

Abstract
Nuclear reprogramming of adult somatic tissue enables embryo-independent generation of autologous, patient-specific induced pluripotent stem (iPS) cells. Exploiting this emergent regenerative platform for individualized medicine applications requires the establishment of bioequivalence criteria across derived pluripotent lines and lineage-specified derivatives. Here, from individual patients with type 1 diabetes (T1D) multiple human iPS clones were produced and prospectively screened using a battery of developmental markers to assess respective differentiation propensity and proficiency in yielding functional insulin (INS)-producing progeny. Global gene expression profiles, pluripotency expression patterns, and the capacity to differentiate into SOX17- and FOXA2-positive definitive endoderm (DE)-like cells were comparable among individual iPS clones. However, notable intrapatient variation was evident upon further guided differentiation into HNF4α- and HNF1β-expressing primitive gut tube, and INS- and glucagon (GCG)-expressing islet-like cells. Differential dynamics of pluripotency-associated genes and pancreatic lineage-specifying genes underlined clonal variance. Successful generation of glucose-responsive INS-producing cells required silencing of stemness programs as well as the induction of stage-specific pancreatic transcription factors. Thus, comprehensive fingerprinting of individual clones is mandatory to secure homogenous pools amenable for diagnostic and therapeutic applications of iPS cells from patients with T1D.
Introduction
Ectopic expression of a set of defined stemness factors triggers reprogramming of adult somatic tissue and generation of induced pluripotent stem (iPS) cells that deploy comprehensive traits of embryonic stem (ES) cells.1,2,3 Embryo-independent iPS cell-based technology allows derivation of autologous patient-specific PS cells,4,5,6,7,8 with ensuing specification of disease-relevant lineages offering, in principle, authentic cell-based platforms for personalized diagnostic and therapeutic applications. In fact, iPS clones have been established from diverse human disease conditions.9,10,11,12,13 Recapitulation of disease phenotypes through redifferentiation of patient-specific iPS cells underscores the utility of nuclear reprogramming and bioengineered lineage specification for individualized disease modeling and drug screening.14,15,16
Type 1 diabetes (T1D) is caused by immune-mediated destruction of insulin (INS)-producing pancreatic β-cells. T1D pathobiology implicates multiple genetic loci and environmental influences in disease susceptibility.17 Several genes located in the major histocompatibility complex on chromosome 6p21.3 have been identified as major genetic factors, while other genes, such as INS, CTLA4, and PTPN22, make additional contributions to the disease pathogenesis.17 Extensive studies in spontaneous T1D rodent models, including the nonobese diabetic mouse strain, have also identified combinations of major histocompatibility complex and other susceptibility genes responsible in the development of T1D.18 Notably, the nonobese diabetic mouse strain is nonpermissive for derivation of ES cells,19,20 while iPS cells from nonobese diabetic mice demonstrate an unstable pluripotent state,21 suggesting possible influence of the T1D-predispositing genetic background on achieving and maintaining pluripotency.
For diagnostic and therapeutic applications it is critical to ensure reliable and efficient differentiation of T1D-derived somatic tissue into iPS cells proficient to yield INS-producing islet-like cells. Based on protocols which facilitate conversion of human ES cells into INS-producing islet-like progeny,22,23,24,25,26 human iPS clones have been recently differentiated into INS-producing cells.27,28,29 Generation of INS-producing cells from three iPS clones from two T1D patients (one clone from a 30-year-old and two clones from a 32-year-old) has further verified the utility of T1D-specific iPS cells for experimental and clinical studies.30 Of note, T1D-specific iPS clones exhibited marked clonal differences in differentiation propensity,30 mandating evaluation of suspected intrapatient variation.
Here, to address the influence of the T1D milieu, we recruited T1D patients, generated multiple iPS clones from each individual, and systematically determined respective differentiation propensities. While distinct from healthy controls, derived T1D iPS clones demonstrated similar pluripotency gene expression profiles and comparable differentiation proficiency for definitive endoderm (DE) conversion. Yet, clonal variations became increasingly prominent upon further guided differentiation of iPS progeny into primitive gut tube- and islet-like cells, and differential regulation of pluripotency-associated genes and pancreatic lineage-specifying genes was associated with inconsistent lineage specification. The observed intrapatient variance in differentiation potential within derived T1D-specific pluripotent clones points to the importance of quality control screening for selection of proficient patient-specific lines.
