

Abstract
Gene therapy with ex vivo-transduced hematopoietic stem/progenitor cells may represent a valid therapeutic option for monogenic immunohematological disorders such as Wiskott-Aldrich syndrome (WAS), a primary immunodeficiency associated with thrombocytopenia. We evaluated the preclinical safety and efficacy of human CD34+ cells transduced with lentiviral vectors (LV) encoding WAS protein (WASp). We first set up and validated a transduction protocol for CD34+ cells derived from bone marrow (BM) or mobilized peripheral blood (MPB) using a clinical grade, highly purified LV. Robust transduction of progenitor cells was obtained in normal donors and WAS patients' cells, without evidence of toxicity. To study biodistribution of human cells and exclude vector release in vivo, LV-transduced CD34+ cells were transplanted in immunodeficient mice, showing a normal engraftment and differentiation ability towards transduced lymphoid and myeloid cells in hematopoietic tissues. Vector mobilization to host cells and transmission to germline cells of the LV were excluded by different molecular assays. Analysis of vector integrations showed polyclonal integration patterns in vitro and in human engrafted cells in vivo. In summary, this work establishes the preclinical safety and efficacy of human CD34+ cells gene therapy for the treatment of WAS.
Introduction
Transplantation of autologous hematopoietic stem cells (HSC) genetically modified ex vivo using viral vectors represents a potentially curative treatment for inherited disorders of the hematopoietic system, including primary immunodeficiencies.1,2 Clinical trials for severe combined immunodeficiency (SCID) due to adenosine deaminase deficiency3 or IL2RG-deficiency (X-linked severe combined immunodeficiency (SCID-X1))4,5 have shown that gene therapy with retrovirally transduced HSC results in long-term clinical efficacy. However, gene modification by γ-retroviral vectors, likely in combination with disease-specific factors, resulted in insertional mutagenesis in the clinical trials for SCID-X16,7 and chronic granulomatous disease.8 Self-inactivating lentiviral vectors (LV) may represent a substantial improvement in terms of safety profile while offering a more robust transduction and better transcriptional control of transgene expression in HSC.9 Two recent studies for the treatment of X-linked adrenoleukodystrophy10 and β-thalassemia11 have provided information on the experimental clinical use of LV-transduced CD34+ cells. In the adrenoleukodystrophy trial, highly polyclonal multilineage engraftment of gene corrected cells at substantial levels (15%) and clear therapeutic benefits were observed. In the β-thalassemia study, transduced CD34+ cells failed to engraft in the first patient while the second patient showed oligoclonal HSC engraftment associated with a dominant clone reaching ~15% of the cells and leading to independency from red blood cell transfusion.11
Wiskott-Aldrich Syndrome (WAS) is an excellent candidate for a gene therapy approach in patients lacking a suitable donor or at high risk of complications.12,13 WAS is a rare and severe X-linked immunodeficiency characterized by eczema, platelet defects, recurrent infections, autoimmunity, and increased susceptibility to develop tumors, in particular lymphomas.14,15 The disorder is caused by mutations in the gene encoding for the WAS protein (WASp),16 preferentially expressed in hematopoietic cell lineages and involved in actin cytoskeleton reorganization, leading to different cellular dysfunctions such as migration, signal transduction, and activation.17,18
The recent report that WAS-gene transfer using a γ-retroviral vector resulted in improved platelet counts and immune functions further supports the rationale for a gene therapy strategy for this disease.19,20 On the other hand, the occurrence of leukemia associated to an insertion near the LMO2 gene21 in this trial reinforces the need for a potentially safer vector platform to deliver the WAS transgene.
We have developed an HSC gene therapy approach based on lentiviral-mediated gene transfer of WAS complementary DNA under the control of a 1.6 kb fragment of the endogenous WAS promoter. We previously showed that transplantation of gene corrected WAS-deficient murine HSC in non-lethally irradiated Was−/− mice resulted in long-term multilineage engraftment of WASp-expressing cells and functional correction of lymphocyte and dendritic cells with a lack of observable toxicity over a short- and long-term period.22,23 Moreover, the vector was able to restore in vitro expression of WASp in CD34+ cells and mature cells from WAS patients.24,25
The recently issued European regulation on Advanced Therapy Medicinal Products26 define genetically modified HSC as the Medicinal Product, and guidelines detailing nonclinical aspects of medicinal products containing genetically modified cells are in preparation by regulatory authorities. Crucial parameters to be assessed include the dose of vector required for therapeutic efficacy, the in vitro toxicity of the vector, the profile of vector integration, the dose of transduced cells available at the end of the manufacturing process, the in vivo biodistribution of transduced cells, and the potential for vector release and germline transmission. Although in vitro tests for human HSC are well defined, surrogate in vivo assays may rely on transplantation of transduced CD34+ cells in humanized immunodeficient mouse models.27,28 Several investigators have employed these assays to study the properties of transduced cells as part of their preclinical studies,29,30,31 but not all aspects related to pharmacological and toxicological properties of transduced CD34+ cells have been previously addressed.
The aim of our study was to develop a clinically applicable transduction protocol for human CD34+ cells using a purified good manufacturing practice (GMP)-grade lentiviral vector for the treatment of WAS and to assess the safety and efficacy in relevant in vitro and in vivo preclinical assays. Our results show robust transduction of CD34+ cells from bone marrow (BM) and mobilized peripheral blood (MPB) of healthy donors (HD) and WAS patients, in the absence of detectable toxicity, allowing to validate manufacturing process. Transduced CD34+ cells infused in immunodeficient mice were able to repopulate and differentiate into multiple lineages with a polyclonal profile of vector integrations, in the absence of vector shedding and germline transmission. Results from this and previous studies have provided the rationale and preclinical data to launch a clinical trial for HSC gene therapy of WAS.
