

Abstract
Streptomyces are best known for producing antimicrobial secondary metabolites, but they are also recognized for their contributions to biomass utilization. Despite their importance to carbon cycling in terrestrial ecosystems, our understanding of the cellulolytic ability of Streptomyces is currently limited to a few soil-isolates. Here, we demonstrate the biomass-deconstructing capability of Streptomyces sp. SirexAA-E (ActE), an aerobic bacterium associated with the invasive pine-boring woodwasp Sirex noctilio. When grown on plant biomass, ActE secretes a suite of enzymes including endo- and exo-cellulases, CBM33 polysaccharide-monooxygenases, and hemicellulases. Genome-wide transcriptomic and proteomic analyses, and biochemical assays have revealed the key enzymes used to deconstruct crystalline cellulose, other pure polysaccharides, and biomass. The mixture of enzymes obtained from growth on biomass has biomass-degrading activity comparable to a cellulolytic enzyme cocktail from the fungus Trichoderma reesei, and thus provides a compelling example of high cellulolytic capacity in an aerobic bacterium.
Plant cell walls represent the largest reservoir of organic polymers in terrestrial ecosystems and are the most abundant source of renewable organic energy on Earth. Most of this energy is stored in the recalcitrant polysaccharide cellulose, which is difficult to hydrolyze because of the highly crystalline structure, and in hemicellulose, which presents challenges because of its structural diversity and complexity. In terrestrial ecosystems, cellulolytic microbes help drive carbon cycling through the deconstruction of biomass into simple sugars. The deconstruction is largely accomplished through the action of combinations of secreted glycoside hydrolases (GHs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), and carbohydrate binding modules (CBMs)1,2,3,4.
Actinobacteria in the genus Streptomyces are an ecologically important group, especially in soil environments, where they are considered to be vital players in the decomposition of cellulose and other biomass polymers2,5,6,7. Streptomyces are able to utilize a wide range of carbon sources, form spores when resources are depleted, and produce antimicrobial secondary metabolites to reduce competition6,8. Although a large number of Streptomyces species can grow on plant biomass, only a small percentage (14%) have been shown to efficiently degrade crystalline cellulose9. Furthermore, the secreted cellulolytic activities of only a few species have been biochemically characterized, and still fewer species have been examined to identify key biomass degrading enzymes10,11. Streptomyces reticuli is one of the best-studied cellulose- and chitin-degrading soil-dwelling Streptomyces; functional analyses of several important cellulases and other hydrolytic enzymes have been reported9,12,13. Furthermore, polysaccharide monooxygenase (PMO) activity with cellulose was identified using the CBM33 protein from Streptomyces coelicolor14, which suggests Streptomyces may use both hydrolytic and oxidative enzymes to deconstruct biomass. With the tremendous amount of sequence data collected in the past few years, and despite the view that Streptomyces make important contributions to cellulose degradation in the soil, genome-wide analyses of cellulolytic Streptomyces have not been reported.
In addition to their putative roles in carbon cycling in the soil, Streptomyces may also potentiate biomass deconstruction in insects through symbiotic associations15,16,17,18. Recent work has identified cellulose degrading Streptomyces associated with the pine-boring woodwasp Sirex noctilio, including Streptomyces sp. SirexAA-E (ActE)19. S. noctilio is a highly destructive wood-feeding insect that is found throughout forests in Eurasia and North Africa and is spreading invasively in North America and elsewhere20. While the wasp itself does not produce cellulolytic enzymes, evidence supports the role of a symbiotic microbial community that secretes biomass-degrading enzymes to facilitate nutrient acquisition for developing larvae in the pine tree21. The white rot fungus, Amylostereum areolatum, is the best-described member of this community, and the success of Sirex infestations is thought to arise from the insect's association with this cellulolytic fungal mutualist. However, work with pure cultures has suggested that ActE and other Sirex-associated Streptomyces are more cellulolytic than A. areolatum19.
Here, we report a systems-level analysis of the capacity of ActE for growth on pure polysaccharides and plant biomass, including genomic, transcriptomic and proteomic studies. We also isolated the secreted enzymes and characterized their reactions on a variety of biomass-derived substrates, including a comparison with a commercial enzyme cocktail from Trichoderma reesei, an industrial standard for fungal biomass degradation4. This work provides insights into how highly cellulolytic Streptomyces may contribute to insect-associated symbiotic communities. Furthermore, the genome-wide analysis provides a basis for evaluating the general cellulolytic capabilities of other members of this important genus.
Results
Genomic characteristics of ActE
Table 1 compares the genomic characteristics of ActE with well-known soil-isolated Streptomyces that produce antibiotics and with two model cellulolytic bacteria, Clostridium thermocellum and Cellvibrio japonicus3,22,23. Putative biomass-degrading protein-coding sequences from ActE were identified by BLAST analysis of the finished genome to the Carbohydrate Active enZyme (CAZy) database. Among the 6357 predicted protein-coding genes, 167 have one or more domains assigned to CAZy families, including 119 GHs, 29 CEs, 6 PLs and 85 CBMs. ActE contains 45 different types of GH families, 4 PL families, 7 CE families, and 21 CBM families. The number of total CAZy domains and diversity of CAZy families are comparable to other highly cellulolytic organisms.
Table 1. Comparison of genomic composition.
| ActE | S. coelicolor | S. griseus | C. thermocellum | C. japonicus | |
|---|---|---|---|---|---|
| Genome size (nt) | 7414440 | 8667507 | 8545929 | 3843301 | 4576573 |
| Proteome size | 6357 | 8153 | 7136 | 3173 | 3750 |
| Total CAZy Proteins | 167 | 221 | 132 | 103 | 183 |
| % CAZy Proteinsa | 2.6% | 2.7% | 1.8% | 3.2% | 4.9% |
| Total GHb | 119 | 154 | 80 | 70 | 124 |
| Total PLc | 6 | 11 | 4 | 6 | 14 |
| Total CEd | 29 | 36 | 23 | 20 | 28 |
| Total CBMe | 85 | 98 | 68 | 121 | 134 |
| antiSMASH clustersf | 22 | 24 | 37 | 3 | 4 |
| Genes in clusters | 620 | 718 | 1139 | 89 | 111 |
| % antiSMASH | 9.8% | 8.8% | 16.0% | 2.8% | 3.0% |
aProteins classified as Carbohydrate Active Enzymes (CAZy).
bGH, glycoside hydrolase.
cPL, pectate lyase.
dCE, carbohydrate esterase.
eCBM, carbohydrate binding module.
fPutative antibiotic producing gene cluster.
