

Abstract
Cleft palate is a developmental defect resulting from the failure of embryonic palatal shelves to fuse with each other at a critical time. Immediately before and during palatal fusion (E13–E15 in mice), transforming growth factor β3 (TGFβ3) is expressed in the palatal shelf medial edge epithelium (MEE) and plays a pivotal role in palatal fusion. Using Tgfβ3−/− mice, which display complete penetrance of the cleft palate phenotype, we tested the hypothesis that intra-amniotic gene transfer could be used to prevent cleft palate formation by restoring palatal midline epithelial function. An adenoviral vector encoding Tgfβ3 was microinjected into the amniotic sacs of mouse embryos at successive developmental stages. Transduced Tgfβ3−/− fetuses showed efficient recovery of palatal fusion with mesenchymal confluence following injection at E12.5 (100%), E13.5 (100%), E14.5 (82%), and E15.5 (75%). Viral vectors injected into the amniotic sac transduced the most superficial and transient peridermal cell layer but not underlying basal epithelial cells. TGFβ3 transduction of the peridermdal cell layer was sufficient to induce adhesion, fusion, and disappearance of the palatal shelf MEE in a cell nonautonomous manner. We propose that intra-amniotic gene transfer approaches have therapeutic potential to prevent cleft palate in utero, especially those resulting from palatal midline epithelial dysfunction.
Introduction
Cleft lip/palate is the most common craniofacial developmental defect, affecting ~1 in 700 newborns.1 The etiology of cleft lip/palate is multi-factorial, including both genetic and epigenetic factors. The current treatment for cleft lip/palate entails a multidisciplinary approach involving surgery, extensive dental treatment, and speech therapy. Even after comprehensive and prolonged treatments, patients often have to cope with facial and dentoalveolar deformities, speech impairment, and psychosocial adversity.
Embryonic development of the palate is a temporospatially coordinated process that is shared among mammals.2,3 The secondary palate develops between embryonic days (E) 12.5 and 15.5 in mice and gestational weeks (GW) 6 and 12 in humans. The palatal shelves arise from the maxillary processes at E12.5 in mice (GW6 in humans), and grow vertically along the lateral sides of the tongue. By E13.5 (GW8), the palatal shelves elevate to assume a horizontal position above the tongue. The medial edge epithelium (MEE) of the palatal shelves makes initial contact and adheres to that of the contralateral shelf at E14.5 (GW10). The MEE consists of two cell layers—the inner basal layer of cuboidal cells and the superficial layer of flat peridermal cells.4 As the opposing palatal shelves approximate one another, peridermal cells of the MEE are thought to undergo apoptosis or migration toward the oral and nasal surfaces forming epithelial triangles and exposing basal cells underneath.4,5,6,7 The juxtaposed MEE adhere through the interactions between adhesion molecules on their apical surfaces, forming a midline epithelial seam (MES).8,9,10 The MES rapidly disappears from the midline, and fusion is completed with mesenchymal confluence by E15.5 (GW12).4,11
Although disturbances at any stage of palatal development can lead to failed fusion, dysfunctional MEE may account for a significant portion of cleft palate cases.12 Palatal shelves in a Tgfβ3−/− mouse embryo develop normally and make contact in the midline at E14.5 as in a wild-type embryo. In the absence of transforming growth factor β3 (TGFβ3) expression; however, the palatal shelves fail to fuse, resulting in complete penetrance of the cleft palate phenotype with no other apparent craniofacial anomaly.13,14 The fusion defect is fully rescued in E14.5 Tgfβ3−/− palatal organ cultures by supplementing the culture media with rhTGFβ3.15 Consistent with its primary importance in palatal fusion, TGFβ3 expression is dramatically increased in the MEE at the time of fusion, which begins at E13 and peaks at E14/14.5.16 In humans, TGFβ3 gene variants are associated with increased risk of nonsyndromic cleft lip/palate.17,18,19,20 Other genes associated with defective palatal fusion include interferon regulatory factor 6 (IRF6),21,22,23 poliovirus receptor-related 1 (PVRL1),10 epidermal growth factor receptor (EGFR),24 and APAF1.25
Given the critical importance of transient high-level expression of TGFβ3 in embryonic palatal fusion, we hypothesized that early gestational delivery of the Tgfβ3 gene to the MEE may be sufficient for the prevention of cleft palate in a Tgfβ3−/− mouse. Our rationale for in utero intervention to prevent a developmental anatomical defect is based on the premise that such exploits a window of opportunity to treat the defect before it is manifested beyond an irreversible pathological state. Gene-based approaches have been tested in utero for their therapeutic potential in animal models for a wide range of monogenetic diseases,26 However, there has yet to be a study exploring the potential of such approach to birth defects with structural deformities. In this study, we showed that intra-amniotic delivery of adenoviral vector encoding Tgfβ3 between E12.5 and E15.5 was effective in the prevention of cleft palate by normalizing palatal midline epithelial function in a Tgfβ3−/− mouse embryo.
Results
Characterization of adenovirally expressed TGFβ3
A full-length mouse Tgfβ3 cDNA encoding latent TGFβ3 was used in the construction of adenoviral vectors to allow proper targeting and enzymatic processing of the latent precursor into a smaller bioactive form by endogenous cells. Immunoblot analysis of conditioned media from COS7 cell cultures, which had been infected with null adenovirus (Ad-Null), Ad-TGFβ3, Ad-GFP, Ad-GFP-TGFβ3, confirmed that the virally produced TGFβ3 is a high molecular precursor form and that it was secreted into extracellular milieu and processed into a smaller fragment of 25 kDa, the size corresponding to that of purified bioactive rhTGFβ3 (Figure 1a, arrowhead). Mink lung epithelial (MLE) cell bioassays show TGFβ3 activity in conditioned media collected from COS7 cells infected with Ad-TGFβ3 or Ad-GFP-TGFβ3 (Figure 1b).
Figure 1.

Immunoblot analysis and bioactivity assays for TGFβ3 produced from recombinant adenoviral constructs. (a) Conditioned media were collected from COS7 cell cultures transduced with an empty adenoviral vector (Ad-Null) and the vectors encoding for latent TGFβ3 (Ad-TGFβ3), GFP (Ad-GFP) or GFP, and latent TGFβ3 (Ad-GFP-TGFβ3), and analyzed on an immunoblot with a TGFβ3-specific antibody. The last lane is loaded with fully processed 25 kDa recombinant human TGFβ3 (rhTGFβ3), as indicated with an arrowhead. (b) Bioactivity of virally expressed TGFβ3 was assayed (N = 3), using mink lung epithelial (MLE) cells stably transfected with a luciferase reporter construct under the control of a TGFβ-responsive plasminogen activator inhibitor-1 promoter. Conditioned media collected from COS7 cell cultures, which had been infected with Ad-GFP-TGFβ3, Ad-GFP, Ad-TGFβ3, or Ad-Null, were assayed for luciferase reporter activity in MLE cells. Bovine serum albumin (BSA) and rhTGFβ3 (10 and 100 nmol/l) were used in control assays. GFP, green fluorescent protein; TGFβ3, transforming growth factor β3.