Results
Effect of prestimulation time and multiplicity of infection of LV on transduction efficiency, growth, and progenitor cell content
We aimed at identifying the culture conditions and vector exposure for clinically applicable efficient LV gene transfer into CD34+ cells without toxicity using serum-free medium, early-acting cytokines, and retronectin. We first investigated the effect of different vector doses on human HD CD34+ cells from BM or MPB prestimulated for 24 hours. Cells were analyzed by PCR for vector copy number (VCN), 12–15 days after transduction when only integrated copies are detected by the assay (Supplementary Figure S1). As shown in Figure 1a, we found a dose effect for both vector and number of exposures, reaching the highest transduction efficiency (0.84 ± 0.63) at two rounds of transduction with a multiplicity of infection of 100 (corresponding to a 1 × 108 TU/ml), with a cumulative dose of 2 × 108 TU/ml.
Figure 1.

Optimization of transduction protocol using pre-GMP vectors. (a) VCN on CD34+ cells transduced with 1 or 2 hits of infection with two different MOI (n = 10 for 1 hit and n = 12 for 2 hits). Statistical analysis for significance was performed by Wilcoxon-matched paired test. In gray are indicated VCN of transduced CD34+ cells from WAS patients' BM or MPB. Lines indicate mean values. (b) CD34+ cells derived from healthy donors BM or (c) MPB were transduced with 1 or 2 hits after a prestimulation of 24 hours (BM, n = 11; MPB, n = 10) and 48 hours (BM, n = 6; MPB, n = 5) for 1 hit, and 24 hours (BM, n = 15; MPB, n = 9) for 2 hits. VCN/cell was calculated on cultured cells. (d) Total number of colonies derived from LTC-IC are reported for 1 (n = 5) and 2 hits (n = 5) of transduction at MOI 100, after 24 hours of prestimulation in the presence of cytokines. Results are reported as mean ± SD. *P value < 0.05, **P value < 0.01. BM, bone marrow; LTC-IC, long-term culture-initiating cell; MOI, multiplicity of infection; MPB, mobilized peripheral blood; UT, untransduced; VCN, vector copy number; WAS, Wiskott-Aldrich syndrome.
We then studied the effect of different prestimulation times (24 or 48 hours) on CD34+ cells followed by a single hit of transduction in comparison to two hits of transduction. After a single hit, VCN was similar after 24 or 48 hour prestimulation. Two hits exposure to LV resulted in higher VCN versus one hit for both MPB and BM, with BM being transduced at an increased efficiency with respect to MPB (1.1 versus 0.5, paired t-test; P value = 0.003) (Figure 1b,c).
Expression of CD34 cell surface marker, clonogenic potential, and yield of recovered CD34+ cells after culture did not show significant differences compared to untransduced (UT) cells in all conditions (Supplementary Tables S1 and S2). As expected, there was a tendency toward a decrease in the percentage of CD34+ cells after 48 hours prestimulation in the two hits protocols due to the longer period of culture (Supplementary Table S2).
Finally, we showed that transduced CD34+ cells display a normal content in primitive progenitor cells (long-term culture-initiating cell (LTC-IC)) as compared to UT (Figure 1d). In conclusion, a 60-hour culture and two hits exposure under clinical grade conditions results in efficient transduction of CD34+ cells without detectable impact on growth and differentiation capacity of transduced hematopoietic stem and progenitor cells.
Efficient transduction of WAS patients' CD34+ cells using a clinical grade LV
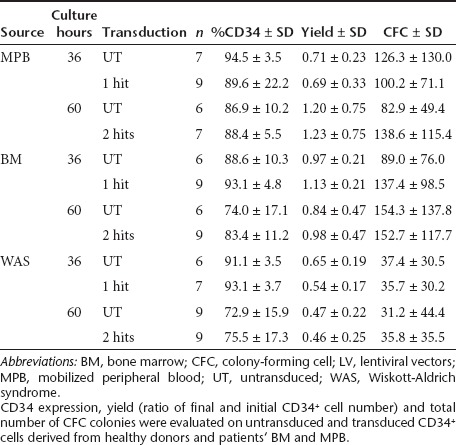
Next, we transferred the optimized protocol to WASp-deficient CD34+ cells obtained from patients' BM or MPB and investigated the efficiency of gene transfer in bulk cultures. Similarly to HD, also WAS CD34+ cells exposed to two hits of infections had a significantly increased VCN as compared to one hit (1.44 ± 0.48 versus 0.64 ± 0.31) (Figure 2a). To verify transduction efficiency at the single progenitor cell level, we measured the percentage of LV positive colony-forming cell (CFC) colonies by quantitative PCR (qPCR). As shown in Figure 2b, the second hit of LV increased the percentage of transduction from 33.5 ± 13.7 to 53.8 ± 15.8 in HD and from 49.0 ± 20.3 to 79.9 ± 14.1 in WAS patients. Intriguingly, WAS patients' CD34+ cells showed a higher VCN and gene transfer into progenitor cells as compared to both HD BM- and MPB-derived cells in all conditions. Importantly, there were no marked differences between transduced and UT cells in terms of growth, CD34 expression, total number of CFC colonies, and proportion of progenitors (erythroid, myeloid or mixed colonies) (Table 1 and data not shown). In WAS BM CD34+ cells, we observed a reduced recovery of cells after thawing and a decreased growth capacity and number of colony-forming cells versus HD CD34+ cells, without difference between UT and transduced cells (data not shown).
Figure 2.

Efficient transduction of CD34+ cells by clinical LV vectors. BM or MPB from healthy donors (squares/white columns) or WAS patients (triangles/black columns) were transduced with 1 or 2 hits using GMP-grade lots of LV vector encoding WAS. (a) VCN/cell were calculated by qPCR on cultured CD34+ cells and (b) single transduced colonies. n for 1 hit trasduction protocol = 14 (6 MPB and 8 BM) and 8 (1 MPB and 7 BM) for HDs and WAS patients respectively, and n for 2 hits protocol = 17 (7 MPB and 10 BM) for HDs and 11 (2 MPB and 9 BM) for WAS patients. Lines indicate median values. *P value < 0.05, **P value < 0.01, ***P value < 0.001. BM, bone marrow; CFC, colony-forming cell; GMP, good manufacturing practice; HD, healthy donors; LV, lentiviral vectors; MPB, mobilized peripheral blood; qPCR, quantitative PCR; VCN, vector copy number; WAS, Wiskott-Aldrich syndrome.