Nearly all publically available Streptomyces genomes encode a relatively high percentage of genes for putative cellulolytic enzymes. Interestingly, ActE and the antibiotic producing Streptomyces, S. griseus and S. coelicolor, shown in Table 1 have similar numbers and compositions of CAZy families, but substantially different genome sizes. However, these antibiotic-producing Streptomyces are not highly cellulolytic (Fig. 1). Relative to S. griseus and S. coelicolor, the ActE genome contains two unique CAZy families but does not possess 16 CAZy families present in these species. However, ActE contains more representatives in 13 CAZy families. Enrichment of certain CAZy families is observed in other highly cellulolytic organisms. For example, C. thermocellum contains 16 genes in the GH9 family alone. It is interesting to consider whether the reduction in total genome size and differences in CAZy composition between ActE and other closely related soil-dwelling Streptomyces might have arisen from evolutionary specialization of ActE, perhaps driven by association with the Sirex-fungal symbiosis.
Figure 1. Growth of ActE in minimal medium containing filter paper as the sole carbon source.
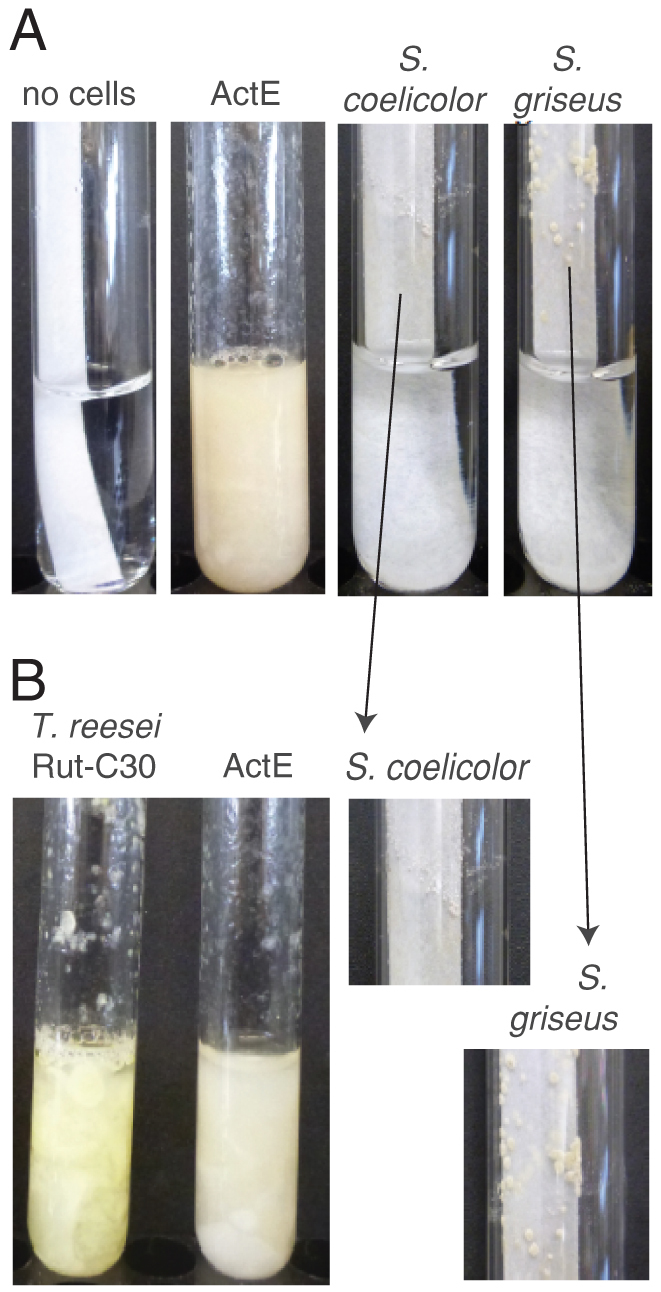
(A) Growth of ActE, Streptomyces coelicolor, and Streptomyces griseus in minimal medium for 7 days at 30°C and pH 6.9. The expanded image shows small colonies of S. coelicolor and S. griseus forming on the surface of the paper. (B) Growth of ActE and Trichoderma reesei Rut-C30 for 7 days at 30°C and pH 6.0.
Secreted proteins from ActE during growth on pure polysaccharides
ActE grew well in minimal medium containing cellulose as the sole carbon source. The growth rate was similar to that of Trichoderma reesei Rut-C30, a model cellulolytic microbe (Fig. 1), and both completely deconstructed filter paper strips in 5–7 days. By comparison, S. coelicolor and S. griseus grew only sparingly as small colonies on filter paper under the same conditions. This difference in growth capabilities among closely related Streptomyces prompted us to further examine ActE, which also grew with a filamentous morphology on polysaccharides including plant biomass (Supplementary Fig. S1).
Reactions of the ActE secretomes
The enzymatic activities of ActE secretomes were compared with a commercial secretome, Spezyme CP. This enzyme cocktail is prepared from T. reesei Rut-C30, and thus provides a useful, routinely available reference point for the capabilities of other cellulolytic organisms. HPLC analysis showed that the ActE cellulose secretome released cellobiose as the primary product during reaction with cellulose (Fig. 2A, 95% of products), which is distinct from the higher proportion of glucose produced by the T. reesei secretome. Similarly, the primary products from xylan and mannan were xylobiose and mannobiose, respectively. Upon accounting for total glucose equivalents released, the ActE secretome obtained from growth on pure cellulose had specific activity that was about half of that provided by Spezyme CP (Fig. 2A, inset). Interestingly, the ActE secretome obtained from growth on pure cellulose had higher specific activity for deconstruction of pure mannan than Spezyme CP (Fig. 2B). Additionally, the ActE secretome obtained from growth on pure xylan had higher specific activity for reaction with pure xylan than Spezyme CP. Cellulose, xylan, and mannan are all abundant in pinewood, thus accounting for the necessity of each of the major catalytic activities detected.
Figure 2. Reactions of ActE secretomes and Spezyme CP.