Intra-amniotic delivery of adenoviral vectors: fetal survival and viral transduction
Ad-GFP-TGFβ3 virus was delivered by intra-amniotic injection to a total of 233 embryos from 27 pregnant dams and the control Ad-GFP to 121 embryos from 14 pregnant dams at five different embryonic stages (E12.5, E13.5, E14.5, E15.5, and E16.5). All injected pregnant mice survived surgery and appeared normal. Overall, the survival rate of embryos determined at the time of harvest (E18.5) ranged from 51 to 100% (181/233) for Ad-GFP-TGFβ3 and 76–93% (101/121) for Ad-GFP, with an average of 78 and 83%, respectively (Table 1). The E12.5 injections, which were performed under ultrasound guidance with 350 nl of virus, resulted in significantly higher survival rates (93% with Ad-GFP and 87% with Ad-GFP-TGFβ3), than the E13.5 injections, which were performed under direct visualization with 5 µl of viral solution (76% with Ad-GFP and 51% with Ad-GFP-TGFβ3). For E13.5–E16.5 injections performed under direct visualization, survival rate generally improved as the age and size of embryos increased.
Table 1. The rate of fetal survival and viral transduction following intra-amniotic injection of Ad-GFP-TGFβ3 and Ad-GFP at different embryonic stages.
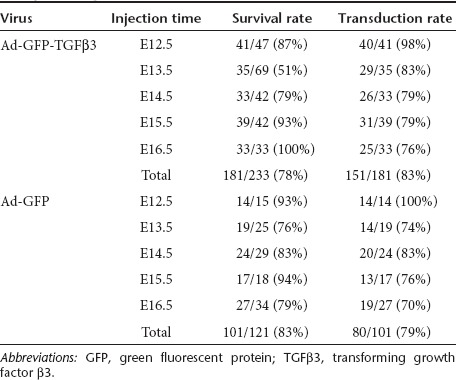
Embryos were examined for viral transduction at the time of harvest. The outer surface of an embryo is covered with a single peridermal cell layer until it begins to slough at around E17. The cornea, however, lacks peridermal covering. In addition, embryonic eyes remain open until E15.5, thus allowing viral access to the corneal epithelium in E12.5–E15.5 injections27 (Figure 2a, arrow). Green fluorescent protein (GFP) expression in the cornea persists until E18.5 and is thus used as a reliable indicator for successful transduction. E12.5 injections performed under the ultrasound guidance resulted viral transduction in 98% (54/55) of embryos, whereas E13.5–E16.5 injections without ultrasound guidance yielded an average transduction rate of 78% (177/227) (Table 1). The injection timing and viral vector used did not influence the rate of successful transduction.
Figure 2.

Adenoviral transduction of mouse embryonic palatal shelves via intra-amniotic administration. Ad-GFP was injected into amniotic cavity at (a,b,f–i) E12.5 or (c–e) 13.5, and the infected embryos were examined at (a,b,c,f–i) E14.5, (d) E15 or (e) E18.5 for the expression of GFP. Viral transduction is readily detectable on (a) cornea (arrow) and epidermis of an infected embryo (b–e) and palatal shelves under a fluorescent stereoscopic microscope. (b) Viral delivery at E12.5 often resulted transduction limited to the anterior half of the palatal shelves, (c) whereas the delivery at E13.5 resulted transduction throughout the entire length of the palatal shelves. A gradual loss of GFP expression along the midline is shown as the palatal shelf fusion progresses from E14.5 to E18.5 (c–e, arrows). (f) A cryosection from an infected embryo shows GFP expression in oral cavity surfaces. The epidermis covering the back of an infected embryo, indicated with a light blue box in a, is shown at higher magnification (in f); viral infection restricted to peridermal cell layer (red arrows). The palatal shelf area, indicated with a red box in g, show viral transduction strictly limited to the superficial peridermal cell layer at higher magnifications (h; red arrows). The area indicated with a yellow box in h is shown at higher magnification (in i) to show that viruses delivered into the amniotic cavity infect the neither basal epithelial cell layer (yellow arrows) nor mesenchyme (asterisk). Under a higher magnification, GFP positive cells indicated with white arrows in h were confirmed peridermal cells that are sloughing off. epi, epidermis; GFP, green fluorescent protein; ns, nasal septum; ps, palatal shelf; t, tongue.
In most of the E12.5 injections, viral transduction, as indicated by GFP expression, was limited to the anterior half of the palatal shelves (Figure 2b, bracket), whereas E13.5 injections resulted in the transduction of surface cells throughout the entire length of palatal shelves (Figure 2c, bracket). The results from later injections at E14.5/E15.5/E16.5 were similar to those of E13.5 injections (data not shown). In all injections, GFP expression appeared robust along the MEE (Figure 2b,c), and gradually faded with the progression of palatal fusion (Figure 2c–e, red arrowheads). Notably, GFP positive cells in the palatal shelf and epidermis were detected only in the peridermal cell layer as shown in the sections prepared from E14.5 embryos injected at E12.5 (Figure 2f,h,i, red arrows). GFP positive cells were not detected in the either epithelial cell layer (Figure 2i, yellow arrows) or mesenchyme (Figure 2i, asterisk) of palatal shelves. Under high magnification, the GFP positive cells indicated with white arrows in Figure 2h were confirmed peridermal cells that were loosened during cryosectioning.
Intra-amniotic injection of Ad-GFP-TGFβ3 rescues cleft palate in Tgfβ3−/− Mice
Tgfβ3−/− mouse embryos transduced with Ad-GFP-TGFβ3 were harvested at E18.5 and scored for palatal fusion as illustrated in Figure 3a. The rescue data from all injections are summarized in Table 2. Intra-amniotic injections at E12.5 and E13.5 induced palatal fusion in all Tgfβ3−/− embryos (14/14); the fusion score for E12.5 injections ranged from 7 to 12, while the score for E13.5 injections was either 11 or 12. Injection at E14.5 induced fusion in 82% (9/11) of Tgfβ3−/− embryos; in this group, all fused palates had scores >9 with the majority (7/9) showing scores of 11–13. It was noted that complete fusion with a score of 13 was achieved only from E14.5 injections. Injection at E15.5 resulted in palatal fusion in 75% of Tgfβ3−/− embryos (6/8); while one embryo (1/6) had the fusion score of 12, the rest (5/6) showed lower fusion scores of 6 or 7. Injection at E16.5 resulted in no rescue (0/8). Control virus, Ad-GFP, presented no rescue effect on palatal fusion in Tgfβ3−/− embryos at any stage (Table 2; Figure 3d,g,j). Tgfβ3+/+ and Tgfβ3+/− embryos showed complete palatal fusion irrespective of the virus type used in the injections (Table 2; Figure 3b,h,k).
Figure 3.

