limited to tumor tissue, such approaches have led to unwanted toxicity and destruction of even normal, healthy cells (11, 17).
Among the few known tumor-specific antigens, perhaps the most widely characterized is a mutated form of EGF receptor (EGFR). The EGFRvIII mutation is a constitutively activated tyrosine kinase central to the oncogenic process that is not found in any normal tissues, but is frequently expressed on the surface of GBM and many other common neoplasms (18). The receptor consists of an in-frame deletion of exons 2–7, the translation of which produces an extracellular junction with a unique glycine residue. EGFRvIII expression is strictly tumor-specific, and its extracellular domain is relatively small, making it an ideal target for the BiTE platform (19).
In this study, we aimed to design, generate, and characterize an EGFRvIII-targeted molecule, bscEGFRvIIIxCD3, resembling the BiTE technology. We performed a series of experiments to evaluate bscEGFRvIIIxCD3 both in vitro and in vivo using a murine model of glioma. Our results show that bscEGFRvIIIxCD3 is both antigen-specific and highly cytotoxic, leading to the activation of T cells and elevated secretion of Th1-type cytokines exclusively in the presence of human EGFRvIII-expressing glioma. Despite conventional notions of CNS immune privilege, systemically administered bscEGFRvIIIxCD3 localized to intracerebral tumors, extending survival in mice with well-established EGFRvIII-expressing glioma, consistent with substantial intratumoral immune cell infiltration.
Taken together, our findings demonstrate that bscEGFRvIIIxCD3 may be used to achieve potent antitumor T-cell responses in the CNS. Moreover, unlike previously described BiTEs, bscEGFRvIIIxCD3 has unparalleled specificity, given its ability to target tumor cells without the risk of cross-reactivity with normal tissues.
Results
Design, Expression, and Purification of bscEGFRvIIIxCD3 by Metal-Affinity Chromatography.
To construct an EGFRvIII-specific bscAb, we selected an affinity-matured anti-human EGFRvIII-specific scFv, MR1-1 (20), and translated it in tandem with an scFv isolated from the anti-human CD3 mAb OKT3 (Fig. 1A). In brief, two scFvs directed against EGFRvIII and CD3 were joined by a flexible five-amino acid (Gly4Ser) linker using overlap extension PCR in the following variable domain orientation: VH-VL-VH-VL. The resultant tandem single-chain bscAb, bscEGFRvIIIxCD3, was expressed in transformed Escherichia coli, isolated from insoluble inclusion bodies, and verified for identity by Western blot analysis (Fig. 1B). Refolded bscAb was purified by metal-affinity chromatography, with a distinct peak observed in the elution profile between 50 and 100 mM imidazole (Fig. 1C). Fractions were collected and tested for purity by SDS/PAGE, indicating a single band of refolded bscAb with a predicted molecular weight of ∼52 kDa (Fig. 1D). In a typical preparation cycle, 100 mg of inclusion bodies from 1 L of culture yielded 1–2 mg of purified bscEGFRvIIIxCD3.
Fig. 1.
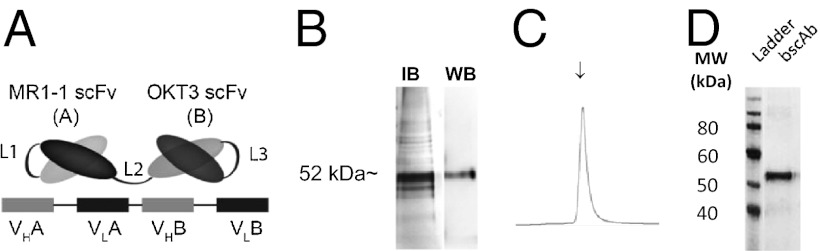
Design, manufacture, and purification of bscEGFRvIIIxCD3 by metal-affinity chromatography. (A) Schema of the bscEGFRvIIIxCD3 design. Linkers L1 and L3 were constructed between the VH and VL domains of their respective scFvs and consisted of (Gly4Ser)3, whereas L2 was constructed as a single Gly4Ser linker between scFvs. (B) BscEGFRvIIIxCD3 was expressed as insoluble inclusion bodies and verified for identity by Western blot analysis. (C) Protein was eluted from an immobilized metal-affinity chromatography column by a stepwise imidazole gradient. (D) SDS/PAGE of the eluted fraction indicated by the arrow in C. Molecular weight markers (in kDa) are shown on the left.