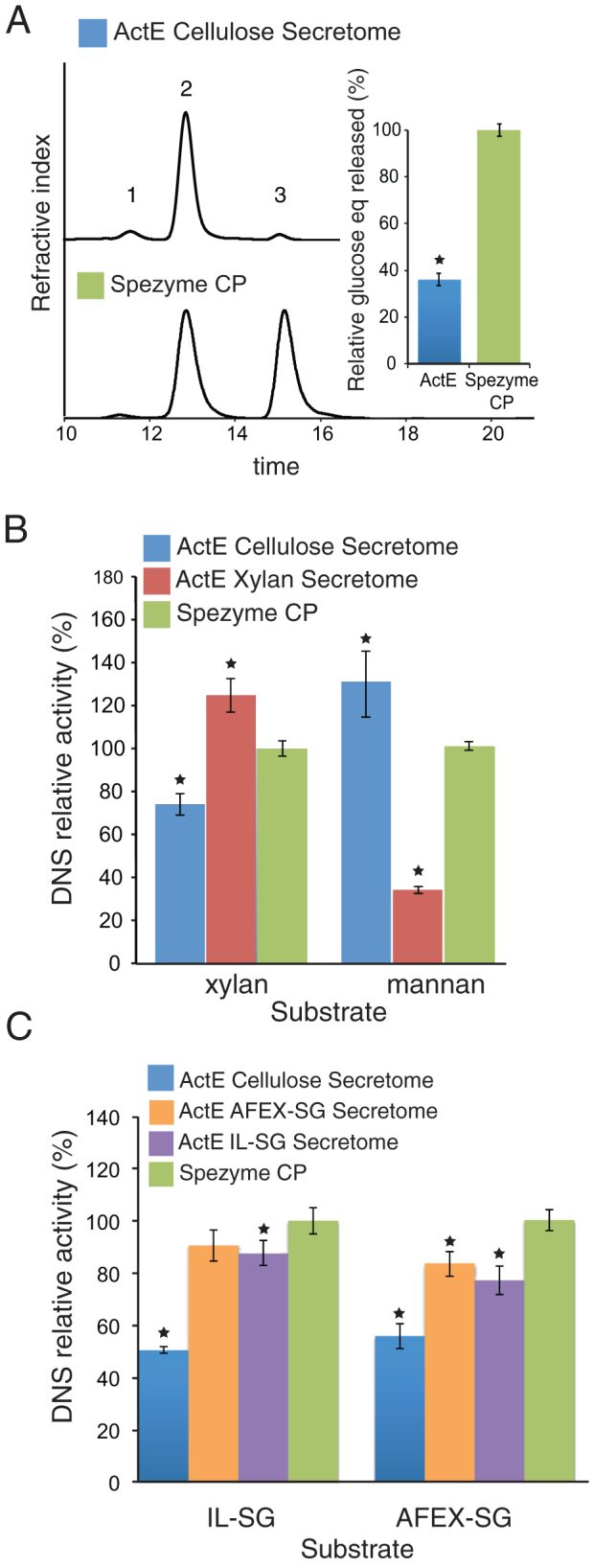
(A) HPLC of sugars released from cellulose (1, cellotriose; 2, cellobiose; 3, glucose) and quantification of glucose equivalent (insert). (B) Reducing sugars released from xylan and mannan by the secretomes of ActE grown on cellulose and xylan. (C) Total reducing sugar released from ionic liquid-switchgrass (IL-SG) or AFEX-switchgrass (AFEX-SG) in reactions of the ActE cellulose, AFEX-SG, and IL-SG secretomes and Spezyme CP. Data represent the mean ± s.d. from three experiments; * indicates P<0.01 compared with Spezyme CP.
Anion exchange chromatography was performed to fractionate the ActE secretome obtained from cells grown on cellulose as the sole carbon source. We identified fractions that hydrolyzed pure polysaccharides by biochemical assays (Supplementary Fig. S2), and confirmed the identity of the protein or proteins contained in these fractions by mass spectrometry (Supplementary Table S1). Where multiple polypeptides were present, the identity of each was confirmed by mass spectrometry to correspond to the indicated gene locus. In several cases, these most likely arise from proteolysis of a single protein found in the secretome. Fractions containing the maximum cellulase activity were highly enriched in SACTE_0236 and SACTE_0237, reducing and non-reducing end cellobiohydrolases from the GH6 and GH48 families, respectively. SACTE_0265 and SACTE_2347 were identified as the major proteins present in fractions associated with xylan and mannan hydrolysis, respectively. A CBM33 polysaccharide monooxygenase (SACTE_3159) was also identified in the ion exchange profile. Moreover, beta-1,3 glucanase activity was identified in fractions that were enriched in SACTE_4755.
When ActE was grown on either ammonia fiber expansion-treated switchgrass (AFEX-SG)24 or ionic liquid-treated switchgrass (IL-SG), the secretomes had ~2-fold increase in specific activity relative to the cellulose secretome and were equivalent to Spezyme CP for reaction with both the AFEX- and IL-treated biomass (Fig. 2C)24. The ActE secretomes retained greater than 60% of maximal activity for the hydrolysis of AFEX- and IL-SG from 30 to 55°C and 35 to 47°C, respectively, which is comparable to recent reports on the temperature profile of secretomes from thermophilic biomass-degrading fungi25 (Supplementary Fig. S3A). The secretomes showed a pH optimum of ~7 for reaction with AFEX-SG and a pH optimum of ~8 for reaction with IL-SG. Moreover, these secretomes retained greater than 60% of maximal activity over the ranges of pH 4.5 to 8.0, and pH 7.0 to 8.0, respectively (Supplementary Fig. S3B). These optimal pH values are considerably higher than observed for Spezyme CP.
Secretome analysis on pure polysaccharides and plant biomass
To identify secreted proteins, supernatants from ActE cultures grown on glucose, cellobiose, cellulose, xylan, chitin, switchgrass, AFEX-SG, and IL-SG were analyzed by LC-MS/MS (Fig. 3, Supplementary Table S2). The proteins were sorted into a descending rank according to spectral counts, and sets whose spectral counts summed to 95% of the total protein in each secretome are shown. Fig. 3A summarizes the percentages of CAZy families in the detected proteins. The glucose secretome had a protein concentration of ~0.03 g/L of culture medium, and among the 136 proteins identified only 3% had a CAZy annotation. Indeed, the majority (>90%) likely originated from cell lysis. In contrast, the polysaccharide secretomes had a protein concentration of ~0.3 g/L of culture medium, a ~10-fold increase from the glucose secretome. Pectate lyase (SACTE_1310), chondroitin/alginate lyase (SACTE_4638), an extracellular solute binding protein (SACTE_4343), bacterioferritin (SACTE_1546), and catalase (SACTE_4439) were observed in all polysaccharide secretomes. The first two proteins, SACTE_1310 and SACTE_4638, have signal peptides and are thus secreted as part of the response needed for growth on polysaccharides.
Figure 3. Composition of ActE secretomes identified by LC-MS/MS.

(A) CAZy genes account for 2.6% of the 6357 predicted protein-coding sequences in the ActE genome. (B) Identity of most abundant proteins in the cellulose secretome proteins is sorted according to decreasing spectral counts (accounting for 95% of total spectral counts); corresponding spectral counts from other secretomes are also shown.