Gene-mediated in utero rescue of cleft palate in Tgfβ3−/− mice. (a) Diagram illustrates the palatal fusion scoring scheme used in this study, which is based on the previously described method.49 The number of ruga pairs included in the fused portion of the palate was counted and a point was assigned to each fused pair (0–9). Also, the pterygoid plate (Ptry) was visually divided into thirds under a dissecting microscope, and one point was assigned to each third that was included in the fused portion of the soft palate (0–3). One additional point was given if fusion was complete at the junction between the primary palate (PP) and secondary palate (SP) (0–1). Thus, complete cleft as shown in the diagram scores 0, whereas completely fused palate would have the maximum score of 13. (b–e) Ad-GFP or Ad-GFP-TGFβ3 was delivered to Tgfβ3−/− (KO) mouse embryos at E14.5 and examined for palatal fusion at E18.5 by SEM. They were compared with uninfected and age-matched wild-type (WT) and Tgfβ3−/− embryos. (f–h) WT and TGFβ3−/− mouse embryos were infected with Ad-GFP or Ad-GFP-TGFβ3 at (f–h) E12.5 or (i–k) E15.5 and examined at E18.5 under a fluorescent stereoscopic microscope. Red brackets (as shown in f,g,i,j) show clefts and the red dotted circle (as shown in f,i) points to the opening between primary and secondary palate. Yellow brackets indicate the areas transduced with viruses (as shown in f–k). Note that E12.5 injections resulted viral transduction in anterior half of the palate compared to more extensive transduction with E15.5 injections. Fusion scores (FS) are shown under each panel (in b–k). GFP, green fluorescent protein; KO, knockout; SEM, scanning electron microscopy; TGFβ3, transforming growth factor β3.
Table 2. Palatal fusion following intra-amniotic delivery of Ad-GFP-TGFβ3 and Ad-GFP to wild-type, Tgfβ3+/− and Tgfβ3−/− mouse embryos.
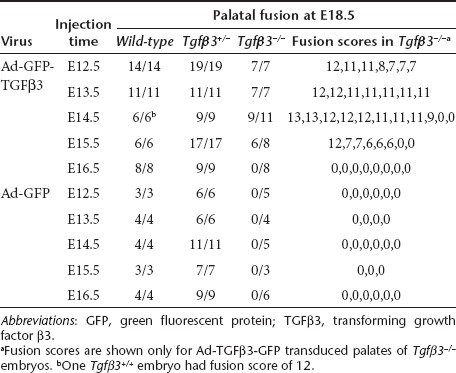
In incompletely rescued palates, clefting of the secondary palate was always present posteriorly (Figure 3f,i, red bracket), indicating that fusion occurred in an anterior to posterior direction. Nine of 33 rescued palates showed complete fusion of the secondary palate with the scores of 12 or 13, and the remainder had fusion in at least anterior ⅔ of the secondary palate with the scores >6 (Table 2). Another location prone to fusion failure was the junction between the primary and secondary palate at the incisive foramen. With a few exceptions from E14.5 injections, most showed a slight opening at this location (Figure 3f, red circle).
During normal palatal development, the nasal septum makes vertical contact and fuses with hard palate anteriorly at the midline (Figure 4a–c). Rescued palates with a fusion score of 13, and a few with the fusion score of 12, demonstrated fusion with the nasal septum (Figure 4e–g). However, for the majority of rescued cleft palates with the fusion score 12 or the scores <12, the vertical fusion was either partially or completely missing (Figure 4i–k). This failure of vertical fusion was accompanied by incomplete fusion between the primary and secondary palates in the anterior palatal segment (Figure 4i). The rescued portion of the secondary palate in Tgfβ3−/− mouse embryos was histologically indistinguishable from the wild-type mouse palate; mesenchymal confluence was achieved with complete disappearance of the midline epithelial seam (Figure 4f–h,j–l).
Figure 4.

Fusion between the secondary palate and the nasal septum in mouse embryos. (a–d) A wild-type mouse embryo (E18.5) that had been injected with Ad-GFP at E14.5 shows that the secondary palate is fused with the nasal septum anteriorly. (e–h) A Tgfβ3−/− mouse rescued with a palatal fusion score of 12 shows vertical fusion with the nasal septum similar to wild-type mice infected with Ad-GFP. (i–l) An example of a complete failure to fuse with the nasal septum is shown in a Tgfβ3−/− mouse rescued with a palatal fusion score of 12. (m) A diagram is shown to indicate where the histological sections were obtained. GFP, green fluorescent protein; TGFβ3, transforming growth factor β3.
Transient transduction of peridermal cells with Ad-GFP-TGFβ3 induces adhesion and disappearance of palatal shelf MEE in Tgfβ3−/− mouse embryos
In wild-type E14.5 embryos, endogenous TGFβ3 was widely detected in oral and nasal epithelia, including the midline epithelial seam, the palatal epithelia on both oral and nasal sides of the palate, and the lower margin of the nasal septum (Figure 5a,b, yellow arrows). As expected, TGFβ3 expression was completely absent in Tgfβ3−/− palates (Figure 5c,d). In Tgfβ3−/− embryos injected with Ad-GFP-TGFβ3 at E12.5 and harvested at E14.5, TGFβ3 was detected only in the peridermal cells along the palatal shelves and the lower border of the nasal septum (Figure 5e,f, yellow arrows). It is noteworthy that virally expressed TGFβ3 was missing in the MES of the Tgfβ3−/− palatal shelves rescued by an intra-amniotic injection of Ad-GFP-TGFβ3 (Figure 5e,f, white arrows). Thus, palatal fusion can occur without TGFβ3 expression in the MES.
Figure 5.

Immunofluorescence localization of endogenous and virally expressed TGFβ3 in E14.5 mouse palatal shelves. (a) Endogenous TGFβ3 is detected in the palatal shelf (PS) epithelium on both oral and nasal sides, in the base of the nasal septum (NS), and the medial epithelial seam (MES) (yellow arrows) in wild-type (WT) mouse. (b) A higher magnification of the MES region indicated with a green box in a shows TGFβ3 expression in all MES cells. (c,d) Uninfected Tgfβ3−/− mouse palate shows complete absence of endogenous TGFβ3. (e) A rescued Tgfβ3−/− mouse palate with Ad-GFP-TGFβ3 (injected at E12.5 and harvested at E14.5) shows virally expressed TGFβ3 in the superficial layer of the palatal shelf and nasal septum epithelium (yellow arrows), but not in the MES (a white arrow). (f) A higher magnification of the green-boxed area in e confirms that the Ad-GFP-TGFβ3 transduced peridermal cells (Pd) are absent in the MES (white arrow). KO, knockout.