Table 1. Characterization of CD34+ cells transduced by clinical LV vectors.
Large scale GMP production of LV-transduced CD34+ cells
To further validate the selected protocol for use in WAS patients, three experiments were performed under large scale GMP conditions on BM CD34+ cells. CD34+ cells (12.5 × 106 cells) from HD were cultured in retronectin-coated bags in serum-free medium supplemented with cytokines and exposed twice to GMP grade purified vector at multiplicity of infection of 100, for a total culture time of 60 hours. As reported in Table 2, CD34 cell surface expression (72.8 ± 4.2%) and yield at the end of culture (1.22 ± 0.20) were in the range of small scale experiments, with no evidence of toxicity on bulk cultures or on clonogenic progenitors. In vitro cultured CD34+ cells remained always cytokine-dependent (data not shown). Analysis of integrated vector by qPCR in transduced cells performed on single CFC colonies, showed robust and reproducible levels of transduction (53–67%). The mean VCN/cell in liquid bulk cultures maintained for 15 days after gene transfer was 0.70 ± 0.10. All quality tests on transduced CD34+ cells were in agreement with the specification of the medicinal drug product, including cell viability, transgene expression, sterility, mycoplasma, endotoxin, replication competent LV, in vitro viral contaminants, and process contaminants (E1A DNA, large T antigen protein and plasmid DNA, free LV in cell supernatant) (Table 2 and data not shown).
Table 2. Large scale validation transduction of CD34+ cells derived from healthy donor BM.

LV gene transfer restores WASp expression in differentiated cells from transduced CD34+ cells
To determine if the clinical grade w1.6W LV vector allowed restoration of WASp expression, we performed transduction on different human cell lines and evaluated the presence of the protein by western blot or flow cytometric analysis. As shown in Figure 3, WASp expression was restored to normal levels after transduction of WASp-deficient untransformed T-cell lines (Figure 3a) and Epstein–Barr virus -transformed B cell line (Figure 3b).
Figure 3.

Restoration of WASp expression in transduced cells. (a) The expression of WASp was evaluated after transduction of untransformed T cells (FACS analyses), (b) EBV-transformed B cell lines (western blot), and (c) CD34+ cells differentiated into myeloid cells (western blot) from normal donors and WAS patient(s). In a, in gray are indicated control cells, black lines indicated stained cells. Numbers represent the percentage of WASp positive cells. BM, bone marrow; EBV, Epstein–Barr virus; FACS, fluorescence-activated cell sorting; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HD, healthy donors; Pt, patient; UT, untransduced; WASp, WAS protein.
We then analyzed WASp expression in transduced CD34+ cells cultured in presence of cytokines promoting the differentiation to the myeloid lineage and specifically to megakaryocytes (MKs). Two hits transduction with w1.6W LV restored WASp expression in BM or MPB CD34+ cells differentiated from three patients, reaching levels comparable to those of HD (Figure 3c) at a VCN of 0.7–1.9. Differentiated MKs from UT and transduced WASp-deficient CD34+ cells were then evaluated for colocalization of WASp with the MKs marker CD41 by confocal microscopy. Remarkably, two hits transduction with the LV WAS allowed expression of WASp in MKs (Supplementary Figure S2).
Normal biodistribution of LV-transduced CD34+ cells in Rag2−/−/γc−/− mice
In order to investigate whether transduction with the w1.6W LV clinical vector affects the ability of human HSC to engraft and differentiate in vivo, we injected transduced HD umbilical cord blood (UCB)-derived CD34+ cells into sublethally irradiated Rag2−/−/γc−/− neonate mice (VCN of CD34+ cells differentiated in vitro 1.8 ± 1.5). Control animals were transplanted with the same amount of UT and unmanipulated (UM) UCB CD34+ cells. After 8 weeks, the biodistribution of human-derived cells was analyzed in different organs by flow cytometry (a representative example of the analysis is shown in Supplementary Figure S3). As shown in Figure 4a, the engraftment of human CD45+ cells was comparable to mice injected with UCB-derived UM and UT cells in all organs tested (BM, thymus, spleen, and liver). As expected, we found a majority of CD19+ cells, and a small proportion of CD34+ progenitor cells and CD13+ myeloid cells in the BM and spleen of transplanted mice (Figure 4b,c). In the thymus, we observed a normal pattern of T-cell differentiation with presence of all subsets (CD4/CD8 double positive cells, single positive) (Figure 4d). No significant differences were observed between different treatment groups either in terms of level of engraftment or differentiation pattern in tested organs. To confirm the engraftment capacity of BM-derived cells, we injected transduced BM CD34+ obtained from large scale transduction validation experiments (VCN: 0.7 ± 0.1), for which UM control cells were not available in sufficient number. As expected, BM-derived cells had lower levels of engraftment with respect to UCB (Figure 4e) with a similar pattern of differentiation (Figure 4f). As shown in Figure 4g, LV-transduced cells were present in all hematopoietic organs, including BM (VCN 1.86 ± 1.31 for UCB and 1.07 ± 0.56 for BM), thymus (VCN 1.11 ± 0.92 for UCB and 1.08 ± 0.56 for BM), spleen (VCN 1.95 ± 1.78 for UCB and 0.48 ± 0.18 for BM), and liver (VCN 1.09 ± 0.87 for UCB and 0.43 ± 0.11 for BM).
Figure 4.

Engraftment of transduced human HSC in Rag2−/−/γc−/− mouse model. Rag2−/−/γc−/− mice were injected after sublethal irradiation with human UCB CD34+ cells either UM, UT or LV transduced. (a) Engraftment of human cells was calculated in terms of CD45+ cell in different organs. The percentage of most representative subpopulations in (b) BM, (c) spleen, and (d) thymus were evaluated by FACS on human CD45+ cells. (e) The percentage of engrafted BM-derived transduced CD34+ cells and (f) subpopulation distribution were evaluated on specified organs. (g) qPCR analysis was performed on cells from different organs or BM. BM, bone marrow; FACS, fluorescence-activated cell sorting; HSC, hematopoietic stem cells; LV, lentiviral vectors; qPCR, quantitative PCR; UCB, umbilical cord blood; UM, unmanipulated; UT, untransduced; VCN, vector copy number.