Purified bscEGFRvIIIxCD3 Binds to T Lymphocytes and to Tumor Cells Expressing EGFRvIII.
Flow cytometry was used to confirm dual specificity of the refolded bscAb against cells expressing the appropriate targets. Analyses revealed binding of bscEGFRvIIIxCD3 to both CD4+ and CD8+ human T cells known to express CD3 within a population of healthy donor peripheral blood mononuclear cells (PBMCs) (Fig. 2A). In addition, bscEGFRvIIIxCD3 was found to bind specifically to U87MG.ΔEGFR glioma cells expressing EGFRvIII, but not to the parental WT U87MG control (Fig. 2B). Binding properties of bscEGFRvIIIxCD3 against individual antigens were further characterized by surface plasmon resonance against recombinant EGFRvIII extracellular domain (ECD) and CD3ε, yielding equilibrium dissociation constants (Kd) of 1.5 × 10−9 M and 6.5 × 10−9 M, respectively (Fig. 2C).
Fig. 2.

Purified bscEGFRvIIIxCD3 binds to T lymphocytes and tumor cells expressing EGFRvIII. (A) BscEGFRvIIIxCD3 binds specifically to CD8+ and CD4+ lymphocytes. (B) BscEGFRvIIIxCD3 also binds specifically to the EGFRvIII-expressing tumor cell line. (C) Surface plasmon resonance reveals dissociation constants for bscEGFRvIIIxCD3 against recombinant target antigens.
BscEGFRvIIIxCD3 Activates T Cells, Resulting in Proliferation and Secretion of Th1-Type Cytokines.
We next tested bscEGFRvIIIxCD3 for its ability to activate T cells in vitro. When incubated with bscEGFRvIIIxCD3 and target cells, CD4+ and CD8+ T cells up-regulated surface expression levels of activation markers CD69 and CD25 (Fig. 3A). Importantly, this required interaction with the EGFRvIII antigen, given that elevated CD69 and CD25 expression was detected only in the presence of EGFRvIII-expressing human glioma cells, not with WT parental controls. We also evaluated the ability of bscEGFRvIIIxCD3 to induce antigen-specific proliferation of T cells, as measured by 3H-thymidine incorporation in vitro. In these experiments, recombinant EGFRvIII ECD was used as a source of cognate antigen, to eliminate the variability in 3H-thymidine incorporation that otherwise would occur owing to proliferation of target glioma cells. When soluble bscEGFRvIIIxCD3 was incubated in the presence or absence of solid-phase BSA, proliferative effects were not detected above baseline levels. However, soluble bscEGFRvIIIxCD3 in the presence of solid-phase EGFRvIII ECD led to significant proliferation that was indistinguishable from that resulting from stimulation with solid-phase bscEGFRvIIIxCD3 alone (Fig. 3B). This finding verifies—as has been well characterized for BiTEs against other antigens—that interaction between soluble bscEGFRvIIIxCD3 and the EGFRvIII target is necessary to potentiate immunologic capping and synapse formation, and that antigen-specificity is a crucial factor mediating this activity (21).
Fig. 3.

BscEGFRvIIIxCD3 activates T cells, resulting in proliferation and secretion of Th1-type cytokines. (A) T cells incubated with bscEGFRvIIIxCD3 and either U87MG.ΔEGFR (black line) or U87MG (gray line) were stained and analyzed for surface expression of activation markers CD69 and CD25. (B) Proliferation of T cells in response to bscEGFRvIIIxCD3 and solid-phase EGFRvIII ECD was measured by 3H-thymidine incorporation. (C) Supernatants from wells containing unstimulated human PBMCs with bscEGFRvIIIxCD3 and either U87MG.ΔEGFR or U87MG were subjected to cytometric bead array analysis for inflammatory cytokines. (D) This analysis revealed elevated secretion of IL-2, IFN-γ, and TNF and polarization to Th1 in the presence of EGFRvIII-expressing tumor cells. Horizontal bars represent a statistical significance of P < 0.001.