TGFβ3 is required for disappearance of the peridermal cell layer of the palatal shelf MEE
Uninjected embryos (Figure 6a–d) and those injected at E13.5 (Figure 6e–h) were examined by scanning electron microscopy for ultrastructural changes associated with fusion process in the palatal MEE (Figure 6). Wild-type Tgfβ3+/+ palates harvested at E14.5 showed the loss of distinct cell boundaries and an appearance of intercellular gaps (Figure 6a,b) which are the characteristic changes occurring during normal palatal fusion.28 This is in contrast to a flat appearance of the MEE with numerous microvilli and intact peridermal cell layer in uninfected E14.5 Tgfβ3−/− palate (Figure 6c,d). Tgfβ3−/− palatal MEE infected with a control vector Ad-GFP at E13.5 and harvested at E18.5 also presented intact peridermal cells (Figure 6e,f). Tgfβ3−/− palates transduced with Ad-GFP-TGFβ3 at E13.5 and harvested at E18.5 (Figure 6g,h), however, were similar in appearance to wild-type Tgfβ3+/+ palates harvested at E14.5 (Figure 6a,b). In rescued palates, peridermal cells retracted away from neighboring cells, and the MEE surface showed pebble-like appearance of exposed basal MEE cells (Figure 6h).
Figure 6.

SEM showing the fate of peridermal cells of the palatal MEE. Uninfected (a,b) wild-type and (c,d) Tgfβ3−/− mouse palates were examined by SEM at E14.5. (e–h) Tgfβ3−/− mouse embryos infected with Ad-GFP or Ad-GFP-TGFβ3 at E13.5 were harvested and examined at E18.5. Boxed areas in a,c,e, and g are shown in high magnification in b,d,f,h, respectively. At E14.5, the wild-type palatal MEE shows pebble-like appearance from the exposed basal cell layer (in b), whereas Tgfβ3−/− palatal MEE presents flat appearance with the intact peridermal cell layer (in d). Tgfβ3−/− mouse palatal shelves infected with Ad-GFP show persistent presence of the intact peridermal cell layer (in f), whereas Tgfβ3−/− palate rescued with Ad-GFP- TGFβ3 shows that peridermal cell layer of the MEE is disappeared, exposing the basal cell layer (in h). GFP, green fluorescent protein; KO, knockout; MEE, medial edge epithelium; SEM, scanning electron microscopy; TGFβ3, transforming growth factor β3; WT, wild-type.
No detectable adverse effect of early gestational exposure to Ad-GFP-TGFβ3 produced on mouse development
In view of the diverse role of TGFβ in the regulation of various cellular activities, we studied potential adverse effects of early gestational exposure to virally produced TGFβ3 on mouse development. Ad-GFP-TGFβ3 (n = 15), Ad-GFP (n = 10), or sterile phosphate-buffered saline (PBS) (n = 14) were intra-amniotically delivered to E13.5 embryos (Tgfβ3+/+), and the mice were monitored for 120 days after birth. All survived and appeared healthy to the end of the monitoring period. No significant differences in weight gain, body length, development, or behavior were observed between the three groups (data not shown). Adenoviral vectors injected into the amniotic cavity can infect a single cell layer of various surfaces of an embryo from its direct exposure to, swallowing of, and breathing in of amniotic fluid. Accordingly, our initial examination of infected embryos at E18.5 revealed that lining cells of the digestive system, lung, periderm, and cornea were transduced with GFP. Microscopic examination of these tissues/organs harvested at postnatal 6 months revealed no detectable gross anomalies, tumors or abnormal growth in any of the three groups (data not shown).
Discussion
This study demonstrates that adenovirus-mediated intra-amniotic delivery of the Tgfβ3 gene restores palatal shelf MEE function and physiological palatal fusion process in Tgfβ3−/− mouse embryos. The rescue only required transduction of the transient peridermal cell layer, suggesting that certain cleft palate cases may be treated in utero by targeting peridermal cells for gene transfer. To our knowledge, this is the first demonstration that a developmental anatomical defect can be corrected in utero with intra-amniotic gene transfer.
The timing of intervention was critically important to avoid cleft palate from becoming permanent. Viral delivery at E12.5, E13.5, and E14.5 were 92% effective in preventing complete fusion failure. Translating these data for human intervention indicates that vector delivery should be planned between GW8 and GW10. Early injection before GW6, which might increase the risk of miscarriage, is unlikely needed. In addition, while viral delivery at E16.5 failed to rescue cleft palate, delivery at E15.5 resulted in fusion extending into ⅔ of the hard palate in 75% of Tgfβ3−/− embryos. The expression of an adenovirally encoded transgene is typically delayed at least 12 hours and begins to peak 24 hours after viral delivery,29 which predicts that viral delivery at E15.5 will not induce TGFβ3 expression until E16 with peak expression at E16.5. Successful rescues from the late injections at E14.5 and E15.5 indicate that the treatment window may extend beyond the physiological timing of palatal fusion at E14.5 (GW12 in humans). Given that most human gene variants associated with cleft palate produce incomplete penetrance of the phenotype, our findings from later injections are especially significant from the therapeutic standpoint; in utero gene transfer may be initiated after a definite diagnosis of fusion failure has been reached. A recent study reported that virtual navigation of a 3D ultrasound data set using the multiplanal mode display (4D software) allowed first-trimester diagnosis of cleft palate in 86% of the cases.30 However, the first-trimester diagnosis of cleft palate still remains a challenge and requires further advancement in imaging technologies. In addition to the timing of intervention, vector dose may also influence the degree of phenotype correction. This study used 350 nl of virus for E12.5 injections, and 5 µl for E13.5–E16.5 injections. Since the amniotic fluid volume roughly doubles between E12.5 (75 µl) and E16.5 (150 µl) with linear regression to fetal growth,31 the vector concentration at E12.5 is about 15-fold lower than that in E13.5 amniotic fluid, correlating with lower rates of viral transduction (Figure 3) and fusion scores (Table 2). The virus concentrations at E14.5, E15.5, and E16.5 are estimated to be 20, 25, and 30% less than that at E13.5, respectively, and thus the most effective rescue, at E13.5, corresponds with the highest vector concentration.