Results
Reprogramming T1D patient-derived cells
Lentiviral vectors encoding OCT4, SOX2, KLF4, and c-MYC were added to transduce epidermal cells from three T1D patients (T1D-1, T1D-2, and T1D-3; 21–38 years old), and one nondiabetic (ND) donor (31-year-old) (Table 1). Under feeder-free conditions, small iPS-like colonies with a sharp-edged, flat, and tightly packed morphology appeared within 2 weeks after vector transduction. T1D- and ND-iPS clones, cultured under feeder-free conditions, exhibited morphology similar to those typical of human PS cells. These T1D- and ND-iPS clones were positive for alkaline phosphatase activity (Figure 1a, Supplementary Figure S1), and expressed human pluripotency-associated markers; SSEA-4, TRA-1-60, TRA-1-81, OCT4, SOX2, KLF4, and NANOG (Figure 1a, Supplementary Figure S2). All tested iPS clones were negative for SSEA-1 expression (Figure 1a, Supplementary Figure S2). Semiquantitative reverse transcription-PCR analysis confirmed induction of endogenous pluripotency-associated genes including OCT4, SOX2, GDF3, TERT, KLF4, c-MYC, and NANOG (Figure 1b). Real-time PCR assays demonstrated the silencing of lentiviral transgene expression in derived iPS clones (Figure 1c, with an exception, clone T1D1#3 maintaining relatively high exogenous KLF4 expression), indicating that derived iPS clones do not depend on exogenous reprogramming factors for sustained pluripotency. Due to technical constraints, the exogenous OCT4 transgene levels were not determined by real-time PCR. Since the four reprogramming factors were expressed from lentiviral vectors of the same vector background, it is likely that vector-derived OCT4 transgene expression was similarly silenced in derived iPS cells. Karyotyping analysis of derived iPS clones revealed cells with normal diploid karyotypes (Figure 1d and Supplementary Figure S3). Only one abnormal karyotype (trisomy 8) was found in iPS T1D-3#3 clone at passage 27 (1 out of 20 cells; Supplementary Figure S3a,b). As the majority of observed cells had normal karyotypes and frequent trisomy 8 has been noted in human iPS cells and ES cells after prolonged passages,31 we conclude that the observed chromosomal abnormality in clone T1D-3#3 is due to continuous in vitro passage.
Table 1. Information of nondiabetic and diabetic individuals.
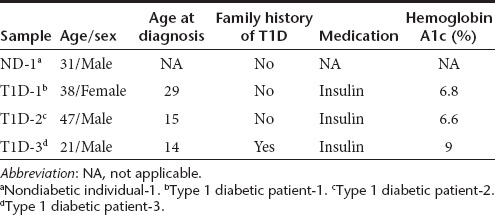
Figure 1.

Generation of iPS cells from patients with type 1 diabetes (T1D). (a) Skin cells from nondiabetic (ND) and type 1 diabetic (T1D) donors were reprogrammed with lentiviral vectors expressing pluripotency-associated factors, OCT4, SOX2, KLF4, and c-MYC. Patient-specific iPS clones cultured under serum- and feeder-free conditions were examined for expression of a series of human pluripotent stem cell markers, including alkaline phosphatase (AP), SSEA1, SSEA4, TRA-1-60, TRA-1-81, OCT4, SOX2, KLF4, and NANOG. (b) RT-PCR analysis was performed to examine expression of key pluripotency-associated genes using total cellular RNA from ND- and T1D-iPS clones. Three clones from ND (ND-1#1, #2, and #3) and three clones each from T1D patients (T1D-1, T1D-2, and T1D-3) were characterized. No cDNA template was used as control (water). (c) Efficient silencing of lentiviral transcripts in induced pluripotent cells. Transgene-specific quantitative real-time PCR demonstrates that the expression of viral transcripts (c-MYC, KLF4, and SOX2) was downregulated in iPS clones. Shown are (Fib) uninfected fibroblasts, (Fib/4f) fibroblasts 3 days after infection with all four viruses, ND iPS clones (ND-1#1, #2, and #3), and T1D-specific iPS clones (T1D1#1, #2, #3; T1D2#1, #2, #3; and T1D3#1, #2, #3). (d) G-banding chromosome analysis of T1D-iPS clone T1D1#2. (e) iPS clones were spontaneously differentiated through embryoid body formation. Pluripotency of derived iPS clones was verified by generation of cells of ectoderm (β-III tubulin, green), endoderm (FOXA2, red), and mesoderm (CD31, green) upon spontaneous differentiation. Nuclei were counterstained by DAPI (blue). Bars = 20 µmol/l. DAPI, 4′,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; iPS, induced pluripotent stem cell; RT-PCR, reverse transcription-PCR.
