

Abstract
The HIV-1 envelope glycoprotein (Env) undergoes conformational transitions consequent to CD4 binding and coreceptor engagement during viral entry. The physical steps in this process are becoming defined, but less is known about their significance as targets of antibodies potentially protective against HIV-1 infection. Here we probe the functional significance of transitional epitope exposure by characterizing 41 human mAbs specific for epitopes exposed on trimeric Env after CD4 engagement. These mAbs recognize three epitope clusters: cluster A, the gp120 face occluded by gp41 in trimeric Env; cluster B, a region proximal to the coreceptor-binding site (CoRBS) and involving the V1/V2 domain; and cluster C, the coreceptor-binding site. The mAbs were evaluated functionally by antibody-dependent, cell-mediated cytotoxicity (ADCC) and for neutralization of Tiers 1 and 2 pseudoviruses. All three clusters included mAbs mediating ADCC. However, there was a strong potency bias for cluster A, which harbors at least three potent ADCC epitopes whose cognate mAbs have electropositive paratopes. Cluster A epitopes are functional ADCC targets during viral entry in an assay format using virion-sensitized target cells. In contrast, only cluster C contained epitopes that were recognized by neutralizing mAbs. There was significant diversity in breadth and potency that correlated with epitope fine specificity. In contrast, ADCC potency had no relationship with neutralization potency or breadth for any epitope cluster. Thus, Fc-mediated effector function and neutralization coselect with specificity in anti-Env antibody responses, but the nature of selection is distinct for these two antiviral activities.
It is well accepted that direct virus neutralization is an important element of antibody-mediated protection against HIV-1 (refs. 1–6 and reviewed in ref. 7). In contrast, less is known about the role of Fc-mediated effector function in the control of HIV-1, although four lines of evidence signal its importance. First, studies in HIV-1–infected people (8–14) and in macaques infected with simian immunodeficiency virus (15, 16) consistently show an inverse correlation between Fc-mediated effector functions, including antibody-dependent cell-mediated cytotoxicity (ADCC) (8, 9) or antibody-dependent cell-mediated viral inhibition (ADCVI), and viral loads or decreased disease progression (17). Second, vaccine-elicited protection both in nonhuman primates (18–21) and in a subset of human subjects in the Vax-004 trial (22) correlates with Fc-mediated effector function often observed in the absence of detectable neutralizing antibodies (18–21). Similarly, there was an inverse relationship between acquisition of HIV-1 and ADCC in the RV144 trial for a subset of subjects who had low to moderate IgA anti-gp120 titers (23). Third, breast milk IgG ADCC responses to gp120 but not to virus neutralization correlated with reduced perinatal transmission of HIV-1 (24). Fourth, passive immunization studies in nonhuman primates (25, 26) showed that abrogation of Fc-mediated effector function diminished the sterilizing protection afforded by the neutralizing mAb b12. These compelling studies show that neutralization alone significantly protects against a simian-human immunodeficiency virus challenge and that Fc-mediated effector function augments this effect. Taken together, these four lines of investigation strongly suggest that Fc-mediated effector function in addition to neutralization contributes to antibody-mediated protection against HIV-1. Thus, it is important to determine the precise relationships among antibody specificity, neutralization, and Fc-mediated effector function in protection against HIV-1.
In this report, we probe these relationships using a panel of human mAbs that recognize transitional epitopes exposed during the earliest stage of viral entry, the interaction of gp120 with CD4. Our studies deliberately focus on antibody responses to epitopes that become exposed during viral entry because passive immunization studies indicate that an antibody has at most a 24-h window to block transmission (ref. 27; reviewed in ref. 28). Thus, transmission-blocking antibodies must block infection by direct neutralization of HIV-1, by Fc-mediated killing of nascently infected cells, or both. Although these two effector functions often are coincident for a given mAb specificity (29, 30), they can be dissociated because nonneutralizing epitopes on both gp120 (12, 31) and gp41 (32) can be ADCC targets. In this report, we probe the relationships among antibody specificity, ADCC, and neutralization using a panel of human mAbs that recognize transitional epitopes exposed on target cells during viral entry.
Results
mAb Isolation and Epitope Cluster Assignment.
A set of 41 CD4-induced (CD4i) mAbs were isolated from five HIV-1–infected individuals and characterized for initial reactivity as detailed in Materials and Methods using recombinant proteins based on the HIV-1Ba-L isolate. All CD4i mAbs showed preferential binding to gp120-CD4 complexes compared with monomeric gp120; none bound trimeric gp140 (SOSIP); and 10 mAbs bound only to gp120-CD4 complexes (Fig. S1). Thus, these 41 CD4i mAbs recognize transitional epitopes that are exposed on HIV-1 envelope glycoprotein (Env) consequent to CD4 binding. Initial epitope specificity assignments were made by competition ELISA using the well-characterized mAbs A32 (33), C11 (33), 17b (34, 35), and 19e (36). mAb A32 recognizes an epitope strongly affected by mutations in mobile layers 1 and 2 of gp120 (37, 38). These layers reorient the seven-stranded β-sandwich platform that harbors the gp41-interactive region relative to the heavily glycosylated gp120 outer domain upon CD4 binding (38). mAb C11 recognizes an epitope strongly affected by mutations in residues of the seven-stranded β-sandwich in addition to a residue in the extended C terminus of gp120 (38, 39). mAbs A32 and C11 are nonneutralizing (Table S1 and refs. 33 and 40), and A32 detects an epitope that is a strong ADCC target in HIV-1–infected people (31). mAb 17b recognizes a well-defined epitope in the classical coreceptor-binding site (CoRBS) (35, 41). mAb 19e (36) recognizes a hybrid epitope involving residues of the CoRBS and CD4 (42). Thus, in contrast to 17b, which binds modestly to monomeric gp120 in the absence of CD4, 19e is strictly complex specific. Both 17b and 19e are neutralizing, depending on the isolate (Table S1 and refs. 35, 36, and 43) and assay format (36), although 19e neutralizes only in the presence of subsaturating concentrations of soluble CD4 (Table S1 and ref. 36).
Three epitope clusters (denoted “A,” “B,” and “C”) are apparent in Table 1 where percent binding values are presented for limiting concentrations of biotinylated proband mAbs and saturating concentrations of unlabeled competitor mAbs. Lower values in Table 1 indicate greater competition. These data illustrate the diversity of specificity among our 41 CD4i mAbs in which nine distinct patterns are apparent among three major epitope clusters.
Table 1.
Epitope cluster assignment by mAb competition
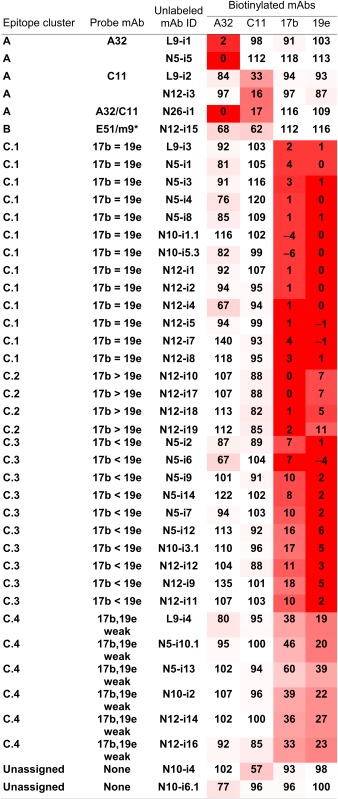
Competition of biotinylated mAbs (top row, last four columns) by unlabeled mAbs (third column) for binding to FLSC in ELISA. To aid comparison, the relative values are shaded in degrees of red corresponding to the degree of competition.
Epitope Cluster A.
In epitope cluster A (Table 1), two mAbs compete for A32, two mAbs compete for C11, and one mAb competes for both. Although C11 and A32 clearly recognize distinct epitopes (38, 44, 45), these mAbs are weakly cross-competitive but in a nonreciprocal fashion (44). Thus, there are at least three distinct patterns of reactivity for cluster A epitopes involving both N- and C-terminal residues on the face of gp120 occluded by gp41 in trimeric Env (38, 45). Although the epitopes recognized by these mAbs are not neutralization determinants (Table S1), they are potent ADCC targets.