The efficacy and safety of fetal gene therapy is critically dependent on accessible cell populations. We have recently described the fetal developmental stage-dependent distribution of transgene expression after intra-amniotic injection of lentiviral vectors.27,32 While early gestational injections administered from E8 to E10 resulted in transgene expression in a number of organs derived from all three germ layers, injections after E12 showed transgene expression limited to the most superficial layer of epithelia of ectodermal and endodermal organs.27,32 Similar to lentiviral transductions, adenoviral vectors delivered between E12.5 and E15.5 transduced only the most superficial layer of epithelial cells, such as the peridermal cell layer covering the epidermis and oral epithelia. The periderm is a transient layer of histologically discernable flattened cells. In the head region, it starts to emerge at late E10 from the upward migration of a subpopulation of ectodermal cells and covers the entire surface by E12 before disappearing from the surface of embryos during the late gestational period.33,34
The role and fate of peridermal cells during palatal shelf fusion remains unclear. Some have suggested that these cells undergo apoptosis or migration toward the oral or nasal side of the palatal shelves to expose the basal layers of the opposing MEE, allowing for the initiation of adhesion.7,35,36 Others have suggested that, unlike those of the epidermis, peridermal cells of the palatal MEE may be actively involved in the initiation of palatal shelf adhesion.37,38,39 Following initial adhesion, peridermal cells must be cleared from the midline for palatal fusion to progress.7,35,36 Ad-GFP-TGFβ3 transduction of Tgfβ3−/− embryos produced the peridermal changes in the palatal MEE that are reminiscent of those observed in wild-type embryos (Figures 5 and 6), suggesting that peridermal expression of TGFβ3 is essential for initial MEE adhesion as well as subsequent formation of the MES. Interestingly, despite the absence of TGFβ3 expression in the MES of injected Tgfβ3−/− embryos, palatal fusion was complete with mesenchymal confluence. Contrary to the currently held view that persistent TGFβ3 expression in palatal MES cells is required for the disappearance of the MES,12 transient expression of TGFβ3 in the peridermal cell layer can orchestrate the entire palatal fusion process. We suggest that TGFβ3 produced by the peridermal cells acts on basal cells of MEE and upregulates the expression of downstream effectors required for the disappearance of MES, such as MMPs and IRF6. Such cell-nonautonomous action of transduced peridermal cells may explain why our approach was able to correct cleft palate with remarkable efficiency.
TGFβ3 is involved in various biological activities such as the epithelial-mesenchymal tissue interaction during lung development13 and scar-free wound healing40 in embryos. TGFβ signaling is also cytostatic/apoptotic.41 The pleiotropic activity of TGFβ3 portends adverse effects when it is ectopically expressed. For example, TGFβ3 may induce apoptosis of embryonic peridermal cells outside the palatal MEE and premature loss of the periderm, resulting in inappropriate epithelial adhesion in various locations. However, there was no evidence for such inappropriate epithelial adhesion in our study. Furthermore, no adverse effect was detected even in postnatal stages as a result of prenatal exposure to TGFβ3 overexpression. The difference between MEE and non-MEE peridermal cells in their responses to TGFβ3 may be partially explained by regional differences in the distribution of TGFβ receptor types.42 The specific effect shown with intra-amniotic TGFβ3 gene transfer, which appears largely limited to the palatal shelf MEE, is desirable in view of the therapeutic efficacy and safety of the approach.
Regarding clinical applicability, the most compelling target condition for the present approach is cleft palate caused by genetic variants leading to loss of TGFβ3 function. TGFβ3 polymorphisms have been associated with an increased risk of nonsyndromic cleft lip/palate.17,18,19,20 Findings from animal studies predict that these gene variations lead to reduced expression or activity of TGFβ3 in the palatal MEE.13,14,15 The therapeutic potential of the TGFβ3 gene may not be limited to conditions originating from reduced TGFβ3 activity. TGFβ3 signaling is believed to be an upstream regulator of other key molecules that have a major influence on the palatal fusion process. For example, Van der Woude syndrome (MIM ID no. 119300), which accounts for about 2% of all cleft lip and palate cases, is an autosomal dominant disorder caused by null or loss of function mutations in IRF6.43 TGFβ3 signaling has been shown to positively regulate IRF6 in the palatal MEE.11,44 Thus, overexpression of TGFβ3 in the MEE could sufficiently increase the expression of functional IRF6 from the normal allele and compensate the haploinsufficiency. Intra-amniotic TGFβ3 gene transfer may also be applicable to cleft palate resulting from certain environmental causes, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin, a known teratogen that induces palatal midline epithelial dysfunction and cleft palate in humans and mice. Addition of TGFβ3 to a mouse palatal organ culture system has been shown to counteract the effects of dioxin on palatal fusion.45 These examples implicate TGFβ3 as a potential therapeutic gene for multiple etiologies of cleft palate.
Many challenging issues, including vector safety and complex ethical issues, need to be addressed before a vectorized approach will be considered a clinical option for cleft palate. Adeno-based vectors have a number of advantages including transduction of dividing and nondividing cells, lack host genome integration, less concern about germ-line transduction/insertional mutagenesis/bystander developmental effects, and high production titer. In this proof-of-concept study, we utilized the first generation adenoviral vector. However, clinical use of this vector is limited by endogenous innate and adaptive immune responses. For gene therapy requiring transient viral transduction, such as therapy for cleft palate, the third generation adenoviral vector, also known as helper-dependent or “gutted” adenoviral vector, or adeno-associated viral vector with reduced immunogenicity may be considered. Though a lentiviral vector has been used to test the efficacy of TGFβ3 in the reduction of cutaneous scar formation in a mouse model,46 cleft palate correction does not require sustained transgene expression, precluding the need for a lentiviral approach.
Prenatal correction of cleft palate, either by gene therapy or surgical intervention, offers benefits that are not attainable through conventional postnatal therapy. It allows recapitulation of normal ontogeny of palatal development, thus avoiding any other irreversible consequences associated with cleft palate. Though applicable to a range of cleft palate cases, a major drawback of fetal surgery is the high rate of fetal morbidity and mortality.47 Gene transfer by intra-amniotic injection should be minimally invasive and less traumatic. With further advances in gene therapy vectors, prenatal prevention of cleft palate will have a chance to be realized.
Materials and Methods
Mice. Tgfβ3tm1Doe C57BL/6J mice were kindly provided by Dr Yang Chai (University of Southern California, Los Angeles, CA). Heterozygous Tgfβ3tm1Doe C57BL/6J (Tgfβ3+/−) female and male mice were time-mated within a 12-hour window and the mid-time point of the window was designated as E0. For genotyping, tail DNA from embryos was analyzed as previously described,6 using the RED Extract N-Amp PCR Kit (Sigma-Aldrich, St Louis, MO) and PCR primers derived from Tgfβ3 intron 5 (5′-TGGGA GTCAT GGCTG TAACT-3′) and intron 6 (5′-CACTC ACACT GGCAA GTAGT-3′). The PCR amplicons expected from the wild-type Tgfβ3 allele is about 400 bp and that from the mutated allele is 1,300 bp. PCR consisted of 31 cycles of 95 ºC for 20 seconds, 56 ºC for 25 seconds, and 72 ºC for 1 minute, followed by one cycle of 72 ºC for 10 minutes. Animal procedures for this study had been approved by the Institutional Animal Care and Use Committee at the Children's Hospital of Philadelphia. Animal care was in accordance with the National Institutes of Health “Guide for Care and Use of Laboratory Animals”.