Derived patient-specific iPS clones were further assayed for the ability to spontaneously differentiate into cells of three germinal layers in vitro. When iPS cells were cultured in suspension, all T1D- and ND-iPS clones formed embryoid bodies (EBs). Immunostaining of EB-derived adherent cells revealed generation of cells prototypic of the ectoderm (β-III tubulin), endoderm (FOXA2) or mesoderm (CD31), (Figure 1e, Supplementary Figure S4a). Moreover, upon subcutaneous injection into immunodeficient mice, T1D- and ND-iPS clones formed tumors within 1 month. Tumors, reaching 1–2 cm in diameter, were harvested for histological analysis. Hematoxylin and eosin staining of tumor sections demonstrated heterogeneous tissues representing ectodermal, endodermal, and mesodermal lineages (Supplementary Figure S4b), verifying the pluripotent differentiation propensity in vivo. These results demonstrate that T1D- and ND-iPS clones are pluripotent, and are able to form cells of three germ layers both in vitro and in vivo.
Global gene expression profiles of derived iPS clones
To further assess the similarity of derived iPS clones, global gene expression profiles of ND- and T1D-iPS clones were determined and compared against primary human keratinocytes32 and H9 human ES cells (GSM551202; GEO DataSets, Boston, MA). Scatter plot analysis demonstrated that the transcriptome of ND- and T1D-iPS clones showed higher similarity to those of H9 human ES cells than primary keratinocytes (Figure 2a,b). Heat map analysis of differentially expressed genes further confirmed that gene expression patterns of derived iPS clones were similar to those of human ES cells, but highly divergent from basal human keratinocytes (Figure 2c). The transcriptome of derived iPS clones were strikingly similar to each other. Two iPS clones, T1D-1#1 and T1D-1#3, showed some variations in gene expression patterns (Figure 2c). We then performed principal component analysis (PCA) of the array data with a 37,788 probe set. Except for the two iPS clones T1D-1#1 and T1D-1#3, 10 iPS clones from four subjects grouped tightly together, illustrating the strong intrapatient and interpatient homogeneity among patient-derived iPS clones. The data set from a human ES cell line clusters closely with the iPS cluster, while primary human keratinocytes form a distinct cluster (Figure 2d). From this we conclude that global gene expression profiles support a high degree of similarity in transcriptome between ND-, T1D-iPS cells, and human ES cells. The diversity noted between human ES cells and our 10 iPS clones (excluding T1D-1#1 and T1D-1#3) could be due to differences in culture conditions. The ES cells used to derive comparative microarray data were cultured with feeder cells, while our experimental clones were derived and maintained under feeder-free conditions.
Figure 2.

Microarray-based gene expression profiles of nondiabetic (ND)- and type 1 diabetic (T1D)-induced pluripotent stem (iPS) cells. (a) Global gene expression patterns were compared between primary human keratinocytes (HKs) and T1D iPS clone (T1D-1#2), or between (b) human embryonic stem (ES) cells (H9) and ND iPS clone (ND-1#2) or T1D-iPS clones (T1D-1#2, T1D-2#2, and T1D-3#2). (c) Heat map analysis of HKs (ND1, ND2, and T2D), iPS cell clones (ND iPS clones, ND-1#1, #2, #3; T1D iPS clones, T1D-1#1, #2, #3; T1D-2#1, #2, #3; and T1D-3#1, #2, #3), and human ES cells (HESC). Expression of genes that are differentially expressed between HK and iPS clones. (Top) Hundred human iPS cell—enriched probe sets; (bottom) top hundred keratinocytes enriched probe sets. The color key (below) indicates the color code gene expression in log2 scale. (d) Principal component analysis (PCA) of the ND- and T1D-iPS clone's microarray data. The PCA plot illustrates the principal components of three ND-, nine T1D-iPS clones, three keratinocytes, and HESC. iPS clones derived from different individuals were grouped together, and keratinocytes were clearly separated from iPS clones.