Fig. 1A depicts ADCC curves for the cluster A mAbs using target cells sensitized with gp120 of the HIV-1Ba-L isolate. ADCC strength is indicated by two parameters, plateau levels of cytotoxicity and the mAb concentration (in nanomolars) required for 50% maximal plateau cytotoxicity (EC50). Over a large series of studies, we have found that cluster A mAbs consistently mediate the highest levels of plateau cytotoxicity, which ranges from ∼30–60% absolute cytotoxicity across individual experiments. Thus, to compare ADCC activity among experiments, we normalize plateau cytotoxicity values using either C11 or N12-i3 as positive controls. It should be noted that small experimental deviations above 100%, representing the statistical variation in our ADCC method, often are seen in dose–response curves. When this approach is used, all cluster A mAbs mediate potent ADCC with median EC50s of 0.15 ± 0.19 nM (SD) and median plateau cytotoxicity values of 101.10 ± 5.6% (SD). Because these mAbs cross-compete for A32, C11, or both, it appears that they recognize a cluster of at least three epitopes on the face of gp120 that is buried on trimeric Env but consequently is exposed to CD4 triggering. Epitope cluster A is depicted on a model of CD4-triggered gp120 in Fig. 2A where it maps to an electronegative surface of CD4-triggered gp120 that is occluded on the untriggered Env trimer by gp41 (46). Although the precise epitope boundaries recognized by cluster A mAbs are not yet defined, these mAbs share the common property of being electropositive by isoelectric focusing (Fig. 2B).
Fig. 1.

(A–C) ADCC curves for mAbs described in Table 1. ADCC was performed as described in Materials and Methods using CEM-NKr-CCR5 target cells sensitized with gp120 of the HIV-1Ba-L isolate.
Fig. 2.

Properties of cluster A mAbs and cognate epitopes. (A) Cluster A mAbs recognize at least epitopes on the electronegative face of gp120 normally occluded by gp41, which comprises the N- and C-terminal extensions, the seven-stranded β-sandwich, and the flexible topological layer 1 of gp120. The electrostatic potential is displayed at the molecular surface of gp120 and is shown in red for negative, in blue for positive, and in white for apolar values. (B) Cluster A mAbs have basic pI values by isoelectric focusing. Mobilities of purified cluster A, B, and C mAbs are shown for a silver-stained IEF gel (pH 6.0–11.0). Each mAb (2 ug) was electrophoresed over an IEF focus gel, pH 6–11 (Gel Company), which then was silver stained. Lanes 1 and 2 correspond to A32 and C11, respectively. Lanes 3–7 are the cluster A mAbs (L9-i1, N5-i5, L9-i2, N12-i3, and N26-i1, respectively). Lane 8 is the cluster B mAb N12-I15. Lanes 9–11 represent mAbs 17b, 19e, and E51, respectively. The cluster C.1 mAbs N5-i4, N5-i3, N10-i5.3, N12-i1, N12-i2, and N12-i4 are in lanes 12–17, respectively; cluster C.2 mAbs N12-i17, N12-I18, and N12-I1 are in lanes 18–20, respectively; cluster C.3 mAbs N5-I2 and N10-I3.1 are in lanes 21 and 22, respectively; and cluster C.4 mAbs L9-i4 and N10-i2 are in lanes 23 and 24, respectively. Lanes 25 and 26 have the ungrouped mAbs N10-i4 and N10-i6.1, respectively. (C) Crystal structures of Fab A32, Fab N5-i5, Fab N12-i15, and Fab N12-i2. (Top) A ribbon representation of overall structures. Fabs are aligned by superposition of their V domains, and CDR H3s are shown in yellow with a dotted yellow line indicating the disordered H100SYYEPGTSY region of mAb N12-i2. (Middle and Bottom) Head-on views showing the paratope with molecular surfaces displayed (rotated 90° from A). For clarity, only V domains are shown. Molecular surfaces are colored according to CDR contribution (CDR H1, red; H2, green; H3, yellow; L1, orange; L2, magenta; and L3, cyan) (Middle), and the electrostatic potential is displayed and shown in red for negative, blue for positive, and white for apolar values (Bottom). Note: The electrostatic potential is displayed over the N12-i2 molecular surface as observed in the crystal and does not show the contribution of the disordered H100SYYEPGTSY region, which carries additional negative charges of a single glutamic acid and two sulfotyrosines.
Based on these observations, we have obtained the crystal structures for the antigen-binding fragments (Fabs) of two cluster A mAbs, A32 and N5-i5, at resolutions of 1.85 Å and 1.95 Å, respectively (Fig. 2C and Table S2; also see Fig. S3). A continuous and well-defined electron density was observed throughout the entire V domains with all complementarity-determining region (CDR) residues being structurally well ordered as indicated by low average B-factors (Table S2). The general shapes and overall electrostatics of the presumed antigen-binding sites of these mAbs are shown in Fig. 2C. Notably each of the two cluster A mAbs has a strong electropositive paratope region contributed largely by positive charges of CDR H3 (mAb A32) or CDR H2 (mAb N5-i5). The overall positive electrostatic surfaces of the A32 and N5-i5 paratopes are consistent with their binding to epitopes in the electronegative cluster A region of gp120. This observation also is consistent with older literature reporting a general inverse charge relationship between epitopes and paratopes (47). mAbs A32 and N5-i5 have relatively short CDR H3s (13 and 10 residues long, respectively), and their putative antigen-binding sites are relatively flat (Fig. 2C). Taken together, the above data show that mAbs that recognize cluster A epitopes uniformly mediate potent ADCC and that they likely recognize net electronegative epitopes on the surface of gp120.
Epitope Cluster B.
Three additional nonneutralizing mAbs, N12-i15, N10-i4, and N10-6.1, were not clearly assignable to competition groups using A32, C11, 17b, and 19e. N10-i4, and N10-6.1 remain unassigned pending further epitope mapping studies. In contrast, although mAb N12-i15 did not compete for either 17b or 19e, it competed for two other CD4i mAbs, E51 (40, 48) and m9-IgG1 [a mutant derivative of mAb ×5 (49, 50)] that recognize CoRBS epitopes (Table 1) (40, 49, 50). Further epitope mapping of N12-i15 using mutant gp120s (Fig. S2 B and C) showed that this mAb recognizes a conformational epitope that is expressed only on gp120 that is triggered by CD4 and involves the V1/V2 loop in conjunction with an isoleucine at position 420 in the CoRBS (Fig. S2 B and C). Although the epitope recognized by N12-i15 is nonneutralizing (Table S1), it is a potent ADCC target (Fig. 1B). It is denoted as “cluster B” in Table 1. mAb N12-i15 mediates ADCC with an EC50 of 1.17 nM and a plateau cytotoxicity of 103.8%, placing it as slightly less potent than cluster A mAbs in terms of EC50 but equal to them in plateau cytotoxicity. Interestingly, it is the only example of such a mAb in our panel and, as such, might represent an uncommon specificity. We have obtained the crystal structure of N12-i15 Fab at 2.6-Å resolution (Fig. 2C). The asymmetric unit of tetragonal crystal of Fab N12-i15 contained four structurally independent but essentially similar Fab molecules (Fig. S3). A continuous and well-defined electron density was observed for all CDR loops with an average V domain B-factor of 33.0 Å2, similar to the average B-factor for the entire structure (Table S2). mAb N12-i15 lacks the long CDR H3 typically associated with the classical CD4i mAbs, such as 17b, that recognize CoRBS epitopes and interact with their cognate epitopes primarily through relatively long and acidic CDR H3s (48). The presumed antigen-binding site of N12-i15 is formed by the moderately long (16 residues) CDR H3 and the exceptionally long CDR L1 (also 16 residues) [category of κ canonical 4 of long CDR L1 loops as defined by Chothia and Lesk (51)], which stack against each other to form a relatively flat groove that is electronegative throughout. The electronegative patch is buried at the heavy chain variable region/light chain variable region interface, and overall the N12-i15 paratope is electrostatically positive. The conformations of CDR H1 and L1 are stabilized by an extended network of internal hydrogen bonds and by multiple hydrogen bonds and stacking interactions within the V domain.
Epitope Cluster C.