Recombinant adenoviruses. GFP and murine Tgfβ3 cDNAs were cloned into E1 and E3 deleted human adenovirus type 5 vectors under the control of a CMV promoter, generating a recombinant adenoviral GFP-TGFβ3 virus (Ad-GFP-TGFβ3). More specifically, the full-length mouse Tgfβ3 cDNA was cloned downstream of, and in frame with, the GFP cDNA and TaV sequence, a cis-acting hydrolase element derived from the Thosea asigna virus. Subsequently, the GFP-TaV-TGFβ3 fragment was inserted into the pShuttle vector (Agilent Technologies, La Jolla, CA) to achieve bicistronic expression of GFP and TGFβ3. The PmeI linearized recombinant shuttle vector DNA was electroporated into Escherichia coli BJ5183-AD-1 harboring the pAdEasy-1 vector (Agilent Technologies). The homologously recombined vector, pAd-CMV/GFP/TaV/TGFβ3, was PacI-linearized and used to transfect HEK293 cells with Fugene6 reagent (Roche Molecular Biochemicals, Indianapolis, IN). Transfected cells were monitored for GFP expression and processed to prepare a high-titer Ad-GFP-TGFβ3 viral stock, using the Adeno-X Virus Purification Kit (Clontech, Mountainview, CA). The viral elute was adjusted to the storage buffer (20 mmol/l Tris-HCl, 25 mmol/l NaCl, 2.5% glycerol, pH 8), and aliquots were rapidly frozen and stored at −80 ºC. Control GFP (Ad-GFP) and TGFβ3 (Ad-TGFβ3) expressing adenoviral vector, using the CMV promoter, were prepared. Viral titer was determined using the Adeno-X Rapid Titer Method Kit (Clontech). The viral titer was 2 × 1010 pfu/ml for Ad-GFP and 2 × 109 pfu/ml for Ad-GFP-TGFβ3.
Bioactivity assays for Ad-GFP-TGFβ3. To confirm the production, processing, and secretion of virally encoded TGFβ3 protein, conditioned media were collected from COS7 cultures transduced with Ad-GFP-TGFβ3 for 2 days and analyzed on immunoblots, using an anti-TGFβ3 IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Bioactivity of virally expressed TGFβ3 was quantified as previously described by others with minor modifications, using MLE cells stably transfected with a firefly luciferase reporter construct under the control of a TGFβ-responsive plasminogen activator inhibitor-1 promoter (kindly provided by Dr James Chang; Stanford University, Palo Alto, CA).48 Briefly, the MLE cells were plated in a 96-well plate (1.6 × 104 cells/well) and allowed to attach for 3 hours in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (HyClone Serum; Thermo Scientific, Waltham, MA). The cell culture medium was then replaced with conditioned media collected from COS7 cell cultures, which had been infected with Ad-GFP-TGFβ3, Ad-GFP, Ad-TGFβ3, and null adenovirus. The conditioned media were prepared by culturing infected cells in serum-free medium for 3 days. MLE cells were incubated in conditioned medium for 14 hours at 37 ºC, washed with PBS, lysed in the cell lysis buffer from Promega Dual-Luciferase Reporter Assay System (Promega, Madison, WI). Cell lysates were assayed for luciferase reporter activity with the EC3 Imaging System (UVP, Upland, CA). The entire experiment was repeated three times (N = 3).
Intra-amniotic injection of adenoviral vectors. Ultrasound-guided microinjection was used to deliver viral vector to E12.5 mouse embryos as previously described.27 Pregnant female mice were anesthetized with isoflurane (4% for induction, 2% for maintenance) and placed supine on a heated pad at 37 ºC. The abdominal skin was disinfected with alcohol, and a 1 cm ventral midline incision was made through the skin and peritoneum. Exposing one uterine horn at a time, the total number of fetuses was recorded. For intra-amniotic viral injection, a prewarmed sterile ultrasound gel (Aquasonic; Parker Laboratories, Fairfield, NJ) was applied to an exposed uterine horn and a fetus was visualized with a 40 MHz ultrasound probe (Vevo 660, VisualSonics, Toronto, Ontario, Canada). A pulled and beveled glass microcapillary pipette (outer diameter 1.14 mm, inner diameter 0.53 mm; Humagen, Charlottesville, VA) was backfilled with mineral oil (Sigma-Aldrich) and attached to the micropipette holder connected to a programmable three-axis microinjector unit (VisualSonics). The pipette tip was then filled with a viral vector. Under ultrasound visualization, the micropipette was advanced through the uterine wall into the amniotic cavity. For each injection, 350 nl of a 1:10 dilution of Ad-GFP-TGFβ3 or Ad-GFP viral stock was delivered to the amniotic cavity with an automated syringe. Injection was repeated until viral vectors were delivered to all embryos. The incision was then closed in two layers using a 4-0 Vicryl suture and the dams placed under a radiant warmer during recovery. Maximum time for this procedure starting from incision to closure was 30 min.
For E13.5, E14.5, E15.5, and E16.5 embryos, microinjection was performed under direct visualization. Pregnant female mice were anesthetized and uterus was exposed, as described above. A hand-pulled and beveled glass micropipette was attached to the microinjector holder and filled with a viral stock. A 5 µl of a 1:10 dilution of Ad-GFP-TGFβ3 or Ad-GFP viral stock was administered to each amniotic cavity using an automated injection system. Postoperative care of the mice was administered as described above. Maximum time for this procedure was 20 minutes.
Assessment of palatal fusion/fusion score. Embryos were harvested from pregnant mice at E18.5. Viable embryos were counted to assess the postoperative survival rate and embryonic tails were harvested for genotyping. The palates harvested at E18.5 were then observed under a dissecting microscope to record the degree of palatal fusion, according to a previously described fusion scoring system.49 The scoring was done blindly, before knowledge of genotype and treatment. Briefly, the nine pairs of rugae and pterygoid plate were used as references in scoring palatal fusion. The number of ruga pairs included in the fused portion of the palate was counted and a point was assigned to each fused pair. Also, the pterygoid plate was visually divided into thirds under a dissecting microscope, and one point was assigned to each third that was included in the fused portion of the palate. One additional point was given if fusion was complete at the junction between the primary and secondary palate. As a result, complete cleft palate would be scored 0, whereas complete palatal fusion would have the maximum score of 13. Any score between 0 and 13 represents a partially fused palate (Figure 3a).
Efficiency and distribution of viral transduction were evaluated under a fluorescent stereoscopic microscope (MZ16 FA; Leica, Heerburg, Switzerland) by visualizing GFP expression on the outer surface of epidermis, cornea, and palatal surface at E14.5 and 18.5. For embryos harvested at E18.5, GFP expression on the surface of embryos was occasionally undetectable and often shown in patchy distribution due to peeling of the peridermal cell layer. Thus, failed viral transduction was concluded only after confirming the lack of GFP expression in the entire embryonic tissue. Autolyzed embryos and those that could not be successfully genotyped were excluded from our analysis.
Postnatal growth assessment of mouse embryos exposed to Ad-GFP-TGFβ3. Wild-type mouse embryos were exposed to Ad-GFP-TGFβ3 virus at E12.5 or E16.5 as described above. The exposed mice were monitored for growth, behavior, and health for a minimum of 120 days into adulthood, and compared to those that had been exposed to either Ad-GFP or sterile PBS. Initially, a few fetuses were randomly selected and examined under fluorescent stereoscope (Leica) to identify the organs with GFP expression. Those noted to have detectable levels of GFP expression were then given particular attention in later dissections and examinations. Mice in all three groups were killed at the end of the observational period, and their organs were examined for presence of any gross abnormalities under a dissecting stereoscope at ×40 magnification.