Effective antitumor immune responses are known to require the secretion of inflammatory cytokines, particularly those associated with Th1 polarization. To test whether activation of T cells by bscEGFRvIIIxCD3 leads to favorable cytokine production, we used cytometric bead array analysis to analyze the supernatants of cultures in which lymphocytes were incubated with bscEGFRvIIIxCD3 and target cells either expressing or not expressing the EGFRvIII tumor-specific antigen. In the presence of negative control target cells, analyses of culture supernatants revealed minimal secretion of IL-2, IFN-γ, and TNF; however, when incubated with EGFRvIII-expressing glioma, bscEGFRvIIIxCD3 elicited significantly greater T-cell function (Fig. 3C), which, when represented as proportions in a multiplex cytokine panel, demonstrated considerable polarization toward a Th1-associated immune response (Fig. 3D).
BscEGFRvIIIxCD3 Redirects T Cells to Kill EGFRvIII-Expressing Gliomas in Vitro.
We next sought to test the ability of bscEGFRvIIIxCD3 to functionally elicit antigen-specific cytotoxic responses in vitro. In a standard 51Cr release assay using freshly thawed human PBMCs as effector cells, we determined that bscEGFRvIIIxCD3-mediated redirection of T cells is indeed highly cytotoxic against multiple previously characterized human EGFRvIII-expressing tumor cells (Fig. 4A). Importantly, cytotoxicity was strictly dependent on the ability of bscEGFRvIIIxCD3 to recognize and bind to the EGFRvIII antigen on target cells, given that incubation of bscEGFRvIIIxCD3 with matched EGFRvIII-negative tumors in the presence of effector cells did not induce observable target cell lysis. Furthermore, demonstrating the need for dual specificity within the actual therapeutic molecule, target cell lysis was not observed on incubation with a nonspecific bscAb control, bscAbxCD3 (designed from antigen-binding portions isolated from isotype clone MOPC-21 and OKT3), but was maintained against EGFRvIII-expressing cells on incubation with the active molecule, bscEGFRvIIIxCD3 (Fig. 4B). Along with being highly specific, bscEGFRvIIIxCD3 also exerted potent bioactivity against EGFRvIII-expressing tumors at greatly reduced concentrations (Fig. 4C), with dose-dependence observed at E:T ratios as low as 2.5:1 (Fig. 4D).
Fig. 4.

BscEGFRvIIIxCD3 redirects T cells to kill EGFRvIII-expressing gliomas in vitro. (A) Standard 51Cr-release demonstrates specific lysis of U87MG.ΔEGFR over U87MG (Left) and D54-EGFRvIII over D54 (Right) by unstimulated human PBMCs and bscEGFRvIIIxCD3 (E:T ratio, 20:1; incubation time, 18 h; [bscEGFRvIIIxCD3], 10 μg/mL). (B) Specific lysis of the U87MG.ΔEGFR cell line is not observed in the presence of negative control bscAbxCD3. (C and D) Specific lysis of the U87MG.ΔEGFR cell line varies proportionally with bscEGFRvIIIxCD3 concentration (C), and the dose-response relationship is maintained over varying E:T ratios (D). (E) Light microscopy of T cells incubated with U87MG in the presence of bscEGFRvIIIxCD3 did not demonstrate appreciable localization to tumor (Top) compared with wells in which target cells expressing EGFRvIII were included (Middle). Experiments were also performed in which U87MG, U87MG.ΔEGFR (red),and T cells (yellow) were incubated with bscEGFRvIIIxCD3, which demonstrated selective localization of T cells to EGFRvIII-expressing tumor cells in vitro (Bottom). Horizontal bars represent a statistical significance of at least P < 0.05.