In contrast to the cluster A and cluster B mAbs, a majority (33/41) of our mAb panel competed for 17b and 19e, where four distinct subclusters are apparent (Table 1). The structures recognized by these mAbs are denoted as “cluster C epitopes” in Table 1 and Fig. 2 and constitute an unexpectedly diverse set of determinants in terms of specificity and function. Table 1 shows that 13 mAbs (cluster C.1) cross-compete for both 17b and 19e with equal potency. In contrast, four mAbs (cluster C.2) have a slight preference for competing for 17b, 10 mAbs (cluster C.3) have a slight preference for competing for 19e, and six mAbs (cluster C.4) compete, although weakly, for both 17b and 19e. Surprisingly, these small differences in competition are functionally significant in terms of neutralization (Table S1 and Fig. 3).
Fig. 3.

Neutralization of the Clade C Tier 1 pseudovirus MW965.26 by cluster C mAbs. Statistical significance was determined by an unpaired t test using Welch’s correction using GraphPad Prism software. Because none of the mAbs in cluster C.4 neutralized (IC50 >50 μg/mL), one value was set arbitrarily at 49.9 μg/mL, and the remainder of this group was set at 50.0 μg/mL to enable the t test.
It has been reported that structure-based stabilization of gp120 enhances the immunogenicity of CoRBS epitopes that correlates strongly with the increased neutralization of the Tier 1 Clade C pseudovirus, MW965.26 (52). Neutralization of this pseudovirus also discriminated among the subclusters in epitope cluster C identified above (Table 1 and Fig. 3). Neutralization of MW9652.26 was greatest in clusters C.1 and C.2 (Table S1, Fig. 3), where there is strong competition for 17b and 19e (Table 1). Indeed, it is most apparent in cluster C.2, where there is a slight preference in competition for 17b over 19e. Only one (L9-i3) of the 17 mAbs in clusters C.1 and C.2 failed to neutralize this virus (Table S1). In contrast, subclusters C.3 and C.4 exhibited weaker neutralization of MW9652.26 (Table S1 and Fig. 3). Three of 10 cluster C.3 mAbs failed to neutralize MW965.26, and one mAb (N12-i11) did so questionably (IC50 = 49.40 μg/mL; Table S1). None of the cluster C.4 mAbs neutralized this virus. Collectively, these data signal substantial neutralization diversity among classical CoRBS epitopes, with the most potent being those that overlap the classical 17b epitope. Neutralization, in terms of inhibiting both MW965.26 (Fig. 3 and Table S1) and other Tier 1 pseudoviruses, is much less apparent for clusters C.3 and C.4, which are less “17b-like” (Table S1). In fact, cluster C.4 epitopes are nonneutralizing using our current virus panel and assay format (Table S1). It also is interesting that a majority of the cluster C.4 epitopes are present exclusively on gp120-CD4 complexes (Fig. S1, pattern C). However, these data also suggest that neutralizing responses to classical CoRBS epitopes represent selection for paratopes that more closely fit CoRBS epitopes that are conserved on Core-V3 structures, such as those described in refs. 52 and 53.
This suggestion is supported most strongly by the crystal structure of our second most potent MW965.26-neutralizing mAb, N12-i2, from cluster C.1 solved at 1.95-Å resolution (Fig. 2C and Table S2). The hexagonal crystals of Fab N12-i2 contained one Fab molecule in the asymmetric unit in which nine residues (SYYEPGTSYH100h, Kabat numbering) of the CDR H3 β-hairpin region were disordered and missing in the final model (Fig. 2C and Table S2). Structural analysis confirmed that mAb N12-i2 shares a common characteristic of the antigen-binding site with canonical CD4i mAbs recognizing CoRBS-associated epitopes. Its paratope is dominated by a long (25-residue) flexible and acidic CDR H3 (−2 net charge as calculated based on the unmodified amino acids) that additionally is tyrosine-sulfated. HPLC and electrospray ionization mass spectrometry analyses suggested that two tyrosine residues in the disordered segment of CDR H3 are modified by O-sulfation (Fig. S4).
As shown in Fig. 1C, ADCC was observed for each of the subgroups of epitope cluster C, although there was significant intrasubcluster heterogeneity. Three patterns of ADCC are apparent. First, each subcluster had at least one mAb that exhibited 100% plateau cytotoxicity, such as mAb N5-i3 in cluster C.1. Only one of these mAbs was as potent as cluster A mAbs in terms of EC50 (Fig. 1, compare A and C). Second, most of the mAbs exhibited ∼75% plateau cytotoxicity across the subclusters. This result is understandable, because once the gp120-CD4 complex has bound to CCR5, the epitope should become occluded. Our target cells have ∼50,000 molecules of CD4 and 15,000 molecules of CCR5. Thus, we would expect plateau cytotoxicity to be in the range of 75% for fully occluded CoRBS epitopes. Third, each group has mAbs that reproducibly exhibit low plateau cytotoxicity in the range of ∼20%. The reason for this low plateau cytotoxicity is not clear, because all our mAbs were expressed in a common cell line (293T) on the IgG1 backbone, making it unlikely that the differences are caused by glycoform heterogeneity. However, because these low levels of plateau cytotoxicity are seen for only a subset of the cluster C mAbs, it is most likely that these differences represent microheterogeneity in epitope representation after gp120 is bound to cell-surface CD4. Clarification of this issue is underway.
Quantitative Ranking of ADCC Activity.
The studies described above suggest a wide range of ADCC activities among mAbs specific for epitope clusters A–C. This range is illustrated clearly in Fig. 4 where EC50 is plotted vs. the plateau cytotoxicity for our CD4i mAb panel using target cells sensitized with recombinant gp120 of the HIV-1Ba-L isolate. A striking pattern emerges from this analysis: All cluster A and cluster B mAbs localize to the far right region of the plot bounded by percent plateau cytotoxicity of >95% and EC50 <2 nM (the area enclosed by a rectangle in Fig. 4). In contrast, there was marked diversity among the cluster C mAbs, only two of which were in the potent region. The other mAbs ranged in activity from just outside the potent region to very low values of both plateau cytotoxicity and EC50. As expected from Fig. 1C, there was no consistent pattern among epitope cluster C subclusters. These data provide a quantitative potency parameter to define the relative activities of anti-Env mAbs that mediate ADCC. Further, our data also confirm that ADCC responses in vivo (12) are selected in an epitope-selective fashion.
Fig. 4.

Plot of % plateau cytotoxicity vs. EC50 for cluster A, B, and C mAbs. The mAb clusters are denoted as follows: cluster A, black circles; cluster B, red squares; cluster C.1, blue triangles; cluster C.2, blue inverted triangles; cluster C.3, blue diamonds; and cluster C.4, blue circles. The rectangle encloses the potent region.
Cluster A, B, and C mAbs Mediate ADCC During Bona Fide Viral Entry.
The studies described above used target cells passively sensitized with monomeric gp120, and it is possible that epitope exposure in this system does not faithfully represent epitope exposure when virions bind target cells. This issue was addressed by using replication-defective, entry-competent virions spinoculated onto CEM.NKR-CCR5 target cells for ADCC. Fig. 5 shows plateau cytotoxicity levels for a sampling of cluster A, B, and C mAbs at 6.6 nM using target cells sensitized with AT-2–inactivated HIV-1Ba-L (54). Except for the cluster C.1 mAb N12-i4, each CD4i mAb exhibited high levels of plateau cytotoxicity on target cells sensitized with AT-2–inactivated HIV-1Ba-L virions, showing that the epitopes they recognize are exposed stably during viral entry. mAb N12-i4 did exhibit significant plateau cytotoxicity; however, the levels are lower than those observed for other C.1 mAbs on either gp120 (Fig. 1C) or AT-2–inactivated virion-sensitized target cells (Fig. 5). These studies show that CD4i mAbs can mediate significant ADCC on target cells during bona fide viral entry, indicating that they recognize epitopes that are biologically relevant targets of potentially protective antibodies, independently of their ability to neutralize.
Fig. 5.

ADCC activity for representative mAbs from each epitope cluster using CEM-NKr-CCR5 cells sensitized with AT-2–inactivated virions of the HIV-1Ba-L isolate. mAbs were used a saturating concentration of 6.6 nM. Each bar is the average of three replicate analyses.