Histology and immunofluorescence labeling. The heads removed from embryos at the time of harvest were fixed overnight in 10% neutral buffered formalin solution (Sigma-Aldrich) and processed for paraffin embedding (Shandon Excelsior ES; Thermo Electron, Waltham, MA); 5 µm thick serial sections were collected and stained with hematoxylin and eosin.
For immunolabeling of palatal shelves, the heads harvested at E14.5/E15 were dissected to remove the mandible and fixed overnight with 4% paraformaldehyde prepared in a phosphate buffer. Fixed specimens were immersed in 20% sucrose solution (Sigma-Aldrich) in 0.2 mol/l phosphate buffer overnight and embedded in OCT Compound embedding medium (Sakura Finetek, Torrance, CA) by quickly freezing at −180 ºC; 5 µm thick frozen sections were blocked with 25% normal goat serum (NGS) (Invitrogen, Carlsbad, CA) in PBS containing 0.25% Triton-X (PBS-T) for 1 hour at room temperature, and incubated overnight at room temperature with a rabbit anti-TGFβ3 IgG (Santa Cruz Biotechnology) in PBS-T containing 3% NGS. The sections were washed and incubated with goat anti-rabbit IgG conjugated with Texas Red (Invitrogen) in PBS-T containing 3% NGS. Slides were washed in PBS-T and mounted with 60% glycerol (Invitrogen) containing Hoechst (Invitrogen). Immunolabeling was imaged using an inverted fluorescent microscope mounted with a digital camera (IX81; Olympus America, Center Valley, PA).
Scanning electron microscopy. Mouse embryos were intra-amniotically injected with Ad-GFP-TGFβ3 and Ad-GFP at E13.5, and harvested at either E14.5 or 18.5. The heads were dissected to remove the mandible and immediately fixed in Karnovsky's solution. Fixed samples were dehydrated through a graded ethanol series and placed in Freon (1,1,2-Trichloro-1,2,2 Trifluoroethane) (Recycle & Reuse Industries, Mansfield, TX) for critical-point drying. The samples were mounted on aluminum stubs with clay and sputter-coated with gold in an Argon atmosphere, using a Denton Vacuum Desk II Cold Sputter Etch Unit (Denton Vacuum, Cherry Hill, NJ). The palates were then viewed under a Quanta 600 FEG Mark II scanning electron microscope (FEI, Hillsboro, OR).
Acknowledgments
We thank Alison Zajac, Imad Salhab, and John J Dienno for their excellent technical assistance on this project. We are also grateful to Yang Chai (University of Southern California) for providing the TGFβ3+/− mice and his helpful suggestions for the manuscript. This work was supported by the grant from American Cleft and Palate Association, and Mary Downs and Peter Randall Endowed Chair Funds. The authors declared no conflict of interest.
REFERENCES
- Fraser FC. The genetics of cleft lip and cleft palate. Am J Hum Genet. 1970;22:336–352. [PMC free article] [PubMed] [Google Scholar]
- Ferguson MW. Palate development. Development. 1988;103 suppl.:41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- Stanier P., and, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet. 2004;13 Spec No 1:R73–R81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- Fitchett JE., and, Hay ED. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev Biol. 1989;131:455–474. doi: 10.1016/s0012-1606(89)80017-x. [DOI] [PubMed] [Google Scholar]
- Nawshad A, LaGamba D., and, Hay ED. Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT) Arch Oral Biol. 2004;49:675–689. doi: 10.1016/j.archoralbio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Taya Y, O'Kane S., and, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development. 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- Cuervo R., and, Covarrubias L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development. 2004;131:15–24. doi: 10.1242/dev.00907. [DOI] [PubMed] [Google Scholar]
- Martínez-Sanz E, Del Río A, Barrio C, Murillo J, Maldonado E, Garcillán B.et al. (2008Alteration of medial-edge epithelium cell adhesion in two Tgf-beta3 null mouse strains Differentiation 76417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogass M, Bringas P., Jr, and, Shuler CF. Characterization of desmosomal component expression during palatogenesis. Int J Dev Biol. 2000;44:317–322. [PubMed] [Google Scholar]
- Suzuki K, Hu D, Bustos T, Zlotogora J, Richieri-Costa A, Helms JA.et al. (2000Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia Nat Genet 25427–430. [DOI] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P, Jr, Urata MM., and, Chai Y. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev Biol. 2006;297:238–248. doi: 10.1016/j.ydbio.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. Molecular control of secondary palate development. Dev Biol. 2007;301:309–326. doi: 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N.et al. (1995Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction Nat Genet 11415–421. [DOI] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN.et al. (1995Transforming growth factor-beta 3 is required for secondary palate fusion Nat Genet 11409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V, Cui XM, Heisterkamp N, Groffen J., and, Shuler CF. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev Dyn. 1997;209:255–260. doi: 10.1002/(SICI)1097-0177(199707)209:3<255::AID-AJA1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Pelton RW, Hogan BL, Miller DA., and, Moses HL. Differential expression of genes encoding TGFs beta 1, beta 2, and beta 3 during murine palate formation. Dev Biol. 1990;141:456–460. doi: 10.1016/0012-1606(90)90401-4. [DOI] [PubMed] [Google Scholar]
- Lidral AC, Romitti PA, Basart AM, Doetschman T, Leysens NJ, Daack-Hirsch S.et al. (1998Association of MSX1 and TGFB3 with nonsyndromic clefting in humans Am J Hum Genet 63557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Hao L, Li S, Bailey LB, Tian Y., and, Li Z. MTHFR, TGFB3, and TGFA polymorphisms and their association with the risk of non-syndromic cleft lip and cleft palate in China. Am J Med Genet A. 2010;152A:291–298. doi: 10.1002/ajmg.a.33113. [DOI] [PubMed] [Google Scholar]
- Ichikawa E, Watanabe A, Nakano Y, Akita S, Hirano A, Kinoshita A.et al. (2006PAX9 and TGFB3 are linked to susceptibility to nonsyndromic cleft lip with or without cleft palate in the Japanese: population-based and family-based candidate gene analyses J Hum Genet 5138–46. [DOI] [PubMed] [Google Scholar]