As visualized by light microscopy, bscEGFRvIIIxCD3 did not appear to affect the behavior of lymphocytes against EGFRvIII-negative glioma cells in vitro (Fig. 4E, Top). However, in the presence of EGFRvIII expression on the surface of matched glioma, lymphocytes visibly localized to the surface of tumor cells after the addition of bscEGFRvIIIxCD3, implying that the cytotoxic effects observed in vitro may occur via contact-mediated mechanisms (Fig. 4E, Middle). Importantly, under conditions in which PKH26-labeled EGFRvIII-positive (red) and matched EGFRvIII-negative (bright field) glioma cells were cultured together in close proximity, bscEGFRvIIIxCD3-redirected carboxyfluorescein diacetate succinimidyl ester-labeled T cells (yellow) were seen to localize only to EGFRvIII-expressing tumors. These results lend further credence to the ability of the BiTE therapeutic platform to selectively distinguish between even closely associated, adjacent cells based on target antigen expression in vitro (Fig. 4E, Bottom).
Antitumor Response Produced by bscEGFRvIIIxCD3 Is Specific to EGFRvIII-Expressing Tumors in Vivo.
To investigate the in vivo activity of bscEGFRvIIIxCD3, we created orthotopic xenograft models using the human malignant glioma cell lines U87MG and U87MG.ΔEGFR. Recent studies have demonstrated that GBMs are infiltrated by immune cells (22, 23); thus, to model this scenario preclinically, NOD scid gamma (NSG) mice were inoculated intracranially with a mixture of tumor cells and unstimulated human PBMCs. After implantation, bscEGFRvIIIxCD3 was administered via daily tail vein injections. Under these conditions, mice implanted with tumors expressing only the WT EGFR did not exhibit a significant survival benefit (Fig. 5A). Similarly, in mice implanted with intracerebral tumors expressing EGFRvIII, treatment with the nonspecific control bscAbxCD3 did not prolong survival compared with mice that received vehicle control (Fig. 5B). In contrast, infusion with bscEGFRvIIIxCD3 in this setting achieved durable, complete cure in six of eight mice without apparent toxicity; this effect was also potent and dose-dependent, yielding prolonged survival at doses as low as 1 μg/d, with comparable results observed in repeated experiments (P < 0.01) (Fig. 5C, Upper). Although less dramatic, significant antitumor effects were also observed on treatment of very late-stage, well-established tumors in mice treated with bscEGFRvIIIxCD3 and peripheral PBMCs just days before death of controls (Fig. 5C, Lower). This result was achieved using a cumulative dose of bscEGFRvIIIxCD3 that was ∼3- to 10-fold lower than the pharmacologic equivalent of current, clinically approved antibody therapies. Histological examination of tumors obtained from mice treated with PBS revealed no evidence of infiltrating immune cells and instead showed invasive glioma tissue extending along peritumoral blood vessels (Fig. 5D, Upper Left). In contrast, brain tissue from mice treated with bscEGFRvIIIxCD3 exhibited marked perivascular cuffing (Fig. 5D, Upper Right, box), as well as diffuse persistence of infiltrating lymphocytes amid regions of intracerebral tumor necrosis (Fig. 5D, Bottom, arrows). Taken together, these data demonstrate that systemic administration of bscEGFRvIIIxCD3 can potently activate T cells in vivo to achieve significant antitumor effects against EGFRvIII-expressing tumors in the CNS.
Fig. 5.

Antitumor response produced by bscEGFRvIIIxCD3 is specific to EGFRvIII-expressing tumors in vivo. NSG mice (n = 8) were implanted i.c. with 1 × 105 tumor cells and unstimulated human PBMCs at a ratio of 1:1. Mice implanted with U87MG (A) or U87MG.ΔEGFR (B) were treated with bscEGFRvIIIxCD3 or control bscAbxCD3 by daily i.v. infusion (arrows). (C, Upper) To assess dose-response, bscEGFRvIIIxCD3 was administered to mice at indicated doses. (C, Lower) For delayed treatment, mice were implanted i.c. with U87MG.ΔEGFR alone, and treatment was started on day 10 after tumor implantation. These mice were immune-reconstituted peripherally with 2 × 107 unstimulated human PBMCs by i.p. injection on day 10, followed by infusion with bscAb. (D, Upper Left) Representative section of i.c. tumor and local invasion surrounding an adjacent vessel from an animal receiving PBS. (D, Upper Right) In contrast, in mice treated with bscEGFRvIIIxCD3, PBMCs are seen to exit peritumoral i.c. vessels toward tumor tissue (box). (D, Lower) Tumor tissue in mice treated with bscEGFRvIIIxCD3 and receiving PBMCs i.p. shows diffuse tumor-associated mononuclear infiltrate and areas of necrosis (arrows).