Discussion
The studies described above reveal significant diversity in specificity and function among CD4i epitopes and their cognate antibodies. This finding is in accordance with previous predictions (55) and findings from a number of published studies (41, 42, 56–58) but contrasts with the suggestion that CD4i antibodies are monolithic in terms of specificity and function (59). Nevertheless, the degree of diversity among our panel of anti-CD4i mAbs was even greater than previously suspected. We were able to divide the mAbs into three major epitope clusters (clusters A, B, and C). However, even within each cluster there was significant diversity in specificity and function. This diversity is discussed below.
Epitope Cluster A.
Cluster A mAbs are nonneutralizing, but they are uniformly potent mediators of ADCC. Epitope cluster A is defined by cross-competition in our mAb panel using two known mAbs, A32 and C11 (33, 37, 38, 39, 45), as probands. mAb A32 recognizes an epitope involving topological layers 1 and 2 of gp120 (37, 38, 45). mAb C11 recognizes an epitope in the seven-stranded β-sandwich and C-terminal extension of gp120 (38, 39, 45). Although these epitopes are clearly distinct, A32 can partially compete for biotinylated-C11 binding to gp120 but not vice versa (44). Three distinct specificities are apparent in cluster A: Two mAbs strongly cross-compete for A32, two mAbs strongly cross-compete for C11, and one mAb strongly cross-competes for both. Collectively, these observations strongly suggest that the face of gp120 occluded in trimeric Env by gp41 is a potent ADCC target, regardless of epitope fine specificity. It should be noted that recent studies using plasmas from HIV-1– infected patients suggested that the A32 epitope is a potent ADCC target in vivo (31). Our studies confirm this conclusion and suggest further that at least two other epitopes on the same face of gp120 are equally potent ADCC targets. Our studies further indicate common physical chemical properties shared among the cluster A epitopes or their cognate mAbs that are responsible for ADCC potency.
In this regard, relatively little is known about the structures of cluster A epitopes save for small-scale mutagenesis studies of the determinants recognized by A32 and C11 (37, 38, 39, 45). Precise definition of the physical chemical basis of potent ADCC by cluster A mAbs awaits structural analysis of mAb-gp120 complexes, preferably through X-ray crystallography. Until then, our studies reveal common physical chemical properties of cluster A mAbs (and, by inference, cluster A epitopes) that might be key elements of ADCC potency. As shown in Fig. 2A, epitope cluster A maps to an electronegative face of gp120 that is occluded by gp41 in trimeric Env. This region becomes exposed after CD4-triggering during bona fide viral entry (Fig. 5) and Env-mediated cell-to-cell fusion (57, 58) where it is a potent ADCC target (Fig. 5). All cluster A mAbs examined to date have strongly basic isoelectric points determined by isoelectric focusing (IEF) (Fig. 2B) or by X-ray crystallography (Fig. 2C). In the latter case, crystal structures of both A32 and N5-i5 confirm strongly basic paratope regions. We propose that electrostatic interactions play a major role in ADCC potency of cluster A epitopes regardless of epitope fine specificity. A formal test of this hypothesis awaits the solution of cluster A mAb-gp120 structures by X-ray crystallography. Taken together, these results suggest unifying physical chemical principles for the ADCC potency of anti-gp120 antibodies. If further studies show that antibodies specific for cluster A epitopes contribute to protective immunity against HIV-1, this physical chemical information should help guide vaccine design.
Epitope Cluster B.
Epitope cluster B is defined by a single mAb, N12-i15, which is unique among the mAbs in our panel. mAb N12-i15 is similar to cluster A mAbs, in that it mediates potent ADCC (Fig. 1 A and B), and it falls into the potent zone shown in Fig. 5. It is also nonneutralizing in the TZM-bl cell assay (Table S1). Interestingly, cluster B is not exposed on either monomeric gp120 or trimeric Env until the conformational change is triggered by CD4 binding. Thus, N12-i15 is strictly specific for gp120-CD4 complexes (Fig. S1, pattern C), like 19e (42), but it does not cross-compete for this mAb (Table 1). Surprisingly, N12-i15 cross-competes for two other CD4i mAbs, E51 (40) and IgG1-m9 (×5) (Fig. S2A) (50, 60), that have overlapping reactivity with 17b and 19e but clearly recognize distinct epitopes. This unusual reactivity, along with the selective exposure of the cluster B on gp120-CD4 complexes, suggests that elements of the CoRBS contribute to this epitope. This suggestion was confirmed by mutagenesis studies in which the I420R mutation that abrogates CoRBS binding and the classical CoRBS-associated 17b epitope (34, 35) altered binding of N12-i15 to gp120-CD4 complexes (Fig. S2C). In this case, the binding was reduced by approximately 40-fold and was not abrogated as observed for 17b (Fig. S2C). Deletion mutagenesis showed that the N12-i15 epitope also involves elements of the V1/V2 loop (Fig. S2B). These studies show that N12-i15 recognizes a complex structure involving elements in the outer domain of gp120 that rearrange consequent to CD4 binding during viral entry. More precise definition of this structure is underway, although, given the regions involved in the epitope, it is unlikely that we will be able to characterize it by X-ray crystallography. The structure of N12-i15 Fab reveals a paratope region that comprises both electronegative and electropositive patches (Fig. 2B), resulting in an overall basic charge that is intermediate between the cluster A mAbs and the classical CoRBS mAb 17b [Fig. 2B; compare lane 8 (N12-i15) with lanes 1–7 (cluster A mAbs) and lane 9 (17b)]. The unique specificity and potent ADCC activity of N12-i15 has prompted an ongoing search for new antibodies in this group.
Epitope Cluster C.
Cluster C comprises mAbs that recognize CoRBS epitopes defined by competition with 17b and 19e, revealing a previously unsuspected complexity. We were able to categorize cluster C mAbs into four subclusters based on differential competition with these two mAbs. Cluster C.1 and C.2 include mAbs that completely inhibit 17b. Cluster C.1 mAbs also strongly block 19e binding to a full-length single-chain gp120-CD4 monomer, FLSC, whereas cluster C.2 mAbs competed for 19e slightly less than cluster C.1 mAbs. Thus, cluster C.1 and C.2 mAbs are representative of classical CoRBS mAbs, in that they have essentially the same neutralization spectrum as 17b (Table S1). In contrast, the neutralization spectrum was narrower for cluster C.3 and C.4 mAbs (Table S1), as best exemplified by neutralization of the Clade C pseudovirus MW965.26 that has been used as a measure of increased neutralization breadth for CoRBS-specific antibodies elicited by structurally constrained gp120 (52). Although ∼95% of the cluster C.1 and C.2 mAbs neutralize this virus, only 60% of the cluster C.3 mAbs and none of the cluster C.4 mAbs clearly neutralized this virus (Table S1). Neutralization of the Clade B Tier 2 virus QH0692.42 also differentiated cluster C.1 and cluster C.2 mAbs from cluster C.3 and C.4 mAbs (Table S1). Thus, epitope cluster C comprises at least four fine specificity groups in which decreasing neutralization breadths and potencies correlate with decreasing ability to compete for 17b and 19e. These data show the considerable diversity in fine specificity and neutralization among mAbs that recognize CoRBS epitopes.
The preferential neutralization of the Tier 1 Clade C pseudovirus MW965.26 and the Tier 2 Clade B pseudovirus QH0692.42 by cluster C.1 and C.2 mAbs suggests a bottleneck in somatic maturation of the neutralizing antibody response to classical CoRBS epitopes. To date, this spectrum of neutralization appears to be the maximum for this type of CoRBS-specific antibodies, as apparent in our data (Table S1) and in the literature (35, 40, 52, 61) and as supported by the observation that the “best” neutralizing antibodies of this category have long, tyrosine-sulfated CDR H3s (48–50, 62, 63). We were able to solve the crystal structure of our second most potent C.1 mAb, N12-i2, and found that it also has a 25-residue CDR H3 that is disordered in a segment that is tyrosine sulfated (Fig. 2C and Fig. S4). When our results are considered in the broader context of the literature, mAb N12-i2 and similar mAbs reported by others (35, 40, 52, 61) represent the maturational limit of neutralization breadth and potency that can be attained by somatic hypermutation of this epitope C subcluster. Thus, mAbs such as N12-i2 probably represent the endgame in terms of neutralization breadth and potency for this category of antibody. It should be noted that screening protocols using recombinant proteins might favor the repeated isolation of such mAbs. Thus broadly neutralizing antibodies specific for CoRBS-associated structures might be identified by alternative screening strategies. Consistent with this view, genetic signature analysis correlated neutralization breadth with reactivity to elements of the CoRBS (64); a broadly neutralizing mAb that recognizes part of the CoRBS was identified recently by screening on cell surface envelope trimers (65).