- Reutter H, Birnbaum S, Mende M, Lauster C, Schmidt G, Henschke H.et al. (2008TGFB3 displays parent-of-origin effects among central Europeans with nonsyndromic cleft lip and palate J Hum Genet 53656–661. [DOI] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y.et al. (2002Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes Nat Genet 32285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugessur A, Shi M, Gjessing HK, Lie RT, Wilcox AJ, Weinberg CR.et al. (2009Genetic determinants of facial clefting: analysis of 357 candidate genes using two national cleft studies from Scandinavia PLoS ONE 4e5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Jiang R., and, Dixon MJ. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum Mol Genet. 2009;18:2632–2642. doi: 10.1093/hmg/ddp201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen PJ, Chin JR, Shum L, Slavkin HC, Shuler CF, Derynck R.et al. (1999Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure Nat Genet 2269–73. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA., and, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Dejneka NS, Surace EM, Aleman TS, Cideciyan AV, Lyubarsky A, Savchenko A.et al. (2004In utero gene therapy rescues vision in a murine model of congenital blindness Mol Ther 9182–188. [DOI] [PubMed] [Google Scholar]
- Endo M, Henriques-Coelho T, Zoltick PW, Stitelman DH, Peranteau WH, Radu A.et al. (2010The developmental stage determines the distribution and duration of gene expression after early intra-amniotic gene transfer using lentiviral vectors Gene Ther 1761–71. [DOI] [PubMed] [Google Scholar]
- Martínez-Alvarez C, Tudela C, Pérez-Miguelsanz J, O'Kane S, Puerta J., and, Ferguson MW. Medial edge epithelial cell fate during palatal fusion. Dev Biol. 2000;220:343–357. doi: 10.1006/dbio.2000.9644. [DOI] [PubMed] [Google Scholar]
- Schneider H, Adebakin S, Themis M, Cook T, Douar AM, Pavirani A.et al. (1999Therapeutic plasma concentrations of human factor IX in mice after gene delivery into the amniotic cavity: a model for the prenatal treatment of haemophilia B J Gene Med 1424–432. [DOI] [PubMed] [Google Scholar]
- Martinez-Ten P, Adiego B, Illescas T, Bermejo C, Wong AE., and, Sepulveda W. First-trimester diagnosis of cleft lip and palate using three-dimensional ultrasonography. Ultrasound Obstet Gynecol. 2011;40:40–46. doi: 10.1002/uog.10139. [DOI] [PubMed] [Google Scholar]
- Cheung CY., and, Brace RA. Amniotic fluid volume and composition in mouse pregnancy. J Soc Gynecol Investig. 2005;12:558–562. doi: 10.1016/j.jsgi.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Endo M, Zoltick PW, Peranteau WH, Radu A, Muvarak N, Ito M.et al. (2008Efficient in vivo targeting of epidermal stem cells by early gestational intraamniotic injection of lentiviral vector driven by the keratin 5 promoter Mol Ther 16131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne C, Tainsky M., and, Fuchs E. Programming gene expression in developing epidermis. Development. 1994;120:2369–2383. doi: 10.1242/dev.120.9.2369. [DOI] [PubMed] [Google Scholar]
- M'Boneko V., and, Merker HJ. Development and morphology of the periderm of mouse embryos (days 9-12 of gestation) Acta Anat (Basel) 1988;133:325–336. doi: 10.1159/000146662. [DOI] [PubMed] [Google Scholar]
- Waterman RE., and, Meller SM. Alterations in the epithelial surface of human palatal shelves prior to and during fusion: a scanning electron microscopic study. Anat Rec. 1974;180:111–135. doi: 10.1002/ar.1091800111. [DOI] [PubMed] [Google Scholar]
- Waterman RE, Ross LM., and, Meller SM. Alterations in the epithelial surface of A-Jax mouse palatal shelves prior to and during palatal fusion: a scanning electron microscopic study. Anat Rec. 1973;176:361–375. doi: 10.1002/ar.1091760311. [DOI] [PubMed] [Google Scholar]
- Martínez-Alvarez C, O'Kane S, Taya Y., and, Ferguson MW. Palate development in the TGF-beta 3 knockout mouse. Low vacuum scanning electron microscopy reveals changes in the medial edge epithelium. Int J Dev Biol. 1996. pp. 115S–116S. [PubMed]
- Martínez-Alvarez C, Bonelli R, Tudela C, Gato A, Mena J, O'Kane S.et al. (2000Bulging medial edge epithelial cells and palatal fusion Int J Dev Biol 44331–335. [PubMed] [Google Scholar]
- Vaziri Sani F, Kaartinen V, El Shahawy M, Linde A., and, Gritli-Linde A. Developmental changes in cellular and extracellular structural macromolecules in the secondary palate and in the nasal cavity of the mouse. Eur J Oral Sci. 2010;118:221–236. doi: 10.1111/j.1600-0722.2010.00732.x. [DOI] [PubMed] [Google Scholar]
- Lu L, Saulis AS, Liu WR, Roy NK, Chao JD, Ledbetter S.et al. (2005The temporal effects of anti-TGF-beta1, 2, and 3 monoclonal antibody on wound healing and hypertrophic scar formation J Am Coll Surg 201391–397. [DOI] [PubMed] [Google Scholar]
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B.et al. (2009A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis Cell 13787–98. [DOI] [PubMed] [Google Scholar]
- Dudas M, Nagy A, Laping NJ, Moustakas A., and, Kaartinen V. Tgf-beta3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev Biol. 2004;266:96–108. doi: 10.1016/j.ydbio.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Little HJ, Rorick NK, Su LI, Baldock C, Malhotra S, Jowitt T.et al. (2009Missense mutations that cause Van der Woude syndrome and popliteal pterygium syndrome affect the DNA-binding and transcriptional activation functions of IRF6 Hum Mol Genet 18535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight AS, Schutte BC, Jiang R., and, Dixon MJ. Developmental expression analysis of the mouse and chick orthologues of IRF6: the gene mutated in Van der Woude syndrome. Dev Dyn. 2006;235:1441–1447. doi: 10.1002/dvdy.20598. [DOI] [PubMed] [Google Scholar]
- Thomae TL, Stevens EA., and, Bradfield CA. Transforming growth factor-beta3 restores fusion in palatal shelves exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem. 2005;280:12742–12746. doi: 10.1074/jbc.M410780200. [DOI] [PubMed] [Google Scholar]
- Waddington SN, Crossley R, Sheard V, Howe SJ, Buckley SM, Coughlan L.et al. (2010Gene delivery of a mutant TGFß3 reduces markers of scar tissue formation after cutaneous wounding Mol Ther 182104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopulos NA, Papadopoulos MA, Kovacs L, Zeilhofer HF, Henke J, Boettcher P.et al. (2005Foetal surgery and cleft lip and palate: current status and new perspectives Br J Plast Surg 58593–607. [DOI] [PubMed] [Google Scholar]
- Yalamanchi N, Klein MB, Pham HM, Longaker MT., and, Chang J. Flexor tendon wound healing in vitro: lactate up-regulation of TGF-beta expression and functional activity. Plast Reconstr Surg. 2004;113:625–632. doi: 10.1097/01.PRS.0000101529.47062.34. [DOI] [PubMed] [Google Scholar]
- Cui XM, Shiomi N, Chen J, Saito T, Yamamoto T, Ito Y.et al. (2005Overexpression of Smad2 in Tgf-beta3-null mutant mice rescues cleft palate Dev Biol 278193–202. [DOI] [PubMed] [Google Scholar]