Guided differentiation of T1D-specific iPS cells into pancreatic progenitors
Next, we employed an established step-wise differentiation protocol28 to test the proficiency of derived iPS cells in generating pancreatic progenitor cells in vitro. Since DE formation is a prerequisite for pancreatic lineage development, we first evaluated the efficiency of DE induction from T1D- and ND-iPS cells using SOX17 and FOXA2 expression as DE markers. Treatment with Activin A and Wnt3a facilitated induction of DE cells with strong SOX17 and FOXA2 expression (Figure 3b, Supplementary Figure S5). Fluorescence-activated cell sorting analysis revealed that 71–95% of iPS progeny were positive for SOX17, similarly 80–99% of cells were positive for CXCR4 (Figure 3c, Supplementary Figure S6), in line with a robust DE induction from derived iPS clones through Activin A and Wnt3a treatment. Pluripotency marker OCT4 expression level was decreased from 99 to 5% after DE induction (Figure 3c, Supplementary Figure S6).
Figure 3.

Differentiation of nondiabetic (ND)- and type 1 diabetic (T1D)-induced pluripotent stem (iPS) clones into definitive endoderm and foregut endoderm cells. (a) Schematic diagram of the differentiation process. iPS clones were differentiated into insulin-producing endocrine cells through definitive endoderm, primitive gut tube, posterior foregut, and pancreatic progenitor stages. (b) iPS cells were treated with activin A and Wnt 3a for 3 days. iPS-derived cells were immunostained with antibodies against SOX17 and FOXA2 to verify induction of definitive endoderm cells. SOX17 (green) expressing cells coexpressed FOXA2 (red). Nuclei were counterstained with DAPI (blue). (c) Flow cytometric analyses were performed to determine SOX17, CXCR4, and OCT4-positive cell populations. iPS-derived definitive endoderm cells were dissociated and stained with anti-SOX17, anti-CXCR4, and anti-OCT4 antibodies. Cells stained with an IgG isotype control were used as negative control. Over 80% of iPS progeny expressed definitive endoderm marker SOX17, 80–90% of cells were CXCR4-positive and 5–20% of cells expressed OCT4. More than 98% of undifferentiated iPS clones were OCT4 positive. (d) iPS progeny on differentiation at day 5 were stained with primitive gut tube markers HNF4A and HNF1B. Induction of primitive gut tube markers was verified by high levels of HNF4A (green) and HNF1B (green) expression. Of note, HNF4A-positive cells were also positive for SOX17 (red) and HNF1B. Nuclei were counterstained with DAPI (blue). (e) iPS progeny differentiated for 9 days were stained for foregut endoderm markers. Detection of pancreatic transcription factors HNF1B (green), HNF6 (red), and PDX1 (red) verified the induction of foregut endoderm cells. Nuclei were counterstained with DAPI (blue). Bars = 20 µmol/l. One clone from ND (ND-1#3) and three clones from T1D patients (T1D-1#3, T1D-2#3, and T1D-3#2) were analyzed. CYC, KAAD-cyclopamine; FGF, fibroblast growth factor; HGF, hepatocyte growth factor; IGF, insulin-like growth factor; ILV, indolactam V; RA, all-trans retinoic acid.