Analyses of vector shedding, mobilization, and germline transmission of LV-transduced CD34+ cells
To assess the safety of the drug product, we first performed experiments to investigate whether infectious vector particles were able to remain associated with gene corrected cells and potentially transduce bystander cells, as shown previously in a mouse model.32 To exclude the occurrence of in vivo generation of replication competent lentivirus (RCL), vector mobilization or shedding to nontarget cells including germline, we followed different approaches exploiting the model of immunodeficient mice transplanted with transduced CD34+ cells.
To investigate vector shedding and germline transmission, we designed specific experiments collecting gonads and brains of male mice (n = 7) injected with human BM- or UCB-derived transduced CD34+ cells and perfused with saline solution, before harvest, to reduce organ contamination with circulating hematopoietic cells. As negative control, brains were used. No vector signal was detected by qPCR (sensitivity threshold: <16 copies in 105 cells) in these organs, indicating the absence of detectable vector shedding to nonhematopoietic tissues, including the germline (data not shown).
To exclude the transfer of packaging function or generation of RCL, we then measured gag p24 HIV by enzyme-linked immunosorbent assay (ELISA) in the plasma of 17 mice transplanted with LV-WAS transduced CD34+ cells. All animals tested were negative except for a mouse that displayed a low-level of protein (0.4 ng/ml). HIV-1 gag sequence was detected by PCR on the BM of positive mice, but there was no evidence of gag integrated in the infused CD34+ cells and the RCL culture assays were negative both on infused CD34+ cells and on BM from the transplanted mouse. More detailed analyses using specific PCR primers indicated the presence of gag-pol packaging plasmid sequences in the original configuration, but not recombined with viral long terminal repeat or transfer vector sequences in the mouse BM (data not shown).
To assay the potential mobilization or shedding of vector cells to bystander cells, we designed a novel PCR assay (B2-SINE) that distinguishes between LV integrated in murine cells versus human cells. This method has a high sensitivity (up to 0.25% transduction, 35 transduced genomes into 14,000 UT ones) and specificity (no signal is detected in 100% human transduced cells) (Supplementary Figure S4). No evidence of vector sequences integrated in the murine genome from human to murine cells was reported in murine tissues (Supplementary Figure S5a), including BM from the mouse that was found positive for p24 (Supplementary Figure S5b).
LV insertion profile from in vitro cultured CD34+ cells and human cells engrafted in vivo are polyclonal
In order to study the integration profile of w1.6W LV vector and the clonal repertoire of transduced cells, we collected insertion sites in vitro and in vivo through the combination of ligation-mediated/linear amplification-mediated PCR (LM/LAM-PCR) and pyrosequencing. We analyzed the distribution of the vector in CD34+ cells from a pool of four HD BM and transduced human UCB in vitro and in vivo upon engraftment in Rag2−/−/γc−/− mice. The insertion profile in vitro was typical of the LV vectors with significant preference for transcriptional units (from 74.5 to 77.0% of integrations) as compared to in silico generated random insertions (n = 100,000, 40.7%) (P < 0.01, z-test for two proportions) (Supplementary Table S3).
This general bias was maintained in vivo, 8 weeks after transduced cell infusion in Rag2−/−/γc−/− mice. As for the frequency of integration sites in proximity of genes involved in “Cancer” disease category from IPA software (Ingenuity Systems, http://www.ingenuity.com) we did not found any enrichment in vivo as compared to random frequency (6.3% in both HD and WAS patients versus 16.1% in random in silico insertions dataset; hit “Cancer” genes are listed in Supplementary Table S4). The number of unique insertions retrieved was in agreement with the overall profile of the LAM-PCR products run on Spreadex gels (Elchrom Scientific, Zurich, Swizerland), showing a polyclonal repertoire both in vitro in human CD34+ cells from HD and WAS patients (Figure 5a) and in vivo upon infusion in Rag2−/−/γc−/− mice (Figure 5b). In addition, by comparing the insertions in vivo in BM and thymus from individual treated mice or in BM, thymus, and spleen from a pool of three mice, we observed the presence of shared identical integrants among the different hematopoietic compartments analyzed (Figure 5c).
Figure 5.

Analysis of w1.6 LV integration profile in human transduced CD34+ cells. LV integration analysis was performed by LAM-PCR on (a) BM-transduced CD34+ cells from healthy donors (left panel, n = 3) and patients (right panel, n = 2), (b) as well as transduced human UCB in vitro and in vivo upon engraftment in Rag2−/−/γc−/− mice. LAM-PCR products are shown on Spreadex gels are shown. (c) Shared identical integrations in BM and thymus of two individual mice (11 and 13) (upper circles) and in BM, thymus (Thy), and spleen (Spl) from a pool of three mice (lower circles). BM, bone marrow; gDNA, genomic DNA; IC, internal control; LAM-PCR, linear amplication-mediated PCR; LV, lentiviral vectors; Pt, patient; UCB, umbilical cord blood.
Discussion
Transplant of ex vivo gene corrected HSC could represent a valid therapeutic option for patients affected by WAS. Our results indicate that the clinical grade WAS-encoding LV under the control of its own promoter can transduce at high efficiency human CD34+ progenitor cells from HD and WAS patients' in a dose-dependent manner, and drive protein expression in differentiated cells. The two hit protocol allowed us to reach 1–2 copies of vector per cells, shown to be required to achieve functional correction of the WAS defect,23,24 and was thus selected for validation and clinical development.
No detectable toxic effect on the growth and in vitro or in vivo differentiation of CD34+ cells were observed. We cannot rule out the two hits protocol (60 hours culture), may reduce the in vivo engraftment properties of hematopoietic stem and progenitor cells with respect to one hit protocol. It should be noted that the selected protocol is shorter than the one previously adopted in γ-retroviral vector-based trials. Long-term studies in humans will allow to determine the engraftment capacity of transduced CD34+ cells at the moment of the infusion.