It also should be noted that four cluster C mAbs (N5-i9, N12-i9, N12-i11 of cluster C.3, and N10-i2 of cluster C.4) that share the common property of being strictly specific for gp120-CD4 complexes (Fig. S1, pattern 3) are neutralizing. We recently showed that 19e, another pattern 3-type neutralizing mAb, is specific for a hybrid epitope comprising residues from both gp120 and CD4 (42). Thus the epitopes recognized by the pattern 3 mAbs in cluster C also may recognize hybrid epitopes and may neutralize postattachment. Interestingly, these cluster C mAbs neutralize without pretriggering with soluble CD4, which is required for 19e neutralization (Table S1 and ref. 36). Studies are underway to characterize the epitopes recognized by the four cluster C mAbs with neutralizing activity to determine whether they comprise residues from both gp120 and CD4. It is interesting that most of the pattern 3 cluster C mAbs fall into clusters C.3 and C.4, which are either poorly neutralizing or nonneutralizing. If these mAbs also recognize hybrid epitopes (42), it is possible that the response to the CD4 moiety selects against neutralization potency. This hypothesis is under investigation.
Although there was a consistent relationship between epitope cluster C fine specificity and neutralization, none was observed for ADCC (Fig. 1C). Values for EC50 and plateau cytotoxicity ranged from two cluster C.1 and C.3 mAbs that have potencies approximately equal to those of cluster A and B mAbs (Fig. 4) to 14 mAbs from all cluster C subclusters with plateau cytotoxicities <40% and EC50s in the 1 nM to >1,000 nM range. The rest of the mAb panel had intermediate values of plateau cytotoxicity and EC50. There was no consistent pattern of ADCC activity among the subclusters that correlated with fine specificity determined by 17b and 19e mAb competition. This degree of diversity was unexpected and remains unexplained. There are two possibilities. First, there might be Fc-glycoform heterogeneity among the mAbs. This possibility is less likely, because all mAbs were expressed in 293T cells using the same IgG1 heavy-chain backbone. Preliminary glycoform analyses have not revealed obvious differences among the mAbs. In addition, the differences in plateau cytotoxicity are hard to explain by differences in glycoforms, which usually have a greater effect on EC50s. Second, there might be microheterogeneity in epitope-exposure dynamics within each subcluster. We are exploring this possibility by determining temporal dynamics of epitope-exposure intensity using single-particle imaging of viral entry. Regardless of the ultimate outcome of these studies, it is very clear that epitope clusters A and B are much more consistently potent than epitope cluster C as determined by either EC50 or plateau cytotoxicity.
In summary, the studies described above reveal unexpected diversity in specificity and function for CD4i mAbs. Although occasional CD4i mAbs can be broadly neutralizing (64, 65), our data show that an array of CD4i mAbs with limited neutralization breadth and potency nevertheless can produce a broad spectrum of Fc-mediated effector function via recognition of epitopes on the gp41-interactive face of gp120 (epitope cluster A) and/or within elements of the CoRBS (epitope clusters B and C). This information sets the stage for future studies to determine the relative contributions of neutralization and Fc-mediated effector function to vaccine-elicited protection against HIV-1. This information should be particularly useful because two very recent studies further implicate a role for Fc-mediated effector function in the prevention of HIV-1 acquisition.
The first study found an inverse correlation between ADCC in two assay formats and acquisition in a subset of RV144 vaccinees that had low to moderate IgA anti-Env antibody responses (23). Notably, A32-like cluster A mAbs that mediate ADCC were isolated readily from a subset of RV144 subjects (66). In the second study, ADCC activity of breast milk IgG correlated with decreased maternal–child transmission of HIV-1 (24). Taken together with an earlier study showing an inverse relationship between ADCVI and HIV-1 acquisition in a subset of Vax-004 subjects (22), substantial data in humans now indicate that Fc-mediated effector functions play a role in reducing HIV-1 acquisition. Our studies shed light on the range of epitopes that are potential targets of potent Fc-mediated effector responses and should aid in further refinements of HIV-1 vaccine candidates.
Materials and Methods
Research Subjects.
Five HIV-1–infected individuals were selected for this study. Volunteers NVS5, NVS10, and NVS12 are part of our Natural Viral Suppressor (NVS) cohort, a group of HIV-1–infected individuals who suppress HIV-1 replication to <400 copies/mL in the absence of antiretroviral therapy (67–71). These individuals have average CD4 counts of 569 cells/μL, 991 cells/μL, and 1,124 cells/μL, respectively, over 13, 11, and 7 y of infection, respectively. Volunteer NVS26 was diagnosed with acute retroviral syndrome with 9,976 HIV-1 RNA copies/mL and was placed immediately on antiretroviral therapy for 5 y before halting therapy. NVS26 has maintained an average CD4 count of 752 cells/μL and <75 HIV-1 RNA copies/mL for more than 4 y after stopping therapy (the time point of the sample). LongT9 is an individual with HIV infection who has maintained an average CD4 count of 1,246 cells/μL and an average of 6,568 HIV-1 RNA copies/mL for almost 10 y The University of Maryland Institutional Review Board approved the use of human subjects, and all individuals provided informed consent.
mAb Isolation.
mAb isolation was carried out as described in ref. 68. Briefly, culture supernatants from memory B cells activated at limiting dilution were screened by ELISA for binding to monomeric gp120, a disulfide-stabilized gp140 SOSIP trimer (72, 73), and a single-chain gp120-CD4 monomer, FLSC (74), all based on the HIV-1Ba-L isolate. This strategy permits the rapid initial assignment of epitope specificity to CD4i (selective reactivity with FLSC), CD4-binding site (CD4bs) (selective reactivity with gp120 and SOSIP gp140), gp120 (reactivity with all three proteins), or gp41/oligomer (selective reactivity with SOSIP gp140) (Fig. S1). Initial epitope-specificity assignments were confirmed using mutagenized fragments of gp120 or gp41 and synthetic peptides. Using this approach, we were able to categorize 61 unique mAbs into CD4i (41 mAbs), CD4bs (10 mAbs), V3 (8 mAbs), and gp41 (2 mAbs) reactivities. The CD4i mAbs were selected for further study. All mAbs, including mAbs used as positive controls [A32 (44, 75), C11 (44, 39), 17b (34, 35), 19e (36), E51 (40) PG9 (76), PG16 (76), and b12 (77)] were expressed from plasmid clones in 293T cells using an IgG1 backbone for heavy-chain variable regions and either a κ- or λ-chain expression vector for light-chain variable regions. mAbs were purified from culture supernatants by protein-A chromatography. IgGm9 (50) was generously provided by Dimiter Dimitrov (National Cancer Institute, Bethesda, MD). The anti-respiratory syncytial virus mAb Synagis (Medimmune) was used as a negative control in some experiments. It was purchased from the University of Maryland Hospital Pharmacy.
ELISA.
ELISAs were performed using an antigen-capture format (78) as described previously (68, 79).
ADCC Assays.