DE cells were then differentiated into gut tube endoderm cells through activation with FGF10 and CYC. iPS progeny upon FGF10 and CYC treatment expressed high levels of primitive gut tube markers HNF4A (hepatocyte nuclear factor 4α) and HNF1B (hepatocyte nuclear factor 1 homeobox B) (Figure 3d, Supplementary Figure S7). Of note, signals for HNF4A and HNF1B colocalized with SOX17, confirming that primitive gut tube cells were derived from SOX17-positive DE cells (Figure 3d, Supplementary Figure S7). Colocalization of HNF1B and HNF4A signals was also observed (Figure 3d). Upon further differentiation with FGF10, CYC, and retinoic acid, iPS-derived gut tube cells began to express high levels of posterior foregut markers, PDX1 (pancreatic and duodenal homeobox 1) and HNF6 (hepatocyte nuclear factor 6) (Figure 3e, Supplementary Figure S8). PDX1- and HNF6-expressing cells were also positive for HNF1B, suggesting derivation of posterior foregut from primitive gut tube cells. A subset of iPS progeny were negative for PDX1 or HNF6, indicating the generation of heterogeneous cell populations (Figure 3e, Supplementary Figure S8). Posterior foregut cells upon guided differentiation in the presence of FGF10, indolactam V, and GLP-1 generated PDX1-positive pancreatic progenitors (Supplementary Figure S9a). PDX1-positive cells were found in all iPS clones tested, indicating a reproducible pancreatic differentiation potential of T1D-specific iPS clones.
Generation of pancreatic hormone-producing islet-like cells from T1D-specific iPS cells
Further guided differentiation generated pancreatic islet-like cells through a pancreatic endoderm stage. iPS-derived islet-like cells, on immunocytochemistry, expressed INS. In islet-like clusters, INS signals colocalized with those of C-peptide, confirming de novo INS synthesis, rather than INS uptake from the medium (Figure 4a). Glucagon (GCG)- and somatostatin (SST)-positive cells were also found predominantly in islet-like clusters (Figures 4b and 3c). Islet-like clusters appeared to be a mixture of immature and mature β-cells; a subset of INS-producing cells coexpressed GCG or SST, characteristics of immature islet cells, whereas others coexpressed the mature β-cell marker, PDX1 (Figure 4d). Quantification, by flow cytometry, of INS-positive cell populations within differentiated patient-specific iPS-derived islet-like clusters demonstrated that 2–3% of the cell population was INS-positive (Figure 4e). Rat insulinoma cell line INS-1 was used as a positive control for staining (Figure 4e) and undifferentiated iPS clones as negative control (Supplementary Figure S10b). OCT4 expression was downregulated after differentiation to 0.04–2.0% (Supplementary Figure S10c). When T1D patient-specific iPS progeny was tested for the capability of glucose-responsive C-peptide secretion, T1D- and ND-iPS–derived cells secreted more C-peptide in response to high glucose stimulation (Figure 4f). These data demonstrate the proficiency of T1D-specific iPS cells to yield INS-producing cell types. Importantly, however, a remarkable intrapatient clonal variation was evident in the generation of INS-positive cells. Most iPS clones could be differentiated into SST- and GCG-producing cells (Supplementary Figure S9b), but failed to generate INS-positive cells. Indeed, among 12 iPS clones tested, only four clones (ND-1#3, T1D-1#3, T2D-2#3, and T1D-3#2) were reproducibly guided into INS-producing cells.
Figure 4.

Successful differentiation of type 1 diabetes (T1D)-specific induced pluripotent stem (iPS) cells into pancreatic hormones-expressing cells. (a) Expression of pancreatic hormones insulin, C-peptide, glucagon (GCG), somatostatin (SST), and PDX1 were detected in differentiated cells at day 26. Insulin (red) expression colocalized with C-peptide (green) expression. (b,c) Some of the insulin-positive cells were also positive for GCG (green) or SST (green). PDX1 (green) expression was also detected in insulin-positive cells. Nuclei were counterstained with DAPI (blue). Bars = 20 µmol/l. (d) Flow cytometry analysis demonstrated ~2% of iPS progeny positive for insulin. Staining with the secondary antibody alone was used as a control. Rat insulinoma cell line INS-1 was used as positive control for insulin staining. (e) Generation of glucose-responsive, insulin-producing cells was confirmed by human C-peptide secretion after high glucose stimulation. (f) The amounts of C-peptide secretion upon high glucose stimulation was measured by C-peptide ELISA assay. Error bars indicate SD. Statistical significance tested by Student's t-test. DAPI, 4′,6-diamidino-2-phenylindole; ELISA, enzyme-linked immunosorbent assay; ND, nondiabetic.