Antitumor Efficacy of bscEGFRvIIIxCD3 Is Abrogated by Targeted EGFRvIII Blockade.
Despite early evidence of success in the clinic, antibody-redirected T-cell platforms recently have been shown to induce lethal “on-target” toxicity when their cognate antigen is not only present in tumors, but also expressed in normal, healthy tissues (e.g., ErbB-2, carcinoembryonic antigen, CD19) (13–16). To address this issue in our model—and to provide additional controls demonstrating the specificity of our approach—we sought to determine whether competitive inhibition of EGFRvIII binding could directly abrogate biological activity of bscEGFRvIIIxCD3 both in vitro and in vivo. The rationale for this hypothesis was based on our data showing that bscEGFRvIIIxCD3 is not effective in the absence of EGFRvIII expression on tumors, and that similarly, a control bscAbxCD3 is not effective in the presence of EGFRvIII-expressing tumors.
To perform specific EGFRvIII blockade, we used a previously published soluble peptide (PEPvIII) corresponding to the extracellular epitope for EGFRvIII-specific antibodies (18). Of note, this peptide is currently in clinical trials as an EGFRvIII-targeted vaccine, and thus has great translational potential in addition to providing biological principle. Using this approach, our data demonstrate that EGFRvIII blockade by PEPvIII successfully disrupts binding to EGFRvIII-expressing tumor targets in vitro. Importantly, PEPvIII inhibition was specific, because peptide-blocked bscEGFRvIIIxCD3 retained the ability to bind the CD3 complex on both CD4+ and CD8+ T cells (Fig. 6A). Despite continued anti-CD3 binding, addition of the cognate PEPvIII peptide was sufficient to completely abolish bscEGFRvIIIxCD3-mediated antitumor effects against EGFRvIII-expressing gliomas in vitro (Fig. 6B). Moreover, EGFRvIII blockade significantly reduced the efficacy of bscEGFRvIIIxCD3 in vivo against EGFRvIII-expressing tumor in the brain (Fig. 6C). Taken together, these results validate the precise target specificity of the bscEGFRvIIIxCD3 construct and definitively establish the requirement for dual binding. In addition, our finding that a soluble cognate peptide can be used to selectively inhibit therapeutic effects of bscEGFRvIIIxCD3 may offer a useful approach to enhancing the safety profile of other bscAbs that target antigens expressed in normal, healthy tissues.
Fig. 6.

Antitumor efficacy of bscEGFRvIIIxCD3 is abrogated by targeted EGFRvIII blockade. (A) EGFRvIII blockade does not affect the ability of bscEGFRvIIIxCD3 to bind T cells, but significantly abrogates binding to EGFRvIII-expressing gliomas in vitro. (B) Standard 51Cr release demonstrates specific lysis of U87MG.ΔEGFR in the presence of bscEGFRvIIIxCD3 and effector cells (E:T ratio 20:1; incubation time 18 h; [bscEGFRvIIIxCD3] 10 μg/mL), which is completely inhibited in the presence of soluble PEPvIII cognate peptide (blockade ratio 1:1). Horizontal bar indicates a statistical significance of P < 0.001. (C) NSG mice (n = 8) were implanted i.c. with 1 × 105 U87MG.ΔEGFR and unstimulated human PBMCs at a ratio of 1:1. Daily i.v. infusions with bscEGFRvIIIxCD3, coinfusion with PEPVIII blockade, or PBS vehicle control began 3 h after implantation and continued for 5 d.