Fig. 3 and Supplementary Table S2 demonstrate that 22 proteins accounted for 95% of the total spectral counts during growth on cellulose; two-thirds were from CAZy families. The five most abundant proteins, in order and representing ~85% of the total spectral counts, were reducing and non-reducing exoglucanases (SACTE_0236 and SACTE_0237), a CBM33 polysaccharide monooxygenase (SACTE_3159), an endoglucanase (SACTE_0482), and a β-mannosidase (SACTE_2347). The first four proteins encode a non-redundant set of enzymes that likely provide the essential activities required for utilization of crystalline cellulose22. Among the 22 most abundant proteins, there were representatives from 9 different GH families, two CE families, two PL families, and two additional CMB33 proteins. Collectively, these secreted proteins represent ~20% of the CAZy composition in the ActE genome.
There were substantial differences in the composition of the xylan and chitin secretomes as compared to the cellulose secretome (Fig. 3, Supplementary Table S2). In the xylan secretome, 92 proteins comprise 95% of the detected spectral counts. Twenty GHs from 18 different CAZy families were included, along with 1 CE4 and 2 PL family proteins. Thus, growth on xylan elicits secretion of representatives from half of the total CAZy families found in the ActE genome. The broad distribution of hemicellulytic enzymes in the xylan secretome contrasts with the considerably less diverse composition of the chitin secretome, which consists of 7 representatives from GH18 (e.g., chitinase, endo beta-N-acetylglucosaminidase), 2 from GH19 (e.g., chitinase, lysozyme), and 1 chitinolytic CBM33 (Supplementary Table S2). While chitinolytic CAZy families account for two-thirds of the proteins secreted during growth on chitin, they represent only ~6% of the diversity of CAZy families found in the genome. These results document the substantially different substrate-specific responses of ActE during growth on different polysaccharides.
The secretomes isolated from cells grown on switchgrass, AFEX-SG, and IL-SG contained the highly abundant secreted proteins identified in the purified cellulose and xylan experiments and some additional proteins. These additional proteins likely reflect cellular response to the more complex composition of polysaccharides present in the biomass samples. The increased diversity of proteins present in the biomass secretome also increased the efficiency of reaction with plant biomass (Fig. 2C). In total, the biomass secretomes contained 31 different CAZy families that contributed to the total spectral counts (~70% of the CAZy families present in the ActE genome), thus representing coordinated and extensive use of CAZyme families present in the ActE genome for biomass utilization.
Gene expression analysis during growth on purified polysaccharides and plant biomass
Gene expression profiles were determined for ActE grown on purified polysaccharides and plant biomass by whole genome microarrays (Figs. 4 and 5, Supplementary Figs. S4 to S9). Genome-wide gene expression was analyzed as a functional annotation network composed of ActE genes (circles) connected to predicted functional groups (triangles; KEGG or CAZy). In Fig. 4, the network was annotated with genome-wide microarray expression data to indicate genes that were differentially expressed when ActE was grown on either AFEX-SG or glucose, and further annotated to indicate normalized expression levels observed during growth on AFEX-SG. While many aspects of metabolism are modestly changed in response to these different carbon sources, the CAZy and ABC transporter categories were substantially enriched in differentially expressed genes (Fig. 4, green circles). Furthermore, pentose sugar metabolism, sulfur metabolism, and some amino acid biosynthesis pathways (e.g. aromatic amino acids) were also highly induced during growth on AFEX-SG relative to other carbon sources (Supplementary Figs. S4 to S9). In contrast, ribosomal, secondary metabolite, and DNA repair genes showed little change in expression across the conditions examined. Within the CAZy functional group, there was a large induction of genes that contained both a GH domain and a CBM2 domain. Among the 11 genes in the ActE genome that contain a CBM2 domain, 6 were induced greater than 4-fold during growth on AFEX-SG. Furthermore, 9 of the 11 CBM2 containing proteins were identified in the secreted proteome (Fig. 3).
Figure 4. Genome-wide changes in expression during growth of ActE on AFEX-treated switchgrass (AFEX-SG) versus glucose.

Nodes are genes (circles) or KEGG/CAZy functional categories (yellow triangles); edges indicate that the gene belongs to the indicated functional group as defined by either KEGG or CAZy analysis. Gene node sizes reflect expression intensity determined by microarray from growth on AFEX-SG as a log2 ratio, where the genome-wide average transcriptional intensity was ~10.5 for both substrates. Node colors represent expression changes as the log2 ratio of AFEX-SG/glucose transcript intensities.
Figure 5. Expression of ActE CAZy genes on various carbon sources.

(A) Hierarchical clustering of expression for 167 CAZy genes from the ActE genome during growth on the indicated substrates. (B) Identity of CAZy genes with distinct changes in expression observed in group 1 CAZy genes during growth in different carbon sources. Information for additional groups is provided in the Supplementary Information.
Given the large number of differentially expressed CAZy genes identified in the network analysis, we analyzed the expression of this group of genes in cultures grown on different carbon sources (Fig. 5). As with other cellulolytic organisms, there was strong correlation between the content of the secreted proteomes and the most highly expressed genes. Of the 167 ActE genes containing CAZy domains, 68 genes (Fig. 5, group 1) showed distinct increases in expression when grown on different polymeric substrates, 14 genes (group 2, see Supplementary Fig. S10) did not show any appreciable level of expression, and 85 genes (group 3, see Supplementary Fig. S11) showed moderate changes in expression with the different substrates. A significant fraction of these genes contained translocation signals for either the Sec or twin-arginine translocation pathways, and genes encoding structural polypeptides for these translocation pathways were also highly expressed. Besides correlation with secreted proteins, the transcriptomic studies also gave insight into co-regulated gene clusters that potentially encode functional units for utilization of different polysaccharides by ActE. In the following, the 130 genes with normalized expression intensities in the top 2% of all genes are described.
During growth on cellulose, four CAZy genes (SACTE_0236, SACTE_0237, SACTE_3159, and SACTE_0482) showed >15-fold increase in transcript abundance (Fig. 5), and the corresponding proteins were highly enriched in the secreted proteome. None of these four were obviously placed in a gene cluster, and the two most highly expressed genes, SACTE_0236 and SACTE_0237, while adjacent on the chromosome, were transcribed in opposite directions. Nevertheless, these four most highly expressed genes and three others that showed >5-fold increase in transcript abundance (SACTE_3717, SACTE_6428, SACTE_2347, Table 2) were associated with a conserved 14 bp palindromic promoter sequence, TGGGAGCGCTCCCA (the CebR binding element). CebR proteins are LacI/GalR-like transcriptional regulators shown to provide transcriptional control of gene expression in response to the presence of cellobiose or other small oligosaccharides in S. griseus, S. reticuli, and Thermobifida fusca26,27,28. Likewise, the genes (SACTE_2285 to SACTE_2289) encoding a CebR regulator (SACTE_2285), a GH1 protein (β-glucosidase), a two-protein cellobiose transporter system, and an extracellular solute binding protein were associated with a CebR binding element and were also among the most highly expressed genes during growth on cellulose. These latter five genes have 75% or greater sequence identity with the cellobiose utilization operon identified in S. griseus and S. reticuli26,29. There were only 15 genes annotated as hypothetical or domain of unknown function (12%) up-regulated during growth on cellulose, a considerably smaller percentage of these than in the entire genome (27%).