Differential regulation of pancreatic lineage-specifying and pluripotency-associated factors in individual iPS clones
To understand the molecular mechanisms of the observed intrapatient variations in differentiation propensities of iPS cells, we analyzed the expression of stage-specific transcription factors at each differentiation step. In line with immunostaining, FOXA2 and SOX17 transcripts were detected by quantitative PCR in all iPS clones treated with Activin A and Wnt3a (Figure 5a, Supplementary Figure S10a), although expression levels of FOXA2 were relatively low (Supplementary Figure S10a). Similarly, at day 9 of differentiation, transcripts of primitive gut tube marker HNF4A were detected in all clones, but the expression levels varied depending on clones (Figure 5a). Clonal variations were more prominent at day 18. T1D-2#2, T1D-2#3, and T1D-3#3 clones did not express some of the key pancreatic endoderm markers including HNF6 and NKX6.1 in this timepoint, while ND-1–derived clones demonstrated higher levels of NKX6.1 induction (Figure 5b). HB9 expression was detected in all the clones differentiated. Most striking differences were observed in iPS progeny in the final stage of differentiation. Reverse transcription-PCR analysis demonstrated variable pancreatic gene expression patterns in individual clones at day 26. In accordance with immunostaining, four clones (ND-1#3, T1D-1#3, T1D-2#3, and T1D-3#2) expressed INS gene as well as other pancreatic genes tested. Three clones (ND-1#1, T1D-2#1, and T1D-3#3) expressed all other genes, except INS. Three clones (T1D-1#2, T1D-2#2, and T1D-3#1) did not express two genes including INS and NKX6.1 or GLUT-2, while two clones (ND-1#2 and T1D-1#1) failed to express three genes (PDX1/GCG/INS and PDX1/NKX6.1/INS). Thus, lack of INS gene induction was frequently associated with absence of NKX6.1 and PDX1 transcripts in fully differentiated cells. Next, we assessed the influence of guided differentiation on the levels of pluripotency-associated factors, OCT4, SOX2, KLF4, NANOG, and c-MYC, in T1D-specific iPS clones. Most clones demonstrated marked downregulation of OCT4 transcripts upon differentiation, while four clones, ND-1#1, ND-1#2, T1D-1#2, and T1D-1#3, showed sustained OCT4 expression even after 26 days of differentiation (Figure 6). NANOG expression levels were downregulated after step-wise differentiation (Figure 6), while SOX2 expression was downregulated in all the clones except ND-1#1 (Figure 6). In contrast to those pluripotent genes, most iPS clones showed sustained expression of c-MYC upon differentiation (Figure 6). Among them, ND-1#2 and T1D-1#1 showed over twofold increase in c-MYC transcripts. Similarly, KLF4 expression generally persisted after differentiation and three clones (ND-1#1, ND-1#2, and T1D-3#2) showed upregulation of KLF4 upon guided differentiation (Figure 6).
Figure 5.

Differential regulation of stage-specific pancreas developmental genes in differentiated type 1 diabetes (T1D)-specific induced pluripotent stem (iPS) cells. RNA was isolated from differentiated iPS progeny at each differentiation stage and quantitative PCR analysis was performed to detect the induction of key stage-specific pancreatic factors. (a) Specific primers were used to detect expression of definitive endoderm marker (SOX17), posterior foregut markers (HNF4A), (b) pancreatic progenitors (HB9 and NKX6.1), and (c) pancreatic endocrine cells (PDX1, INS, NKX6.1, GLUT-2, SST, and GCG). The expression levels were normalized to those of GAPDH. Green bars represent before differentiation, red bars after differentiation. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GCG, glucagon; ND, nondiabetic; SST, somatostatin.
Figure 6.

Expression of pluripotency-associated genes after step-wise differentiation to insulin-producing cells. The levels of OCT4, SOX2, c-MYC, KLF4, and NANOG transcripts in type 1 diabetes (T1D)-specific induced pluripotent stem (iPS) cells before (green bars) and after (red bars) differentiation was examined by real-time PCR analysis. The expression levels were normalized to those of GAPDH. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ND, nondiabetic.