ADCC assays were carried out using the rapid fluorescence ADCC method described in ref. 80 modified to reduce prozone effects. CEM-NKr-CCR5 target cells were sensitized with recombinant gp120 of the HIV-1Ba-L isolate as described in ref. 80 (in most studies) or with inactivated HIV-1 as described below. The sensitized target cells were incubated with mAb dilutions for 15 min and were washed with culture medium before the addition of peripheral blood mononuclear effector cells from healthy donors at a final ratio of 50:1. The effector and target cells were pelleted by centrifugation and incubated for 3 h at 37 °C followed by fixation and cytolysis determined by flow cytometry as described in ref. 80. In one series of studies, the CEM-NKr-CCR5 target cells were sensitized by spinoculation for 2 h at 12 °C exactly as described in ref. 81 with AT-2–inactivated HIV-1 of the Ba-L isolate (54) at a multiplicity of infection (MOI) of 5. This MOI was calculated using analytical information provided by Jeffery Lifson (National Cancer Institute at Frederick, Frederick, MD), who generously supplied this preparation. This highly purified virus preparation has been characterized extensively for the presence of stable (i.e., nonshedding) Env trimers in which the gp120:gp41 ratios remain 1.0 during storage and after freeze–thaw cycles (82). Absolute cytotoxicities ranged from ∼30 to 60% at plateau in each experiment regardless of the mode of sensitization. Absolute cytotoxicity values were normalized using the mAbs C11 or N12-i3, because they consistently yield maximum values of absolute cytotoxicity among the many mAbs we have tested. These mAbs recognize overlapping epitopes in the seven-stranded β-sandwich of gp120 (see below and refs. 38 and 45). Values of plateau cytotoxicity were taken as the peak value in the plateau region of the dose–response curve. The concentrations of mAb (in nanomolars) mediating EC50 were determined by nonlinear curve fitting of the dose–response curves using internal algorithms of GraphPad Prism. In the few instances in which a true plateau in the dose–response curve was not achieved, the peak cytotoxicity value was used, and EC50s were determined by nonlinear curve fitting as above. In all such cases, potencies were weak, and the values determined did not adversely affect the rank ordering of the mAbs.
Virus Neutralization Assays.
Standardized virus neutralization assays were carried out using a standardized TZM-bl assay and HIV-1 pseudotyped viruses as described in ref. 83. In addition, neutralization of CD4-triggered HIV-27312A in TZM-bl cells was performed as described (36).
X-Ray Crystallography: Crystallization Experiments.
Fabs were produced from purified IgG1 (10 mg/mL) by proteolytic digestion with immobilized papain (Pierce) and were purified using protein A (GE Healthcare), followed by gel filtration chromatography on a Superdex200 16/60 column (GE Healthcare) in a buffer containing 0.3 M NaCl, 5 mM Tris (pH 7.2), and 0.02% NaN3. Fab molecular weights were determined by electrospray ionization MS. Initial vapor-diffusion sitting-droplet crystallization trials at room temperature were set up robotically (OryxNano Protein Crystallization Robot; Douglas Instruments) using Fabs at a concentration of 10 mg/mL, commercially available Crystal Screens from Hampton Research, and Classics Suites from Qiagen (0.2-μL droplets mixed in a 1:1 ratio). Crystallization hits from robotic screening were optimized and scaled up manually using a hanging-drop vapor diffusion method in a 24-well plate format. Final crystallization conditions are shown in Table S3. Crystals were soaked briefly in reservoir solution supplemented with 25–30% (vol/vol) glycerol and were flash-frozen in the nitrogen steam at 100 K.
Data Collection, Structure Solution, and Refinement.
Complete datasets to 1.85-Å (Fab A32), 1.95-Å (Fab N5-i5 and N12-i2), and 2.6-Å (Fab N12-i15) resolutions were collected at beamlines BL7-1 and BL9-1 of the Stanford Synchrotron Radiation Lightsource on an ADSC QUANTUM 315 and MAR325 detector, respectively. Data were integrated and reduced with HKL2000 (84), and structures were solved by molecular replacement with MrBUMP (85) and Phaser (86) from the CCP4 suite (87). MrBUMP was used to search the Protein Data Bank (PDB) to obtain the best-fitting model of variable domain, and then the constant and variable domains were used as separate searching models in Phaser. Structures of Fab N12-i2 and Fab N12-i15 were solved using coordinates extracted from PDB entry 1RZ8 (48), and the structures of Fab A32 and Fab N5-i5 were solved based on PDB entry 1N7M (88). Initial models were improved with several cycles of translation, libration, and screw-rotation (TLS)-restrained refinement implemented in Refmac (89) and Phenix (90) and were coupled with manual refitting and rebuilding with COOT (91). Data collection and final refinement statistics are shown in Table S2.
Structure Validation and Analysis and Figures.
The quality of the final refined models was monitored using the program MolProbity (92) and the PDB validation server (http://deposit.pdb.org/validate/). Structural alignments were performed using the Dali server and the program lsqkab from ccp4 Package (87). Molecular graphics were generated using Pymol (www.pymol.org).
Homology Modeling of CD4-Triggered gp120.
Homology modeling of CD4-triggered gp120 was carried out using ICM-Pro software (Molsoft) to build a model of CD4-triggered gp120. Initial modeling used the full-length gp120 sequence of HIV-1Ba-L and a YU2-Core V3 structure [PDB ID 2B4C (62)] followed by additional modeling using a gp120 core structure that includes the N- and C-termini [PDB ID 3JW0 (45)]. The structure was completed by using a built-in loop-modeling algorithm to model the V1/V2 loop. The electrostatic surface was calculated the REBEL electrostatics algorithm of the ICM package.
Supplementary Material
Acknowledgments
We thank Ms. Rachita Salkar, Ms. Qiang Lin, Ms. Christine Obreicht, Ms. XinWei Zhao, Ms. Ilia Joelle Prado, and Mr. Wensheng Huang for outstanding technical support and Drs. Dimiter Dimitrov and Jeffrey Lifson of the National Cancer Institute for the kind gifts of reagents. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the US Department of Energy Office of Biological and Environmental Research, by the National Institutes of Health (NIH) National Center for Research Resources, Biomedical Technology Program (P41RR001209), and by the National Institute of General Medical Sciences. We thank the University of Maryland X-ray Crystallography Shared Service for crystallographic resources. This work was supported by grants from The Bill and Melinda Gates Foundation (to R.C.G. and G.K.L.) and by National Institute of Allergy and Infectious Diseases (NIAID)/NIH Grants R01 AI-084830 and R01 AI-087181 (to G.K.L). M.M.S. was supported by NIAID/NIH K23 Career Development Grant K23 AI-084580. M.S.S. was supported by Grant 1032144 from The Bill and Melinda Gates Foundation.
Footnotes
The authors declare no conflict of interest.
Data deposition: The crystallography, atomic coordinates, and structure factors reported in this paper have been deposited in the Protein Data Bank (PDB), www.pdb.org (PDB ID codes 3TNM, 3QEG, and 3QEH).
See Author Summary on page 19 (volume 110, number 1).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1217609110/-/DCSupplemental.