Table 2. Analysis of upstream DNA sequence elements in ActE genes upregulated during growth on cellulose.
| Locus | Catalytic domain | CBM | Annotated function | Sequencea | Rankb | Fold changeb |
|---|---|---|---|---|---|---|
| SACTE_0236 | GH48 | CBM2 | 1,4-beta cellobiohydrolase | TGGGAGCGCTCCCA | 1 | 21.7 |
| SACTE_0237 | GH6 | CBM2 | 1,4-beta cellobiohydrolase | TGGGAGCGCTCCCA | 2 | 17.3 |
| SACTE_3159 | CBM33 | CBM2 | Cellulose-binding domain | TGGGAGCGCTCCCA | 3 | 16.2 |
| SACTE_0482 | GH5 | CBM2 | Endo-1,4-beta-glucosidase | TGGGAGCGCTCCCA | 4 | 15.4 |
| SACTE_2288 | Transport systems inner membrane component | TGGGAGCGCTCCCA | 5 | 11.2 | ||
| SACTE_3717 | GH9 | CBM2 | 1,4-beta cellobiohydrolase | TGGGAGCGCTCCCA | 6 | 9.7 |
| SACTE_6428 | CBM33 | Chitin-binding, domain 3 | GGGAGCGCTCCCA | 9 | 7.9 | |
| SACTE_2347 | GH5 | CBM2 | Beta-mannosidase | TGGGAGCGCTCCCA | 11 | 5.0 |
| SACTE_2287 | Transport systems inner membrane component | TGGGAGCGCTCCCA | 15 | 4.3 | ||
| SACTE_2289 | Family 1 extracellular solute-binding protein | TGGGAGCGCTCCCA | 19 | 3.9 | ||
| SACTE_0352 | GCN5-related N-acetyltransferase | TGGGAGCGCTCCCA | 22 | 3.6 | ||
| SACTE_2286 | GH1 | Glycoside hydrolase 1 | GGGAGCGCTCCCA | 27 | 3.4 | |
| SACTE_0483 | CBM2 | Cellulose-binding family protein | GGGAGCGCTCCCA | 503 | 1.6 | |
| SACTE_0562 | GH74 | CBM2 | Secreted cellulase (endo) | TGGGAGCGCTCCCA | 5759 | 0.7 |
| SACTE_2285 | LacI family transcriptional regulator (CebR) | TGGGAGCGCTCCCA | 6229 | 0.6 | ||
aPredicted binding sequence element found upstream from gene locus.
bRanking and fold change in expression intensity detected by microarray for ActE genes when grown on cellulose relative to glucose.
Several characteristics distinguished expression during growth on either xylan or chitin. First, unique sets of genes were induced, as there was only 14% and 10% overlap, respectively, when compared to cellulose. Second, ~33% of the top 2% of genes expressed during growth on either xylan or chitin were annotated as hypothetical or domain of unknown function, which greatly exceeds the unknown fraction in the cellulose secretome. During growth on xylan, two clusters of genes were up-regulated. One extended from SACTE_0357 to SACTE_0370, encoding proteins from the GH11, GH13, GH42, GH43, GH78, GH87, and CE4 families, a LacI-like transcriptional regulator, a secreted peptidase, and two sets of inner membrane transporters and associated solute binding proteins. Alternatively, during growth on chitin, three CBM33 proteins were up-regulated (SACTE_0080, SACTE_2313, SACTE_6493), and two of these had an immediately adjacent gene encoding a GH18 (SACTE_6494) or GH19 (SACTE_0081) that was up-regulated.
When ActE was grown on biomass samples, 14 additional CAZy genes were uniquely up-regulated, and the corresponding proteins were identified in the proteomic analysis of biomass secretomes (Figs. 3 and 4). A gene cluster extending from SACTE_5858 to SACTE_5864 was uniquely up regulated during growth on biomass. Among these genes, SACTE_5860 and SACTE_5862 are annotated as a twin-arginine translocation pathway protein and an ABC transporter, respectively, while the rest are annotated either as hypothetical protein or as domain of unknown function.
Discussion
Symbiotic cellulolytic fungi and bacteria help a diverse array of eukaryotes access the energy stored in plant biomass and are fundamental to terrestrial energy flow and ecosystem function. However, our understanding of symbiotic cellulose degradation is limited to a few well-studied model organisms, most of which survive only in special anoxic digestive organs of the host. For example, ruminants carry communities of anaerobic microbes in their rumen that deconstruct biomass, facilitating nutrient acquisition from plant biomass for the host30,31. Insects also obtain nutrients from plant biomass by forming symbiotic relationships with cellulolytic microbes. Termites are a classic example, in which microbes deconstruct the plant biomass in the insect's hindgut32. Alternatively, leaf-cutter ants and other fungus-farming insects cultivate stable communities of externally associated aerobic microbes in specialized gardens33,34. The insects provide plant biomass to these fungal and bacterial communities, which in exchange help convert the plant material into usable nutrients for their host. Recently, aerobic Streptomyces were isolated from the microbial communities associated with the herbivorous woodwasp Sirex noctilio and they were shown to be efficient at degrading cellulose and plant biomass19. While Streptomyces have long been considered important decomposers in the soil and may also be common cellulolytic symbionts associated with herbivorous insects, genome-wide studies characterizing the cellulolytic capacities of these ubiquitous bacterial species have been lacking. In this study, we have detailed the highly efficient biomass-degrading ability of an insect-associated Streptomyces, ActE, through a combination of biochemical, genomic, transcriptomic and proteomic analyses.
Biochemical assays of the ActE secretome showed comparable specific activity to a commercial enzyme cocktail prepared from the secretome of T. reesei Rut-C30, the major progenitor organism of biofuels enzymology. The secretomes of ActE grown on biomass contain enzymes with endocellulase, exocellulase, 1,3-beta-glucanase, mannanase, xylanase, esterase, pectate lyase, and other activities. The presence of endo- and exocellulases distinguishes ActE from C. japonicus, whose genome lacks one exocellulase that is usually associated with growth on cellulose22. The vigorous reactions of the ActE secretome with xylan and mannan are consistent with the abundance of arabinoxylan and glucomannan in pinewood. Furthermore, production of cellobiose, xylobiose, and mannobiose as the primary polysacccharide deconstruction products may preferentially channel energy to only select members of Sirex/Amylostereum community. Moreover, the high activity with 1,3-beta-glucan (callose) suggests a capability to attack the protective polymers produced by the pine tree during wound response and/or the cell wall of fungi present in the larval galleries.