Discussion
It has been widely demonstrated that bscAbs of the BiTE class can be used to generate potent T-cell immune responses against tumors outside the CNS. Importantly, our findings in the present study demonstrate that similar responses may be achieved against established tumors in the brain. An additional advance reported here is the targeting of EGFRvIII with BiTE technology, which allows bscEGFRvIIIxCD3 to target tumors specifically without cross-reactivity against antigens expressed normal tissues.
In general, bscAbs designed to redirect the immune system against cancer have an extensive history marked by several shortcomings. Whereas some alternative bscAb constructs have resulted in prohibitive toxicity owing to nonspecific T-cell activation, others have been hampered by low potency, in many cases requiring extremely high E:T ratios and high concentrations of the therapeutic agent, as well as T-cell prestimulation or costimulation to achieve acceptable preclinical efficacy (12). In comparison, our present findings demonstrate that bscEGFRvIIIxCD3 has the ability to safely and specifically mediate antitumor responses that are both dose-dependent and efficacious at low E:T ratios (2.5:1) without the need for additional T-cell stimulation. As an indication of the potency of bscEGFRvIIIxCD3, a cumulative dose of only 5 μg (∼0.25 mg/kg) in vivo was sufficient to significantly prolong survival in mice with EGFRvIII-expressing tumors. This dose is roughly equivalent to 0.02 mg/kg for the average 60-kg adult (24); in stark contrast, recommended therapeutic doses of currently approved antitumor mAbs range from 2 mg/kg (for Herceptin) (25) up to 10 mg/kg (for Avastin) (26), highlighting the potential for vast improvement over currently available antibody treatments for solid tumors.
Previous studies have established that bscAbs of the BiTE class potentiate antitumor immune responses solely in the presence of cognate antigen expression by target cells. This is related in part to the fact that the soluble anti-CD3 scFv moiety alone is not in itself directly tumoricidal, nor does it elicit functional T-cell activation or cytokine release compared with its cross-linking mAb counterparts (27). As such, our in vitro and in vivo data corroborate that bscEGFRvIIIxCD3 is indeed dependent on target cell EGFRvIII surface expression, as indicated by the inability of bscEGFRvIIIxCD3 to mediate appreciable antitumor effects in the absence of EGFRvIII expression on matched tumor cell lines. Furthermore, the need for dual specificity was confirmed by the absence of antitumor activity both with the use of a control bscAbxCD3 and on EGFRvIII-specific blockade with cognate PEPvIII peptide. Importantly, these data also support peptide blockade as a useful strategy for eliminating unintentional T-cell activation against normal tissues. Given our findings, it may even be reasonable to create larger peptide constructs designed to not enter the CNS, which in theory would reduce systemic toxicity while permitting intracerebral efficacy of antibody-redirected T-cell therapies.
One unexpected finding in the present study is the ability of systemically administered bscEGFRvIIIxCD3 to treat tumors in the CNS. Several previous attempts have been made to validate immunotherapy as a viable treatment modality for tumors in the brain; however, in the great majority of published studies, beneficial antitumor immune responses have been limited to animal models in which vaccines were administered before tumor challenge. In contrast, it generally has been difficult to demonstrate successful treatment of intracerebral tumors after implantation, which might help explain the consistently poor clinical translation of immune-based interventions for patients with high-grade gliomas. Such considerations should be taken into account when interpreting the results of the present study, in which survival advantages were observed in the setting of even late-stage, well-established tumors. These results provide evidence that bscEGFRvIIIxCD3 may be considerably more effective on clinical translation compared with other strategies that have been tested in animal models of tumor challenge.
To test bscEGFRvIIIxCD3 in vivo, we chose an NSG mouse model, which has the distinct advantage of evaluating drug candidates in an animal system using human tumor tissue. Indeed, initial preclinical studies of the CD19-targeted BiTE now in clinical trials were originally performed in immunocompromised mouse models, the main limitations of which have been described elsewhere (28). In brief, one of the major drawbacks of this approach includes the need to supply human effector T cells, which is further compounded by the fact that human cells often have a limited functional half-life in the murine background. Even taking into account these considerations, however, it may be reasonable to conclude that our results in fact underestimate the full potential of bscEGFRvIIIxCD3, especially given that—in a realistic clinical scenario—circulating polyclonal T-cell populations should be present in great numbers and persistent throughout the body.