Discussion
Present study provides a systematic blueprint for generation and characterization of multiple iPS clones from individuals with or without T1D, and determines patient- and clone-specific differentiation propensity and proficiency to yield functional INS-producing progeny. Derivation of iPS cells from T1D patients was reproducible and T1D-specific iPS clones maintained pluripotency under feeder-free conditions. All iPS clones tested demonstrated the capacity to differentiate into SOX17- and FOXA2-positive DE cells. Although clonal variations became increasingly prominent upon further guided differentiation of iPS progeny into primitive gut tube- and islet-like cells, one iPS clone from each patient were capable of differentiating into INS-producing islet-like cells. Derivation and differentiation of T1D-specific iPS cells into disease-relevant cell types paves the way for novel individualized medicine applications for T1D.
Disease-specific iPS cells allow, in principle, generation of large numbers of disease-relevant, genetically matched cells which could provide a unique platform for diagnostic and therapeutic applications. Successful derivation of disease-specific iPS cells and their differentiation into disease-relevant cell types have been reported from patients with various human diseases.9,10,11,12,13,30,33 Importantly, several studies demonstrated that patient-specific iPS cells recapitulate disease phenotypes,14,15,16 verifying the potential applications of patient-specific iPS cells for disease modeling. T1D-specific iPS cells have been reported from three patients to date,11,30 and three iPS clones from two patients have been successfully differentiated into INS-producing cells.30 However, as only few disease-specific iPS clones and an ES cell line, rather than iPS clones from a ND individual, was used as a control,30 the reproducibility of the differentiation proficiencies among T1D-specific iPS clones remain to be determined. The present study demonstrates the feasibility and reproducibility of multiple iPS clones derivation from T1D patients. Selected T1D-specific iPS clones were proficient in yielding glucose-responsive, INS-producing cells upon guided differentiation, in turn providing a platform whereby to model disease and eventually lead to novel cell replacement therapy. In modeling T1D, a complex genetic trait involving immunological reaction, both iPS-derived β-like cells and the reconstruction of the autologous immune response would be required to recapitulate patient-specific T1D progression. Nevertheless, an iPS-based system would enable detailed analysis of patient-specific immune-mediated destruction of β cells at a cellular level.
Despite the promise of iPS technologies, recent studies with standard mouse and human iPS lines have demonstrated potential barriers to diagnostic and therapeutic applications of patient-specific iPS cells. For instance, the reprogramming process and subsequent expansion of iPS cells can induce genetic and epigenetic abnormalities,34,35,36,37 whereas atypical gene expression patterns in iPS progeny can induce T-cell–dependent immune response in syngeneic recipients.38 In addition, substantial differences in spontaneous differentiation propensities have been demonstrated among human ES cell lines and standard iPS lines.39,40 Variations in differentiation propensities for INS-producing cells have also been noted among human iPS lines.27,28,29 However, these studies focused on few standard iPS lines and, therefore, the variations among patient-specific iPS clones, especially for their pancreatic differentiation propensities, remain to be determined. Here, immunostaining and quantitative PCR analysis of differentiating iPS cells at various timepoints revealed differential induction of stage-specific pancreatic genes among iPS clones. Almost all clones were able to induce key pancreatic differentiation factors, such as PDX1 and NKX6.1, at some stage, but strong variations were evident in the degree of expression and the timing of induction. For instance, NKX6.1 transcript was present on differentiation at day 26, but not on day 18, in iPS clone T1D-2#3, while other clones showed induction of NKX6.1 on day 18. Although PDX1 protein expression was detected on day 9 in all iPS clones by immunostaining, PDX1 expression was not detected in iPS clones ND-1#2 and T1D-1#1 on day 26. Notable correlation was observed between the lack of PDX1 and/or NXK6.1 induction on day 26 and the failure to induce INS-producing cells. In addition, notable variations in regulating pluripotency gene expression were observed before and after step-wise differentiation. Successfully differentiated clones also downregulated pluripotency genes, except one clone, T1D-3#2 where KLF4 expression was upregulated. Thus, differential regulation of pancreatic lineage-specifying genes and pluripotency-associated genes underlined the clonal variability of T1D-specific iPS clones.