During growth on biomass, ~75% of the CAZy families present in the ActE genome are expressed, and in many cases, these genes are part of gene clusters that may be suitable for adaption to biotechnological processes. Interestingly, the dominant architecture for these enzymes is the fusion of a catalytic domain with a CBM2 domain. This is similar to the architecture used by T. reesei, whose enzymes also primarily consist of a single catalytic domain fused to a CBM1 domain.
GH61 and CBM33 proteins are now identified as copper-dependent polysaccharide monooxygenases35,36,37 that function with extracellular oxidoreductases38. There are six CBM33 genes in the ActE genome, and this study provides insight into their function. Three (SACTE_3159, SACTE_6428, SACTE_2313) were highly expressed and secreted during growth on cellulose, while three were observed only during growth on chitin (SACTE_0080, SACTE_2313, SACTE_6493). SACTE_3159 is unique among the six ActE CBM33 proteins, as it is fused to a CBM2 domain. It is also the most highly expressed CBM33 protein during growth on cellulose. Since genes encoding cellobiose oxidase and cellobiose dehydrogenase are not present in ActE, it is possible that an extracellular oxidoreductase is provided by other members of the Sirex community, in analogy to the combination of a Thermoascus aurantiacus GH61 and a Humicola insolens cellobiose dehydrogenase38. A. areolatum, a white rot basidiomycete from the Sirex community, is one reasonable source for these enzymes. The detection of a high level of catalase in the secreted proteome further suggests the overall environment in the insect-microbe community is oxidizing, with additional implications for how cellulose and lignin may be attacked.
The ActE genome contains 1738 genes (~30%) annotated as hypothetical or domain of unknown function. It is interesting that up to one-third of genes expressed during growth on xylan and chitin were in these classes. This result implies a relative lack of understanding regarding proteins involved in utilization of these abundant polysaccharides by Streptomyces. Furthermore, several genes with unknown function were uniquely expressed during growth on biomass and provide a basis for future studies to improve understanding of natural biomass utilization.
Cellulolytic microorganisms have developed different enzymatic approaches to overcome the complexity of plant cell walls. Cellulolytic anaerobic bacteria such as C. thermocellum use a multi-protein complex called the cellulosome for biomass deconstruction39,40, while other anaerobes use non-cellulosomic free enzymes25 or cell-surface bound enzymes31. Aerobic cellulolytic fungi such as Phanaerochaete and Postia use a rich repertoire of both oxidative and hydrolytic enzymes to deconstruct biomass, and genome-enabled studies of their cellulolytic capacities are available4,35,36,37,41. This research has identified that an insect-associated Streptomyces isolated from an ecological niche defined by invasive attack on plant biomass has vigorous capabilities for attack of plant polysaccharides. Gene clusters that encode solute binding proteins, transporters, and extracellular enzymes were present and highly expressed during growth on the respective pure polysaccharide substrates, providing insights into the building blocks needed to install enhanced cellulolytic capacity into less reactive industrial strains of Streptomyces. Interestingly, the genes involved in extracellular aspects of cellulose utilization were not organized into extensive gene clusters but instead appear to be controlled by a CebR-like regulatory system distributed throughout the genome. In contrast xylan utilization appears to have a more organized structure also under control of a CebR-like regulatory system. Thus, genetic manipulation of CebR-like regulation in ActE may increase expression of hydrolytic and oxidative enzymes for biomass deconstruction.
Bacteria from the genus Streptomyces are best known for industrial production of antibiotics, and two well-known examples, S. coelicolor and S. griseus, have been intensively studied to better understand their capacity for producing antibiotics. Similar to these, ActE contains 22 clusters of genes potentially involved in secondary metabolite production (Table 1). However, none of the secondary metabolite genes found in ActE were expressed under the growth conditions investigated here (Fig. 4, Supplementary Figs. S4 to S9). While many Streptomyces, including S. coelicolor, S. griseus and others grow on plant polysaccharides6, they have considerable phenotypic differences observed during growth on pure crystalline cellulose compared to ActE (Fig. 1).
In conclusion, we have shown that an aerobic Streptomyces bacterium associated with Sirex woodwasps secretes a mixture of enzymes with high specific activity for biomass deconstruction. Thus, ActE potentially makes profound contributions to nutrient acquisition in the Sirex-Amyelosterum mutualism. Given the associations of Streptomyces with other wood feeding insects34, the present results suggest the contributions of externally-associated Streptomyces to insect-mediated biomass deconstruction are a widespread, important, and a heretofore underappreciated factor shaping carbon flow in terrestrial ecosystems. In contrast, to symbiotic cellulolytic bacteria that reside in the guts of animals (including insects such as termites), ActE is an externally propagating, aerobic bacterium that can secrete a potential mixture of hydrolytic and oxidative enzymes enzymes into the pinewood galleries created by its host. ActE thus provides a new example for how aerobic bacteria contribute to the deconstruction of plant biomass in Nature.
Methods
Genome analysis
The complete genome sequence of Streptomyces sp. SirexAA-E (ActE, taxonomy ID 862751) was determined by the Joint Genome Institute, project ID 4086644. Gene annotation models were predicted using Prodigal42, examined using Artemis43, and are available at NCBI with the following accession numbers, GenBank: CP002993.1; RefSeq: NC_015953.1. Carbohydrate-active enzymes were annotated by comparison of all translated open-reading frames to the CAZy database2. We collected CAZy annotated genes from the CAZy database (www.cazy.org). We then used BLASTP to compare all ActE protein-coding sequences to the CAZy database and to the pfam database (ftp://ftp.ncbi.nih.gov/pub/mmdb/cdd/little_endian/Pfam_LE.tar.gz). These two annotations were then crosschecked, and proteins annotated by both databases were identified as our final CAZy annotation. Secreted proteins were identified by SignalP, TatP, and SecretomeP analyses. BLAST was used to identify sequence orthologs in other organisms. Secondary metabolite gene clusters were identified by AntiSmash analysis44. CebR boxes were identified by using BLAST comparison of the S. griseus CebR box sequence to the ActE genome26. Networks of expression and functional categories were visualized using Cytoscape45.