WAS patient-derived cells displayed an overall reduced capacity to grow in culture when exposed to cytokines as compared to HD cells, likely as a result of a defect in cell signaling or proliferation. Indeed, a key role for WASp in signaling of the cytokine receptor for stem cell factor (c-Kit) has been identified in murine hematopoietic cells.33 The higher efficiency of gene transfer into WAS CD34+ cells as compared to HD may be related to a higher susceptibility to infection from LV of WASp-deficient cells, possibly related to alterations in entry and post-entry processing of LV due to the absence of the protein. However, we cannot exclude that in vitro corrected WAS cells might have a selective advantage over UT cells, in agreement with the observed in vitro selective advantage for BM cells from WASp-heterozygous mice.33
Since the experimental medicinal product is represented by the autologous transduced CD34+ cells and there is limited clinical experience with the ex vivo use of LV, we designed assays to assess biodistribution and the potential risk of vector shedding, mobilization, and germline transmission using the relevant human target cells in in vitro and in vivo models.27 Our results show that WAS LV transduced healthy HSC migrate to the lymphohematopoietic organs and undergo in vivo short-term differentiation similarly to UT and unmanipulated HSC. Despite their limitations, our data combined to previous observations28,30,31 indicate that immunodeficient mice could represent a suitable model for assessing biodistribution and safety of human CD34+ cells from different sources. The sensitivity and observation time may be further improved by the use of novel immunodeficient strains which allow higher engraftment of human cells.34
Even though viral vector particles might be associated to the drug product at the end of the manufacturing process, our data show that LV distributes along with human hematopoietic cells, remaining integrated in the human genome and is not mobilized or shed to murine cells, as assessed by different approaches. In addition to the classical techniques for the analysis of vector biodistribution, we developed a new technique adapting and improving the B2 SINE-PCR from Follenzi et al.35 Differently from conventional methods, this approach allows to distinguish between episomal and integrated form in unwanted target. In addition, integrated vector can be detected in several tissues including germ lines where silencing of transgene could mask the vector presence. The detection of HIV gag sequences in the BM of one mouse together with the evidence of HIV p24 gag antigen in the plasma raised the remote possibility that a RCL may have been generated, although such an event should have occurred during cell transduction as the virus batch was tested for RCL and shown to be negative. However, the safety features of the third generation packaging system combined with a SIN transfer vector used in this study make it extremely unlikely that a productive viral recombinant is generated during vector production or cell transduction. Indeed, no RCL was detected in the mouse tissue samples by the RCL assay. Thus, the most likely interpretation of the findings is the occurrence of a stable transfection of the packaging plasmid in a rare cell engrafted in the mouse. This is possible given that the plasmids used for the transient transfection of the vector-producer cells represent a major contaminant of the final vector product, even if the downstream purification process removes almost 99.9% of the input. This event was not deemed to be associated with pathology because it did not represent RCL nor it would lead to vector mobilization because the Gag-containing particles released from the cells would lack an envelope to make them infectious as well as a viral RNA, as the transduced SIN vector would be resistant to packaging and mobilization. If such Gag-expressing cells were to be infused in a patient their output would be diluted in the larger body fluids to well below detection and, most likely, the cells would be cleared by viral antigen-directed immunity (at variance with its survival in the immunocompromised SCID mouse).
The analysis of vector integrations in vitro and in vivo showed a strong bias of WAS vector for coding regions without major preference for regions upstream of transcription start sites, a feature typical of lentiviral vectors10,36 (Supplementary Table S3), and proto-oncogenes (Supplementary Table S4). The insertional profile was polyclonal in human CD34+ cells both in vitro and after engraftment and selection in vivo in Rag2−/−/γc−/− mice (Figure 5b), in agreement with the results of preclinical studies of WAS-LV integration in murine cells.37
A gene therapy approach based on a γ-retroviral vector encoding WASp under the control of a strong viral promoter (myeloproliferative sarcoma virus (MPSV)) showed the therapeutic potential20 of this approach but was associated with the occurrence of leukemia in a patient.21 In this regard, the implementation of a LV platform in combination with a moderately active internal promoter should confer a substantially improved safety profile9,38 over the MLV approach.
In summary, our data demonstrate that the GMP grade WAS-encoding LV can efficiently transduce CD34+ cells under validated clinical conditions, leading to restoration of protein expression with no toxicity. This study provides important information about the quality and preclinical safety and efficacy data on transduced HSC for WAS patients complementing previous in vitro and in vivo studies performed in the murine WAS KO model which showed efficient engraftment of vector-transduced cells and immune reconstitution without the occurrence of LV-induced tumors.22,23 Phase I/II gene therapy clinical protocols based on infusion of LV-transduced autologous CD34+ cells combined with a reduced intensity conditioning received approval from regulatory authorities and have recently started by our and other groups.39,40
Materials and Methods
Patients' and healthy donor cells. HDs granulocyte colony-stimulating factor MPB- or BM- derived CD34+ cells were purchased from Lonza (Basel, Switzerland). HD UCB and WAS patients' BM cells were obtained after written informed consent according to standard ethical procedure and with approval of the San Raffaele Institute Bioethical Committee (protocol TIGET-01 and -02). Biological material from six WAS patients (score 3–5), diagnosed on the basis of clinical symptoms and genetic analysis41,42 (Supplementary Table S5), was obtained at the time of diagnostic or therapeutic procedures. WASp expression was not detectable by western blot in patients' cells except for a weak expression observed in patient 3. CD34+ cells were purified from mononuclear cells by positive selection with immunomagnetic beads according to the manufacturer's procedure (Miltenyi Biotec, Bergisch-Gladbach, Germany).