Owing to long-standing notions of immune privilege in the brain, previous studies of bscAbs in CNS malignancies were limited to intralesional treatment in vivo (29), and bscAbs had not been tested for efficacy by systemic administration as reported here. However, a growing body of evidence supports the possibility that EGFRvIII-specific molecules may have a special ability to accumulate in tumor tissue located in the brain, and that this phenomenon may be attributed in part to exclusive expression of the EGFRvIII mutation in malignancy. In human studies, systemically delivered radiolabeled antibodies specific for EGFRvIII have demonstrated high levels of tumor uptake in the brains of patients with gliomas (30). In contrast, antibodies specific for antigens expressed both in the brain and in other areas of the body were detected at relatively low levels intracerebrally, and instead accumulated preferentially in systemic tissues, including the liver, spleen, and bone marrow (31).
Such studies provide evidence that macromolecular-based therapies have the potential to mediate therapeutic effects at an intracerebral tumor site, and that a higher percentage of cerebral penetration may be achieved when targeting tumor-specific antigens like EGFRvIII. Given their greatly reduced molecular weight, bscAbs of the BiTE class may prove even more efficient in their ability to localize intracerebrally, and studies to further investigate this possibility are underway. Moreover, although CNS access is certainly limited to some degree, the relative potency of the BiTE platform at extremely low doses suggests that only small amounts may actually need to reach the tumor to mediate significant therapeutic effects.
We also observed an association between the therapeutic effects of bscEGFRvIIIxCD3 and the accumulation of immune cells in the CNS. Activated T lymphocytes are known to have the ability to penetrate the blood–brain barrier (BBB) even under normal physiological conditions (32). Interestingly, recent data from clinical trials showed that treatment with BiTEs may lead to the peripheral activation of circultating T cells, which in certain studies was temporally associated with subsequent unexplained, but transient, CNS side effects in multiple patients (11, 16). Whether the mechanisms behind these phenomena can be explored to enhance intracerebral localization of T cells—or that of BiTEs attached to the surface of infiltrating T cells—is currently under investigation.
The primary goal of the present study was to propel translation of a treatment for patients with GBM. Further investigation is needed to determine whether the therapeutic benefits reported here can be replicated in human studies, and whether the efficacy of bscEGFRvIIIxCD3 is affected by standard-of-care therapies for GBM, including radiation and temozolomide chemotherapy, a major side effect of which is profound lymphopenia. These areas of research may best be approached in preclinical murine models with syngeneic tumors or in transgenic mice with T cells expressing human CD3 (33). Almost certainly, the relevance of such studies will continue to broaden as this promising drug class progresses through ongoing clinical trials.
Materials and Methods
Detailed information on the mice and cell lines used in this study is provided in SI Materials and Methods.
BscEGFRvIIIxCD3 preparation, flow cytometry, and additional in vitro and in vivo analyses were carried out as described in SI Materials and Methods.
Unless stated otherwise, groups were compared using a two-sample, two-tailed t test to determine statistical significance. Survival curves were estimated for each group using Kaplan–Meier product-limit estimation. Primary comparative analyses of the curves for each group were performed using the generalized Wilcoxon test. Statistical significance was set at P < 0.05.
Supplementary Material
Acknowledgments
We thank Charles Pegram, David Snyder, and Scott Szafranski for their expert technical assistance. This work was supported in part by National Institutes of Health Grants 5R01-CA135272-04 (to J.H.S.), 5P50-NS020023-29 (to D.D.B. and J.H.S.), 3R25-NS065731-03S1 (to J.H.S.), and 1P01-CA154291-01A1 (to D.D.B. and J.H.S.), as well as grants from the Pediatric Brain Tumor Foundation (to D.D.B. and J.H.S.), Ben and Catherine Ivy Foundation (to J.H.S.), Duke Cancer Institute (to J.H.S. and B.D.C.), and Cancer Research Institute (to B.D.C.).