References
- 1.Li A, et al. Synergistic neutralization of a chimeric SIV/HIV type 1 virus with combinations of human anti-HIV type 1 envelope monoclonal antibodies or hyperimmune globulins. AIDS Res Hum Retroviruses. 1997;13(8):647–656. doi: 10.1089/aid.1997.13.647. [DOI] [PubMed] [Google Scholar]
- 2.Mascola JR, et al. Potent and synergistic neutralization of human immunodeficiency virus (HIV) type 1 primary isolates by hyperimmune anti-HIV immunoglobulin combined with monoclonal antibodies 2F5 and 2G12. J Virol. 1997;71(10):7198–7206. doi: 10.1128/jvi.71.10.7198-7206.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mascola JR, et al. Protection of Macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J Virol. 1999;73(5):4009–4018. doi: 10.1128/jvi.73.5.4009-4018.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba TW, et al. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nat Med. 2000;6(2):200–206. doi: 10.1038/72309. [DOI] [PubMed] [Google Scholar]
- 5.Parren PW, et al. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J Virol. 2001;75(17):8340–8347. doi: 10.1128/JVI.75.17.8340-8347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimura Y, et al. Determination of a statistically valid neutralization titer in plasma that confers protection against simian-human immunodeficiency virus challenge following passive transfer of high-titered neutralizing antibodies. J Virol. 2002;76(5):2123–2130. doi: 10.1128/jvi.76.5.2123-2130.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker LM, Burton DR. Rational antibody-based HIV-1 vaccine design: Current approaches and future directions. Curr Opin Immunol. 2010;22(3):358–366. doi: 10.1016/j.coi.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baum LL, et al. HIV-1 gp120-specific antibody-dependent cell-mediated cytotoxicity correlates with rate of disease progression. J Immunol. 1996;157(5):2168–2173. [PubMed] [Google Scholar]
- 9.Forthal DN, et al. Antibody-dependent cellular cytotoxicity independently predicts survival in severely immunocompromised human immunodeficiency virus-infected patients. J Infect Dis. 1999;180(4):1338–1341. doi: 10.1086/314988. [DOI] [PubMed] [Google Scholar]
- 10.Ljunggren K, et al. Antibodies mediating cellular cytotoxicity and neutralization correlate with a better clinical stage in children born to human immunodeficiency virus-infected mothers. J Infect Dis. 1990;161(2):198–202. doi: 10.1093/infdis/161.2.198. [DOI] [PubMed] [Google Scholar]
- 11.Forthal DN, et al. FcgammaRIIa genotype predicts progression of HIV infection. J Immunol. 2007;179(11):7916–7923. doi: 10.4049/jimmunol.179.11.7916. [DOI] [PubMed] [Google Scholar]
- 12.Chung AW, et al. Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc Natl Acad Sci USA. 2011;108(18):7505–7510. doi: 10.1073/pnas.1016048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung AW, et al. Activation of NK cells by ADCC responses during early HIV infection. Viral Immunol. 2011;24(2):171–175. doi: 10.1089/vim.2010.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung AW, et al. Activation of NK cells by ADCC antibodies and HIV disease progression. J Acquir Immune Defic Syndr. 2011;58(2):127–131. doi: 10.1097/QAI.0b013e31822c62b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banks ND, Kinsey N, Clements J, Hildreth JE. Sustained antibody-dependent cell-mediated cytotoxicity (ADCC) in SIV-infected macaques correlates with delayed progression to AIDS. AIDS Res Hum Retroviruses. 2002;18(16):1197–1205. doi: 10.1089/08892220260387940. [DOI] [PubMed] [Google Scholar]
- 16.Sun Y, et al. Antibody-dependent cell-mediated cytotoxicity in simian immunodeficiency virus-infected rhesus monkeys. J Virol. 2011;85(14):6906–6912. doi: 10.1128/JVI.00326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forthal DN, Landucci G. In vitro reduction of virus infectivity by antibody-dependent cell-mediated immunity. J Immunol Methods. 1998;220(1-2):129–138. doi: 10.1016/s0022-1759(98)00152-5. [DOI] [PubMed] [Google Scholar]
- 18.Gómez-Román VR, et al. Vaccine-elicited antibodies mediate antibody-dependent cellular cytotoxicity correlated with significantly reduced acute viremia in rhesus macaques challenged with SIVmac251. J Immunol. 2005;174(4):2185–2189. doi: 10.4049/jimmunol.174.4.2185. [DOI] [PubMed] [Google Scholar]
- 19.Florese RH, et al. Contribution of nonneutralizing vaccine-elicited antibody activities to improved protective efficacy in rhesus macaques immunized with Tat/Env compared with multigenic vaccines. J Immunol. 2009;182(6):3718–3727. doi: 10.4049/jimmunol.0803115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hidajat R, et al. Correlation of vaccine-elicited systemic and mucosal nonneutralizing antibody activities with reduced acute viremia following intrarectal simian immunodeficiency virus SIVmac251 challenge of rhesus macaques. J Virol. 2009;83(2):791–801. doi: 10.1128/JVI.01672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao P, et al. Multiple vaccine-elicited nonneutralizing antienvelope antibody activities contribute to protective efficacy by reducing both acute and chronic viremia following simian/human immunodeficiency virus SHIV89.6P challenge in rhesus macaques. J Virol. 2010;84(14):7161–7173. doi: 10.1128/JVI.00410-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forthal DN, Gilbert PB, Landucci G, Phan T. Recombinant gp120 vaccine-induced antibodies inhibit clinical strains of HIV-1 in the presence of Fc receptor-bearing effector cells and correlate inversely with HIV infection rate. J Immunol. 2007;178(10):6596–6603. doi: 10.4049/jimmunol.178.10.6596. [DOI] [PubMed] [Google Scholar]
- 23.Haynes BF, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366(14):1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mabuka J, Nduati R, Odem-Davis K, Peterson D, Overbaugh J. HIV-specific antibodies capable of ADCC are common in breastmilk and are associated with reduced risk of transmission in women with high viral loads. PLoS Pathog. 2012;8(6):e1002739. doi: 10.1371/journal.ppat.1002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hessell AJ, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449(7158):101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 26.Hessell AJ, et al. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat Med. 2009;15(8):951–954. doi: 10.1038/nm.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishimura Y, et al. Transfer of neutralizing IgG to macaques 6 h but not 24 h after SHIV infection confers sterilizing protection: Implications for HIV-1 vaccine development. Proc Natl Acad Sci USA. 2003;100(25):15131–15136. doi: 10.1073/pnas.2436476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis GK. Challenges of antibody-mediated protection against HIV-1. Expert Rev Vaccines. 2010;9(7):683–687. doi: 10.1586/erv.10.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott CF, Jr, et al. Human monoclonal antibody that recognizes the V3 region of human immunodeficiency virus gp120 and neutralizes the human T-lymphotropic virus type IIIMN strain. Proc Natl Acad Sci USA. 1990;87(21):8597–8601. doi: 10.1073/pnas.87.21.8597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koup RA, et al. Antibody-dependent cell-mediated cytotoxicity directed by a human monoclonal antibody reactive with gp120 of HIV-1. AIDS. 1991;5(11):1309–1314. doi: 10.1097/00002030-199111000-00004. [DOI] [PubMed] [Google Scholar]
- 31.Ferrari G, et al. A HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent ADCC activity and defines a common ADCC epitope in human hiv-1 serum. Journal of Virology. 2011;85(14):7029–7036. doi: 10.1128/JVI.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyler DS, et al. Identification of sites within gp41 that serve as targets for antibody-dependent cellular cytotoxicity by using human monoclonal antibodies. J Immunol. 1990;145(10):3276–3282. [PubMed] [Google Scholar]
- 33.Robinson JE, Yoshiyama H, Holton D, Elliott S, Ho DD. Distinct antigenic sites on HIV gp120 identified by a panel of human monoclonal antibodies. abstr. Q449. J Cell Biochem Suppl. 1992;16E:71. [Google Scholar]
- 34.Thali M, et al. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J Virol. 1993;67(7):3978–3988. doi: 10.1128/jvi.67.7.3978-3988.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiang SH, Doka N, Choudhary RK, Sodroski J, Robinson JE. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res Hum Retroviruses. 2002;18(16):1207–1217. doi: 10.1089/08892220260387959. [DOI] [PubMed] [Google Scholar]