LV. The vector used in this study (w1.6W) is a 3rd generation, self-inactivating vesicular stomatitis virus (VSV.G) pseudotype lentiviral vector constructed on a pCCL backbone, previously described23 (Supplementary Figure S6a). The vector lots were produced by MolMed S.p.a. (Milan, Italy), using a large scale validated process. The vectors were produced following GMP guidelines, except for initial studies of gene transfer protocol optimization, which were research grade. Vector characterization is reported in Supplementary Table S6.
Cell culture and transduction. CD34+ cells were seeded on non-tissue culture treated Retronectin (TaKaRa Bio, Shiga, Japan) coated plates (30 µg/ml) or, for GMP manufacturing validation, in VueLife bags (American Fluoroseal, Gaithersburg, MD) at 1 × 106 cells/ml in serum-free CellGro SCGM Medium (Cell Genix Technologies, Friburg, Germany) in the presence of cells culture grade stem cell factor 300 ng/ml (Amgen, Thousand Oaks, CA), FLT3-L 300 ng/ml, Thrombopoietin 100 ng/ml, and IL-3 60 ng/ml (all from Peprotech, Rocky Hill, NY). Following different prestimulation times, cells were transduced with w1.6W LV at different multiplicity of infection for one or two hits of transduction. For the two hits protocol, a wash period of 10–12 hours was performed between the vector exposures. At the end, cells were analyzed by flow cytometry for the expression of CD34 and CD45 cell surface markers, plated for colony-forming unit cells (CFU-C) and LTC-IC assays or cultured (described in Supplementary Materials and Methods) to perform proviral integration evaluation by qPCR, WASp expression measurement, and integration profile analysis. This culture method promotes the differentiation of CD34+ cells toward myeloid lineage.
Epstein–Barr virus-transformed B cell lines and untransformed T-cell lines were established and transduced as previously described.25
Western blot analysis. Western blot analyses was performed as previously described,24 using anti-hWASP (H250; Santa Cruz Biotechnologies, Santa Cruz, CA), anti-hGAPDH (Chemicon, Temecula, CA), and anti-hActin (Sigma-Aldrich, St Louis, MO) antibodies, followed by secondary HRP-coupled antibodies (DAKO A/S, Glostrup, Denmark).
Mice. Rag2−/−/γc−/− mice on BALB/c background were obtained from the Central Institute for Experimental Animals (Nogawa, Japan). All animals were bred and maintained in a specific pathogen-free animal facility. Procedures were performed according to protocols approved by the Committee for Animal Care and Use of San Raffaele Scientific Institute (Committee Protocol no. 318). Three to four days after birth, mice were irradiated with two doses of 3 and 2.5 Gy, given at 3 hours interval. Three hours after irradiation, mice were injected with 0.3 × 106 UCB cells or 0.7 × 106 BM cells in 25–30 µl phosphate-buffered saline into the temporal vein.
After 8 weeks, mice were euthanized and organs were collected and analyzed for the presence of human CD45+ cells and the expression of different lineage-specific surface markers. For collection of gonads and brain, mice were perfused injecting saline solution for 10 minutes through blood vessels of mice to eliminate hematopoietic contamination from organs, before sacrifice. Cells were analyzed for proviral integration evaluation by qPCR and integration profile by LAM-PCR analyses.
Real-time PCR analysis. In order to evaluate the number of LV copies integrated per genome, a qPCR was performed using specific primer and probes for human telomerase, LV, and murine β-actin. A reference standard was obtained from serially diluted transduced human T-cell line carrying one copy of integrated LV. Results of integrated vector copies were normalized for the number of evaluated genomes. As negative control, samples of UT cells were used. All the reactions were performed according to the manufacturer's instructions and analyzed with an ABI PRISM 7900 sequence detection system (Applied Biosystems, Foster City, CA). Primers and probes sequences were reported in Supplementary Table S7
Flow cytometric analysis. Surface staining of transduced HSC was performed with anti-CD34-PE and CD45-PerCP monoclonal antibodies. Cells collected from the organs of Rag2−/−/γc−/− mice were stained for the expression of surface markers: CD45-APC, CD19-PC7, CD34-PC5, CD3-FITC, CD4-PerCP, CD8-PE, CD13-PE, and CD15-FITC (all from BD Biosciences, San Jose, CA). Detection of WASp expression was performed after permeabilization (Cytofix/Cytoperm kit; BD Biosciences) by a purified anti-hWASp monoclonal antibody (clone 5A5; BD Biosciences) followed by an Alexa 488 conjugated goat anti-mouse antibody (Invitrogen, Carlsbad, CA). Cells were acquired using a FACSCantoII (BD Biosciences, San Jose, CA) and analyzed with FCS Express Software (De Novo Software, Los Angeles, CA).
Evaluation of p24 by ELISA assay and RCL detection. HIV p24 protein concentration was measured in mice plasma collected 7 weeks after UCB-transduced CD34+ cell injection or culture supernatants by Alliance HIV-1 p24 ELISA kit (Perkin Elmer, Waltham, MA) according to manufacturer's instructions.
The RCL assay was performed according to published method.43,44 Briefly, permissive C8166-45 cell line were cocultured with transduced CD34+ cells and cultured for 28 days. At the end of the culture, HIV p24 protein presence was determined by ELISA assay on culture supernatant. Moreover, C8166-45 genomic DNA was extracted to verify the absence of VSV-G DNA by qPCR.
B2-SINE PCR for detecting murine-specific integrations. In order to detect the presence of integrated LV in murine genome after systemic injection of human CD34+ transduced cells, we performed a modified and improved three-step B2 SINE-PCR technique35 (Supplementary Figure S6b). Sequences of primers and PCR conditions are reported in Supplementary Materials and Methods.
Integration profile analyses. Integrations were retrieved by LAM-PCR, as previously described.9,45 Briefly, after a linear PCR amplification of genome-vector junctions, followed by a magnetic capture and second strand reconstitution, a digestion with Tsp509I, HpyCH4IV or MseI was performed. Digestion products were ligated with a restriction site-complementary linker cassette and underwent to two sequential rounds of exponential PCR steps. The LAM-PCR products were tagged with megaprimers and underwent 454 pyrosequencing. Resulting genomic sequences were automatically aligned to the human genome (assembly hg18 of the UCSC genome browser, http://genome.ucsc.edu/).