Biomass substrates
Switchgrass and ammonia fiber expansion (AFEX)-treated switchgrass were obtained from the Great Lakes Bioenergy Research Center. Extensively washed ionic liquid (IL)-treated switchgrass was the generous gift of Dr. Masood Hadi (Joint BioEnergy Institute).
Growth of organisms
ActE, Streptomyces coelicolor, Streptomyces griseus and Trichoderma reesei RUT-C30 were grown at pH 6.9 (pH 6.0 for T. reesei), where 1 L contains: 10.72 g K2HPO4; 5.24 g KH2PO4; 2 g (NH4)2SO4; 0.5 mL of an iron solution (1 mg of FeSO4 dissolved in 1 mL in 0.01 M HCl); 1 mL of 1 M MgSO4 solution; 1 mL of thiamine solution (1 mg/mL); and 5 mL of trace elements solution (SPV-4). The sole carbon source (0.5% w/v) in the medium was glucose, cellulose (either Whatman #1 filter paper or Sigmacell-20, Sigma/Aldrich, St. Louis, MO as indicated), xylan, chitin, switchgrass, AFEX-treated switchgrass or IL-treated switchgrass. Cultures were incubated for 7 d at 30°C with shaking.
Preparation of secretomes
Supernatants obtained from different culture media were prepared by centrifugation of the culture medium for 10 min at 3000 × g, which removed the remaining insoluble polysaccharide and adhered cells. The supernatant fraction was then passed through a 0.20 μm nylon membrane filter (Whatman) in order to remove any remaining cells. For enzymatic assays, the supernatants were concentrated using a 3 kDa cut off ultrafiltration membrane. The concentration of secretome protein was determined by Bradford assay.
Enzyme activity measurements
Reduced sugar assays were carried out by mixing secretome preparations with polysaccharide-containing substrates including cellulose (either Whatman #1 filter paper or Sigmacell-20 as indicated), xylan, chitin, mannan, switchgrass, AFEX pretreated switchgrass, or ionic-liquid pretreated switchgrass24. After incubation in 0.1 M sodium phosphate, pH 6 at 40°C for 20 h, the reducing sugar content was detected by dinitrosalicylic acid assay46 and calibrated by using glucose, xylose, or mannose as standards. Purified polysaccharide preparations had negligible background response in the absence of added enzymes. Cellobionic and gluconic acids were assayed by a coupled enzyme assay (K-GATE system, Megazyme, Bray Ireland). Spezyme CP was obtained from Genencor with batch number #4901522860. The distributions of soluble sugar oligomers obtained from secretome reactions were determined using a Shimadzu Liquid Chromatograph HPLC system (Shimadzu Scientific Instruments, Columbia, MD) equipped with a refractive index detector (RID-10A) and a Phenomenex Rezex RPM-monosaccharide column. The temperature was maintained at 85°C and Milli-Q water was used as the mobile phase at 0.6 mL min−1 flow rate. Glucose, cellobiose, cellotriose, cellotetraose, cellopentaose, and cellohexaose (Sigma) were used as standards. The integrated areas of peaks were analyzed by EZ start 7.2 SP1 software (Shimadzu).
Extracellular protein profiles
Extracellular proteins from culture supernatants were precipitated with trichloroacetic acid (TCA), resuspended in denaturing sample buffer (SDS and 2-mercaptoethanol), and separated by SDS-PAGE in 4–20% gels. Protein bands of interest were excised from the gel, digested with trypsin, desalted with C18 pipette tips (Millipore, Billerica, MA) and identified by MALDI-TOF (MDS SCIEX 4800 MALDI TOF/TOF, Applied Biosystems, Foster City, CA). LC-MS/MS was performed at the University of Wisconsin Biotechnology Center. Samples were prepared by TCA precipitation of 100 ng of total secreted protein from 7-day old culture supernatants. Protein samples were digested with trypsin (sequencing grade trypsin, Promega, Madison, WI) and were desalted using C18 pipette tips (Millipore, Billerica, MA). High-energy collision dissociation (HCD) MS analyses employing a capillary LC-MS/MS were performed on an electrospray ionization FT/ion-trap mass spectrometer (LTQ Orbitrap XL, Thermo Fisher Scientific, San Jose, CA). The MS and MS/MS spectra were searched against the spectra obtained from the ActE proteome using Scaffold (Scaffold_3_00_06, Proteome Software, Portland, OR). Comparisons of secretome composition were carried out using Mathematica v.8.0.4.0 (Wolfram Research).
RNA microarrays
ActE was grown in minimal medium plus the indicated substrate for 7 days. The cell pellet was separated from the culture medium by centrifugation for 10 min at 3000 × g. Microarray experiments were carried out as reported previously23. The total RNA was extracted from the cell pellet and purified. The University of Wisconsin Gene Expression Center carried out the syntheses of cDNA and array hybridizations. Four-plex arrays were constructed by Nimblegen and hybridized with 10 μg of labeled cDNA. ArrayStar (v4.02, DNASTAR, Madison, WI) was used to quantify and visualize data. All analyses were based on three or more biological replicates per carbon source. Quantile normalization and robust multi-array averaging (RMA) were applied to the entire data set. Unless otherwise specified, expression levels are based on log2 values and statistical analysis of the datasets were performed using the moderated t-test.
Author Contributions
T.E.T., A.J.B., C.R.C. and B.G.F. designed experiments. T.E.T., A.J.B. and G.R.L. performed experiments. T.E.T., A.J.B., C.R.C. and B.G.F. wrote the paper.
Supplementary Material
Suppelementary info
Supplemetary Table S2
Acknowledgments
The authors thank, Johnnie A. Walker, Bradon McDonald, Joe Moeller, Robert Zinkle, Grzegory Sabat, Dr. Jonathan Klassen, Dr. Kenneth F. Raffa, and Dr. Aaron Adams for technical supports. The authors also thank Dr. Garet Suen, and Dr. Steve Slater for advice during the preparation of this paper. This work was funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494).