- 36.Decker JM, et al. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J Exp Med. 2005;201(9):1407–1419. doi: 10.1084/jem.20042510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore JP, et al. Exploration of antigenic variation in gp120 from clades A through F of human immunodeficiency virus type 1 by using monoclonal antibodies. J Virol. 1994;68(12):8350–8364. doi: 10.1128/jvi.68.12.8350-8364.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pancera M, et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci USA. 2010;107(3):1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore JP, Willey RL, Lewis GK, Robinson J, Sodroski J. Immunological evidence for interactions between the first, second, and fifth conserved domains of the gp120 surface glycoprotein of human immunodeficiency virus type 1. J Virol. 1994;68(11):6836–6847. doi: 10.1128/jvi.68.11.6836-6847.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiang SH, et al. Epitope mapping and characterization of a novel CD4-induced human monoclonal antibody capable of neutralizing primary HIV-1 strains. Virology. 2003;315(1):124–134. doi: 10.1016/s0042-6822(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 41.Wyatt R, et al. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393(6686):705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 42.Lewis GK, et al. Identification and characterization of an immunogenic hybrid epitope formed by both HIV gp120 and human CD4 proteins. J Virol. 2011;85(24):13097–13104. doi: 10.1128/JVI.05072-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan N, et al. CD4-Induced conformational changes in the human immunodeficiency virus type 1 gp120 glycoprotein: Consequences for virus entry and neutralization. J Virol. 1998;72(6):4694–4703. doi: 10.1128/jvi.72.6.4694-4703.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore JP, Sodroski J. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J Virol. 1996;70(3):1863–1872. doi: 10.1128/jvi.70.3.1863-1872.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finzi A, et al. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell. 2010;37(5):656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wyatt R, et al. Analysis of the interaction of the human immunodeficiency virus type 1 gp120 envelope glycoprotein with the gp41 transmembrane glycoprotein. J Virol. 1997;71(12):9722–9731. doi: 10.1128/jvi.71.12.9722-9731.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sela M, Mozes E. Dependence of the chemical nature of antibodies on the net electrical charge of antigens. Proc Natl Acad Sci USA. 1966;55(2):445–452. doi: 10.1073/pnas.55.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang CC, et al. Structural basis of tyrosine sulfation and VH-gene usage in antibodies that recognize the HIV type 1 coreceptor-binding site on gp120. Proc Natl Acad Sci USA. 2004;101(9):2706–2711. doi: 10.1073/pnas.0308527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moulard M, et al. Broadly cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to gp120-CD4-CCR5 complexes. Proc Natl Acad Sci USA. 2002;99(10):6913–6918. doi: 10.1073/pnas.102562599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang MY, et al. Improved breadth and potency of an HIV-1-neutralizing human single-chain antibody by random mutagenesis and sequential antigen panning. J Mol Biol. 2004;335(1):209–219. doi: 10.1016/j.jmb.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 51.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196(4):901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 52.Dey B, et al. Structure-based stabilization of HIV-1 gp120 enhances humoral immune responses to the induced co-receptor binding site. PLoS Pathog. 2009;5(5):e1000445. doi: 10.1371/journal.ppat.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang CC, et al. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310(5750):1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossio JL, et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol. 1998;72(10):7992–8001. doi: 10.1128/jvi.72.10.7992-8001.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeVico AL. CD4-induced epitopes in the HIV envelope glycoprotein, gp120. Curr HIV Res. 2007;5(6):561–571. doi: 10.2174/157016207782418560. [DOI] [PubMed] [Google Scholar]
- 56.Kwong PD, et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393(6686):648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finnegan CM, Berg W, Lewis GK, DeVico AL. Antigenic properties of the human immunodeficiency virus envelope during cell-cell fusion. J Virol. 2001;75(22):11096–11105. doi: 10.1128/JVI.75.22.11096-11105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finnegan CM, Berg W, Lewis GK, DeVico AL. Antigenic properties of the human immunodeficiency virus transmembrane glycoprotein during cell-cell fusion. J Virol. 2002;76(23):12123–12134. doi: 10.1128/JVI.76.23.12123-12134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Labrijn AF, et al. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol. 2003;77(19):10557–10565. doi: 10.1128/JVI.77.19.10557-10565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang MY, et al. Broadly cross-reactive HIV neutralizing human monoclonal antibody Fab selected by sequential antigen panning of a phage display library. J Immunol Methods. 2003;283(1-2):17–25. doi: 10.1016/j.jim.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 61.Scheid JF, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature. 2009;458(7238):636–640. doi: 10.1038/nature07930. [DOI] [PubMed] [Google Scholar]
- 62.Darbha R, et al. Crystal structure of the broadly cross-reactive HIV-1-neutralizing Fab X5 and fine mapping of its epitope. Biochemistry. 2004;43(6):1410–1417. doi: 10.1021/bi035323x. [DOI] [PubMed] [Google Scholar]
- 63.Huang CC, et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317(5846):1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gnanakaran S, et al. Genetic signatures in the envelope glycoproteins of HIV-1 that associate with broadly neutralizing antibodies. PLOS Comput Biol. 2010;6(10):e1000955. doi: 10.1371/journal.pcbi.1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein F, et al. Broad neutralization by a combination of antibodies recognizing the CD4 binding site and a new conformational epitope on the HIV-1 envelope protein. J Exp Med. 2012;209(8):1469–1479. doi: 10.1084/jem.20120423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bonsignori M, et al. ADCC-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. Journal of Virology. 2012;85(14):11521–11532. doi: 10.1128/JVI.01023-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sajadi MM, Heredia A, Le N, Constantine NT, Redfield RR. HIV-1 natural viral suppressors: Control of viral replication in the absence of therapy. AIDS. 2007;21(4):517–519. doi: 10.1097/QAD.0b013e328013d9eb. [DOI] [PubMed] [Google Scholar]
- 68.Guan Y, et al. Discordant memory B cell and circulating anti-Env antibody responses in HIV-1 infection. Proc Natl Acad Sci USA. 2009;106(10):3952–3957. doi: 10.1073/pnas.0813392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sajadi MM, et al. Epidemiologic characteristics and natural history of HIV-1 natural viral suppressors. J Acquir Immune Defic Syndr. 2009;50(4):403–408. doi: 10.1097/QAI.0b013e3181945f1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sajadi M, Redfield RR. Long-term nonprogressive disease among individuals with untreated HIV infection. JAMA. 2010;304(16):1784–1785. doi: 10.1001/jama.2010.1476. author reply 1785–1786. [DOI] [PubMed] [Google Scholar]
- 71.Sajadi MM, et al. Correlation between circulating HIV-1 RNA and broad HIV-1 neutralizing antibody activity. J Acquir Immune Defic Syndr. 2011;57(1):9–15. doi: 10.1097/QAI.0b013e3182100c1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanders RW, et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J Virol. 2002;76(17):8875–8889. doi: 10.1128/JVI.76.17.8875-8889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iyer SP, et al. Purified, proteolytically mature HIV type 1 SOSIP gp140 envelope trimers. AIDS Res Hum Retroviruses. 2007;23(6):817–828. doi: 10.1089/aid.2006.0261. [DOI] [PubMed] [Google Scholar]
- 74.Fouts TR, et al. Expression and characterization of a single-chain polypeptide analogue of the human immunodeficiency virus type 1 gp120-CD4 receptor complex. J Virol. 2000;74(24):11427–11436. doi: 10.1128/jvi.74.24.11427-11436.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wyatt R, et al. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J Virol. 1995;69(9):5723–5733. doi: 10.1128/jvi.69.9.5723-5733.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walker LM, et al. Protocol G Principal Investigators Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326(5950):285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roben P, et al. Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J Virol. 1994;68(8):4821–4828. doi: 10.1128/jvi.68.8.4821-4828.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moore JP, Wallace LA, Follett EA, McKeating JA. An enzyme-linked immunosorbent assay for antibodies to the envelope glycoproteins of divergent strains of HIV-1. AIDS. 1989;3(3):155–163. doi: 10.1097/00002030-198903000-00006. [DOI] [PubMed] [Google Scholar]
- 79.DeVico A, et al. Antibodies to CD4-induced sites in HIV gp120 correlate with the control of SHIV challenge in macaques vaccinated with subunit immunogens. Proc Natl Acad Sci USA. 2007;104(44):17477–17482. doi: 10.1073/pnas.0707399104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gómez-Román VR, et al. A simplified method for the rapid fluorometric assessment of antibody-dependent cell-mediated cytotoxicity. J Immunol Methods. 2006;308(1-2):53–67. doi: 10.1016/j.jim.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 81.O’Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol. 2000;74(21):10074–10080. doi: 10.1128/jvi.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chertova E, et al. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), Is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J Virol. 2002;76(11):5315–5325. doi: 10.1128/JVI.76.11.5315-5325.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seaman MS, et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol. 2010;84(3):1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Otwinowski Z, Minor W, Charles W, Carter Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;(Academic, New York) doi: 10.1016/S0076-6879(97)76066-X. Vol 276, pp 307–326. [DOI] [PubMed] [Google Scholar]
- 85.Keegan RM, Winn MD. MrBUMP: An automated pipeline for molecular replacement. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt 1):119–124. doi: 10.1107/S0907444907037195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 1):32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anonymous Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 88.Yin J, Andryski SE, Beuscher AE IV, Stevens RC, Schultz PG. Structural evidence for substrate strain in antibody catalysis. Proc Natl Acad Sci USA. 2003;100(3):856–861. doi: 10.1073/pnas.0235873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 90.Afonine PV, et al. Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 11):1153–1163. doi: 10.1107/S0907444910026582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 92.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