Statistical analysis. Statistical analysis was performed using GraphPad Prism software (GraphPad Software, La Jolla, CA), analyzing data sets for significance by Mann–Whitney two-tailed test, unless otherwise stated.
SUPPLEMENTARY MATERIAL Figure S1. VCN evaluation of different time-point in transduced CD34+ cells. Figure S2. Restoration of WASp expression in transduced cells. Figure S3. Engraftment of human HSC in Rag2−/−/γc−/− mouse model. Figure S4. SINE-PCR resolution and sensitivity. Figure S5. No vector sequence integrated in murine bystander cells. Figure S6. Schematic representation of LVV and SINE-PCR method. Table S1. Recovery and CFU numbers from healthy control's BM and MPB CD34+ cells untransduced and transduced with different vector concentration. Table S2. Recovery and CFU numbers of healthy control's BM and MPB-derived CD34+ cells untransduced and transduced after different prestimulation time. Table S3. Insertion profile in in vitro- and in vivo- transduced cells. Table S4. Cancer genes. Table S5. Summary of patients score and mutations. Table S6. Characterization of GMP and pre-GMP vector. Table S7. Sequence of primers for qPCR. Materials and Methods.
Acknowledgments
This work was supported by Fondazione Telethon and the European Commission (CONSERT contract LSBH-CT-2004-005242 and CLINIGENE LSHB-CT2006-018933 and CELL-PID HEALTH-F5-2010-261387) and PERSIST. M.S. and M.R. are employed by MolMed S.p.a.. All the other authors declared no conflict of interest.
Supplementary Material
VCN evaluation of different time-point in transduced CD34+ cells.
Restoration of WASp expression in transduced cells.
Engraftment of human HSC in Rag2−/−/γc−/− mouse model.
SINE-PCR resolution and sensitivity.
No vector sequence integrated in murine bystander cells.
Schematic representation of LVV and SINE-PCR method.
Recovery and CFU numbers from healthy control's BM and MPB CD34+ cells untransduced and transduced with different vector concentration.
Recovery and CFU numbers of healthy control's BM and MPB-derived CD34+ cells untransduced and transduced after different prestimulation time.
Insertion profile in in vitro- and in vivo- transduced cells.
Cancer genes.
Summary of patients score and mutations.
Characterization of GMP and pre-GMP vector.
Sequence of primers for qPCR.
REFERENCES
- Aiuti A., and, Roncarolo MG. Ten years of gene therapy for primary immune deficiencies. Hematology Am Soc Hematol Educ Program. 2009. pp. 682–689. [DOI] [PubMed]
- Fischer A, Hacein-Bey-Abina S., and, Cavazzana-Calvo M. 20 years of gene therapy for SCID. Nat Immunol. 2010;11:457–460. doi: 10.1038/ni0610-457. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L.et al. (2009Gene therapy for immunodeficiency due to adenosine deaminase deficiency N Engl J Med 360447–458. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC.et al. (2010Efficacy of gene therapy for X-linked severe combined immunodeficiency N Engl J Med 363355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J.et al. (2004Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector Lancet 3642181–2187. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E.et al. (2008Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1 J Clin Invest 1183132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H.et al. (2008Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients J Clin Invest 1183143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A.et al. (2010Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease Nat Med 16198–204. [DOI] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C.et al. (2006Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration Nat Biotechnol 24687–696. [DOI] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I.et al. (2009Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy Science 326818–823. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F.et al. (2010Transfusion independence and HMGA2 activation after gene therapy of human ß-thalassaemia Nature 467318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovich AH, Stone JV, Tomany SC, Ireland M, Kollman C, Pelz CJ.et al. (2001Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program Blood 971598–1603. [DOI] [PubMed] [Google Scholar]
- Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, Schulz A, Thrasher AJ, Mazzolari E.et al. (2008Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation Blood 111439–445. [DOI] [PubMed] [Google Scholar]
- Shcherbina A, Candotti F, Rosen FS., and, Remold-O'Donnell E. High incidence of lymphomas in a subgroup of Wiskott-Aldrich syndrome patients. Br J Haematol. 2003;121:529–530. doi: 10.1046/j.1365-2141.2003.04310.x. [DOI] [PubMed] [Google Scholar]
- Ochs HD., and, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117:725–38; quiz 739. doi: 10.1016/j.jaci.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Derry JM, Ochs HD., and, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;79:following 922. [PubMed] [Google Scholar]
- Ramesh N., and, Geha R. Recent advances in the biology of WASP and WIP. Immunol Res. 2009;44:99–111. doi: 10.1007/s12026-008-8086-1. [DOI] [PubMed] [Google Scholar]
- Bosticardo M, Marangoni F, Aiuti A, Villa A., and, Grazia Roncarolo M. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood. 2009;113:6288–6295. doi: 10.1182/blood-2008-12-115253. [DOI] [PubMed] [Google Scholar]
- Boztug K, Dewey RA., and, Klein C. Development of hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Curr Opin Mol Ther. 2006;8:390–395. [PubMed] [Google Scholar]
- Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA.et al. (2010Stem-cell gene therapy for the Wiskott-Aldrich syndrome N Engl J Med 3631918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avedillo Díez I, Zychlinski D, Coci EG, Galla M, Modlich U, Dewey RA.et al. (2011Development of novel efficient SIN vectors with improved safety features for Wiskott-Aldrich syndrome stem cell based gene therapy Mol Pharm 81525–1537. [DOI] [PubMed] [Google Scholar]
- Dupré L, Marangoni F, Scaramuzza S, Trifari S, Hernández RJ, Aiuti A.et al. (2006Efficacy of gene therapy for Wiskott-Aldrich syndrome using a WAS promoter/cDNA-containing lentiviral vector and nonlethal irradiation Hum Gene Ther 17303–313. [DOI] [PubMed] [Google Scholar]