References
- Baldrian P. & Valaskova V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol Rev 32, 501–521 (2008). [DOI] [PubMed] [Google Scholar]
- Cantarel B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37, D233–238 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynd L. R., Weimer P. J., van Zyl W. H. & Pretorius I. S. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev 66, 506–577 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster A. & Schmoll M. Biology and biotechnology of Trichoderma. Appl Microbiol Biotechnol 87, 787–799 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford D. L. Lignocellulose decomposition by selected Streptomyces strains. Appl Environ Microb 35, 1041–1045 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow M. & Williams S. T. Ecology of actinomycetes. Annu Rev Microbiol 37, 189–216 (1983). [DOI] [PubMed] [Google Scholar]
- McCarthy A. J. & Williams S. T. Actinomycetes as agents of biodegradation in the environment--a review. Gene 115, 189–192 (1992). [DOI] [PubMed] [Google Scholar]
- Schlatter D. et al. Resource amendments influence density and competitive phenotypes of Streptomyces in soil. Microb Ecol 57, 413–420 (2009). [DOI] [PubMed] [Google Scholar]
- Wachinger G., Bronnenmeier K., Staudenbauer W. L. & Schrempf H. Identification of mycelium-associated cellulase from Streptomyces reticuli. Appl Environ Microb 55, 2653–2657 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishaque M. & Kluepfel D. Cellulase complex of a mesophilic Streptomyces strain. Can J Microbiol 26, 183–189 (1980). [DOI] [PubMed] [Google Scholar]
- Semedo L. T. et al. Streptomyces drozdowiczii sp. nov., a novel cellulolytic streptomycete from soil in Brazil. Int J Syst Evol Microbiol 54, 1323–1328 (2004). [DOI] [PubMed] [Google Scholar]
- Schlochtermeier A., Walter S., Schroder J., Moorman M. & Schrempf H. The gene encoding the cellulase (Avicelase) Cel1 from Streptomyces reticuli and analysis of protein domains. Mol Microbiol 6, 3611–3621 (1992). [DOI] [PubMed] [Google Scholar]
- Walter S. & Schrempf H. Physiological studies of cellulase (Avicelase) synthesis in Streptomyces reticuli. Appl Environ Microb 62, 1065–1069 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg Z. et al. Cleavage of cellulose by a CBM33 protein. Protein Science 20, 1479–1483 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell D. E., Anderson J. M. & Crosse R. Isolation of facultatively aerobic Actinomycetes from the gut, parent soil and mound materials of the termites Procubitermes-Aburiensis and Cubitermes-Severus. Fems Microbiol Ecol 85, 151–159 (1991). [Google Scholar]
- Pasti M. B. & Belli M. L. Cellulolytic activity of actinomycetes isolated from termites (Termitidae) gut. Fems Microbiol Lett 26, 107–112 (1985). [Google Scholar]
- Pasti M. B., Pometto A. L., Nuti M. P. & Crawford D. L. Lignin-solubilizing ability of Actinomycetes isolated from termite (Termitidae) gut. Appl Environ Microb 56, 2213–2218 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A. et al. Hemicellulose-degrading bacteria and yeasts from the termite gut. J Appl Bacteriol 80, 471–478 (1996). [DOI] [PubMed] [Google Scholar]
- Adams A. S. et al. Cellulose-degrading bacteria associated with the invasive woodwasp Sirex noctilio. ISME Journal 5, 1323–1331 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron M. J. et al. Putative origin of clonal lineages of Amylostereum areolatum, the fungal symbiont associated with Sirex noctilio, retrieved from Pinus sylvestris, in eastern Canada. Fungal Biol 115, 750–758 (2011). [DOI] [PubMed] [Google Scholar]
- Kukor J. J. & Martin M. M. Acquisition of digestive enzymes by siricid woodwasps from their fungal symbiont. Science 220, 1161–1163 (1983). [DOI] [PubMed] [Google Scholar]
- Deboy R. T. et al. Insights into plant cell wall degradation from the genome sequence of the soil bacterium Cellvibrio japonicus. J Bacteriol 190, 5455–5463 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer A. et al. Global gene expression patterns in Clostridium thermocellum as determined by microarray analysis of chemostat cultures on cellulose or cellobiose. Appl Environ Microb 77, 1243–1253 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. et al. Influence of physico-chemical changes on enzymatic digestibility of ionic liquid and AFEX pretreated corn stover. Bioresour Technol 102, 6928–6936 (2011). [DOI] [PubMed] [Google Scholar]
- Tolonen A. C. et al. Proteome-wide systems analysis of a cellulosic biofuel-producing microbe. Mol Syst Biol 6, 461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marushima K., Ohnishi Y. & Horinouchi S. CebR as a master regulator for cellulose/cellooligosaccharide catabolism affects morphological development in Streptomyces griseus. J Bacteriol 191, 5930–5940 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter S. & Schrempf H. The synthesis of the Streptomyces reticuli cellulase (avicelase) is regulated by both activation and repression mechanisms. Mol Gen Genet 251, 186–195 (1996). [DOI] [PubMed] [Google Scholar]
- Deng Y. & Fong S. S. Development and application of a PCR-targeted gene disruption method for studying CelR function in Thermobifida fusca. Appl Environ Microb 76, 2098–2106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosser A. & Schrempf H. A lipid-anchored binding protein is a component of an ATP-dependent cellobiose/cellotriose-transport system from the cellulose degrader Streptomyces reticuli. Eur J Biochem 242, 332–338 (1996). [DOI] [PubMed] [Google Scholar]
- Hess M. et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467 (2011). [DOI] [PubMed] [Google Scholar]
- Suen G. et al. The complete genome sequence of Fibrobacter succinogenes S85 reveals a cellulolytic and metabolic specialist. PLoS One 6, e18814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke F. et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565 (2007). [DOI] [PubMed] [Google Scholar]
- Suen G. et al. An insect herbivore microbiome with high plant biomass-degrading capacity. PLoS Genet 6, e1001129 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott J. J. et al. Bacterial protection of beetle-fungus mutualism. Science 322, 63 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeson W. T., Phillips C. M., Cate J. H. D. & Marletta M. A. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J Am Chem Soc 134, 890–892 (2012). [DOI] [PubMed] [Google Scholar]
- Quinlan R. J. et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. P Natl Acad Sci USA 108, 15079–15084 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaaje-Kolstad G. et al. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 (2010). [DOI] [PubMed] [Google Scholar]
- Langston J. A. et al. Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl Environ Microb 77, 7007–7015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer E. A., Lamed R., White B. A. & Flint H. J. From cellulosomes to cellulosomics. Chem Rec 8, 364–377 (2008). [DOI] [PubMed] [Google Scholar]
- Tamaru Y., Miyake H., Kuroda K., Ueda M. & Doi R. H. Comparative genomics of the mesophilic cellulosome-producing Clostridium cellulovorans and its application to biofuel production via consolidated bioprocessing. Environ Technol 31, 889–903 (2010). [DOI] [PubMed] [Google Scholar]
- Vanden Wymelenberg A. et al. Comparative transcriptome and secretome analysis of wood decay fungi Postia placenta and Phanerochaete chrysosporium. Appl Environ Microb 76, 3599–3610 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K. et al. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 (2000). [DOI] [PubMed] [Google Scholar]
- Medema M. H. et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39, W339–346 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31, 426–428 (1959). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppelementary info
Supplemetary Table S2