Footnotes
Conflict of interest statement: B.D.C., C.-T.K., M.C., D.D.B. and J.H.S. have a patent pending for EGFRvIII as a tumor-specific target for bispecific antibody therapy.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219817110/-/DCSupplemental.
References
- 1.Stummer W, et al. Fluorescence-guided resection of glioblastoma multiforme by using 5-aminolevulinic acid-induced porphyrins: A prospective study in 52 consecutive patients. J Neurosurg. 2000;93(6):1003–1013. doi: 10.3171/jns.2000.93.6.1003. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups National Cancer Institute of Canada Clinical Trials Group Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Imperato JP, Paleologos NA, Vick NA. Effects of treatment on long-term survivors with malignant astrocytomas. Ann Neurol. 1990;28(6):818–822. doi: 10.1002/ana.410280614. [DOI] [PubMed] [Google Scholar]
- 4.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3(9):666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: Moving beyond current vaccines. Nat Med. 2004;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudley ME, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suntharalingam G, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 10.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 11.Bargou R, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321(5891):974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 12.Choi BD, et al. Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin Biol Ther. 2011;11(7):843–853. doi: 10.1517/14712598.2011.572874. [DOI] [PubMed] [Google Scholar]
- 13.Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69(12):4941–4944. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- 14.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: Case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18(4):666–668. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan RA, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klinger M, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119(26):6226–6233. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- 17.Heslop HE. Safer CARS. Mol Ther. 2010;18(4):661–662. doi: 10.1038/mt.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi BD, et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009;19(4):713–723. doi: 10.1111/j.1750-3639.2009.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bluemel C, et al. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother. 2010;59(8):1197–1209. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuan CT, et al. Increased binding affinity enhances targeting of glioma xenografts by EGFRvIII-specific scFv. Int J Cancer. 2000;88(6):962–969. doi: 10.1002/1097-0215(20001215)88:6<962::aid-ijc20>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 21.Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006;43(6):763–771. doi: 10.1016/j.molimm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Yang I, et al. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci. 2010;17(11):1381–1385. doi: 10.1016/j.jocn.2010.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohr J, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin Cancer Res. 2011;17(13):4296–4308. doi: 10.1158/1078-0432.CCR-10-2557. [DOI] [PubMed] [Google Scholar]
- 24.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 25.Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 26.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 27.Le Gall F, Reusch U, Moldenhauer G, Little M, Kipriyanov SM. Immunosuppressive properties of anti-CD3 single-chain Fv and diabody. J Immunol Methods. 2004;285(1):111–127. doi: 10.1016/j.jim.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Dreier T, et al. T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3- bispecific single-chain antibody construct. J Immunol. 2003;170(8):4397–4402. doi: 10.4049/jimmunol.170.8.4397. [DOI] [PubMed] [Google Scholar]
- 29.Grosse-Hovest L, et al. Supraagonistic, bispecific single-chain antibody purified from the serum of cloned, transgenic cows induces T-cell–mediated killing of glioblastoma cells in vitro and in vivo. Int J Cancer. 2005;117(6):1060–1064. doi: 10.1002/ijc.21294. [DOI] [PubMed] [Google Scholar]
- 30.Scott AM, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci USA. 2007;104(10):4071–4076. doi: 10.1073/pnas.0611693104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zalutsky MR, Moseley RP, Coakham HB, Coleman RE, Bigner DD. Pharmacokinetics and tumor localization of 131I-labeled anti-tenascin monoclonal antibody 81C6 in patients with gliomas and other intracranial malignancies. Cancer Res. 1989;49(10):2807–2813. [PubMed] [Google Scholar]
- 32.Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005;26(9):485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn C, et al. Human CD3 transgenic mice: Preclinical testing of antibodies promoting immune tolerance. Sci Transl Med. 2011;3(68):68ra10. doi: 10.1126/scitranslmed.3001830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.