- Marangoni F, Bosticardo M, Charrier S, Draghici E, Locci M, Scaramuzza S.et al. (2009Evidence for long-term efficacy and safety of gene therapy for Wiskott-Aldrich syndrome in preclinical models Mol Ther 171073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré L, Trifari S, Follenzi A, Marangoni F, Lain de Lera T, Bernad A.et al. (2004Lentiviral vector-mediated gene transfer in T cells from Wiskott-Aldrich syndrome patients leads to functional correction Mol Ther 10903–915. [DOI] [PubMed] [Google Scholar]
- Charrier S, Dupré L, Scaramuzza S, Jeanson-Leh L, Blundell MP, Danos O.et al. (2007Lentiviral vectors targeting WASp expression to hematopoietic cells, efficiently transduce and correct cells from WAS patients Gene Ther 14415–428. [DOI] [PubMed] [Google Scholar]
- < http://ec.europa.eu/health/human-use/ advanced-therapies/index_en.htm >
- Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A.et al. (2004Development of a human adaptive immune system in cord blood cell-transplanted mice Science 304104–107. [DOI] [PubMed] [Google Scholar]
- Benhamida S, Pflumio F, Dubart-Kupperschmitt A, Zhao-Emonet JC, Cavazzana-Calvo M, Rocchiccioli F.et al. (2003Transduced CD34+ cells from adrenoleukodystrophy patients with HIV-derived vector mediate long-term engraftment of NOD/SCID mice Mol Ther 7317–324. [DOI] [PubMed] [Google Scholar]
- Ficara F, Superchi DB, Hernández RJ, Mocchetti C, Carballido-Perrig N, Andolfi G.et al. (2004IL-3 or IL-7 increases ex vivo gene transfer efficiency in ADA-SCID BM CD34+ cells while maintaining in vivo lymphoid potential Mol Ther 101096–1108. [DOI] [PubMed] [Google Scholar]
- Cornils K, Lange C, Schambach A, Brugman MH, Nowak R, Lioznov M.et al. (2009Stem cell marking with promotor-deprived self-inactivating retroviral vectors does not lead to induced clonal imbalance Mol Ther 17131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will E, Bailey J, Schuesler T, Modlich U, Balcik B, Burzynski B.et al. (2007Importance of murine study design for testing toxicity of retroviral vectors in support of phase I trials Mol Ther 15782–791. [DOI] [PubMed] [Google Scholar]
- Pan YW, Scarlett JM, Luoh TT., and, Kurre P. Prolonged adherence of human immunodeficiency virus-derived vector particles to hematopoietic target cells leads to secondary transduction in vitro and in vivo. J Virol. 2007;81:639–649. doi: 10.1128/JVI.01089-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani M, Venkatasubrahmanyam S, Sanyal M, Levy S, Butte A, Weinberg K.et al. (2009Wiskott-Aldrich syndrome protein is an effector of Kit signaling Blood 1142900–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G.et al. (2005Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice Blood 1061565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi A, Sabatino G, Lombardo A, Boccaccio C., and, Naldini L. Efficient gene delivery and targeted expression to hepatocytes in vivo by improved lentiviral vectors. Hum Gene Ther. 2002;13:243–260. doi: 10.1089/10430340252769770. [DOI] [PubMed] [Google Scholar]
- Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio A, Cassani B.et al. (2007Hot spots of retroviral integration in human CD34+ hematopoietic cells Blood 1101770–1778. [DOI] [PubMed] [Google Scholar]
- Mantovani J, Charrier S, Eckenberg R, Saurin W, Danos O, Perea J.et al. (2009Diverse genomic integration of a lentiviral vector developed for the treatment of Wiskott-Aldrich syndrome J Gene Med 11645–654. [DOI] [PubMed] [Google Scholar]
- Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, Brugman MH.et al. (2009Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors Mol Ther 171919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy A, Roncarolo MG., and, Thrasher AJ. Development of lentiviral gene therapy for Wiskott Aldrich syndrome. Expert Opin Biol Ther. 2008;8:181–190. doi: 10.1517/14712598.8.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy A., and, Thrasher AJ. Gene therapy for the Wiskott-Aldrich syndrome. Curr Opin Allergy Clin Immunol. 2011;11:545–550. doi: 10.1097/ACI.0b013e32834c230c. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Watanabe C, Liu T, Hollenbaugh D, Blaese RM, Kanner SB.et al. (1997Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype Blood 902680–2689. [PubMed] [Google Scholar]
- Trifari S, Scaramuzza S, Catucci M, Ponzoni M, Mollica L, Chiesa R.et al. (2010Revertant T lymphocytes in a patient with Wiskott-Aldrich syndrome: analysis of function and distribution in lymphoid organs J Allergy Clin Immunol 125439–448.e8. [DOI] [PubMed] [Google Scholar]
- Farson D, Witt R, McGuinness R, Dull T, Kelly M, Song J.et al. (2001A new-generation stable inducible packaging cell line for lentiviral vectors Hum Gene Ther 12981–997. [DOI] [PubMed] [Google Scholar]
- Escarpe P, Zayek N, Chin P, Borellini F, Zufferey R, Veres G.et al. (2003Development of a sensitive assay for detection of replication-competent recombinant lentivirus in large-scale HIV-based vector preparations Mol Ther 8332–341. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Schwarzwaelder K, Bartholomae C, Zaoui K, Ball C, Pilz I.et al. (2007High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat Methods 41051–1057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
VCN evaluation of different time-point in transduced CD34+ cells.
Restoration of WASp expression in transduced cells.
Engraftment of human HSC in Rag2−/−/γc−/− mouse model.
SINE-PCR resolution and sensitivity.
No vector sequence integrated in murine bystander cells.
Schematic representation of LVV and SINE-PCR method.
Recovery and CFU numbers from healthy control's BM and MPB CD34+ cells untransduced and transduced with different vector concentration.
Recovery and CFU numbers of healthy control's BM and MPB-derived CD34+ cells untransduced and transduced after different prestimulation time.
Insertion profile in in vitro- and in vivo- transduced cells.
Cancer genes.
Summary of patients score and mutations.
Characterization of GMP and pre-GMP vector.
Sequence of primers for qPCR.