In contrast to the clear intrapatient variability, interpatient differences were not prominent. This is in part due to the strong intrapatient variations which could mask variations between T1D patients or between ND and T1D subjects. Nevertheless, common trends in iPS clones were consistent from the same donors. Six iPS clones from T1D-2 and T1D-3 showed consistent and high levels of induction of most islet markers; ISL1, NEUROD1, MAFA, PDX1, GLUT-2, SST, and GCG. Further analysis using additional iPS clones from individual donors may reveal interpatient or inter-disease variations.
Individualized iPS applications require the selection of representative iPS clones that reproducibly and reliably differentiate into disease-relevant cell types from each patient. The observed intrapatient variations will impose an additional translational challenge, especially for diagnostic or disease-modeling purposes. In this context, reducing clonal variations would accelerate iPS applications in practice for T1D research. Studies have demonstrated that continuous passaging of the iPS cells diminishes the differences between iPS cells and ES cells, implying that iPS cells lose the epigenetic memory inherited from parental cells upon prolonged passaging.41 This notion suggests increased bioequivalence of individual iPS clones at later passage, and calls for quality assessments after long passage. On the other hand, we also need to pay attention to avoid prolonged in vitro passage, which could lead to accumulation of chromosomal abnormalities. Here, we used relatively mature iPS cells at passage 12–34, with the majority exhibiting normal karyotypes. Importantly, PCA of global gene expression profiles of derived iPS clones demonstrated the homogeneity of 10 out of 12 iPS clones and their similarity to human ES cell data. This observation indicates that our iPS clones have lost their epigenetic memories of parental cells by passage 12–34. Other markers for proper reprogramming, such as silencing of transgene expression42 and in vitro spontaneous differentiation through EB formation, further confirmed consistent reprogramming of patient-derived cells. Nevertheless, these commonly used quality assessments were insufficient to achieve consistent pancreatic differentiation of each iPS clone. Of note, successful induction of DE through spontaneous differentiation of human PS cells has been used as one of the key criteria to evaluate the pluripotency of iPS cells.40 Our data demonstrate that successful DE formation does not guarantee proficiency for the ensuing differentiation process, highlighting the complexity of the problem of clonal variations.
Two of the three iPS clones from patient T1D-1 (T1D-1#1 and T1D-1#3) showed sustained exogenous KLF4 and SOX2 expression (Figure 1c) and did not cluster with other iPS clones in the PCA (Figure 2d). These data suggest that the two clones were not fully reprogrammed. Stringent quality control would eliminate such partially reprogrammed clones as inappropriate for diagnostic and therapeutic applications. Intriguingly, clone T1D-1#3 was the only clone among the three T1D-1–derived clones, able to differentiate into INS-producing cells. This discrepancy not only underscores the challenge in selecting patient-specific iPS clones for clinical applications, but also offers an attractive potential of generating INS-producing cells from partially reprogrammed cells. Previous studies have demonstrated that human iPS cells retain a residual epigenetic memory of tissue of origin and tend to differentiate preferentially into the lineage from which they were originally derived.43,44 Here, iPS clones were isolated from ectodermal lineage, skin-derived keratinocytes. It is plausible that deriving iPS cells from endodermal origin cells, such as heptaocytes, may improve the iPS differentiation propensity into INS-producing cells.
As clonal variability arises from differential regulation of stage-specific pancreatic factors and pluripotency-associated genes, the variations could be minimized by increasing lineage specification and omission of the possible reactivation of integrated, pluripotent transgenes. Significant improvement in β-cell differentiation protocols, which currently achieves only few percent of iPS progeny-producing INS27,28,29,30 would result in a more robust, consistent, and orchestrated induction of pancreatic stage-specifying factors. Efficient pancreatic differentiation would also lead to consistent silencing of pluripotency genes. In addition, reprogramming through non-integrating vector system45,46,47,48 would avoid/minimize sustained expression or reactivation of reprogramming factors, including oncogenic c-MYC, in differentiating iPS cells. We have recently established transgene-free iPS clones from T1D and T2D patients using Sendai viral vectors.32 Investigation of their pancreatic differentiation potential will elucidate the role of reprogramming vector integration in clonal variations of iPS cells.
To secure homogenous pools amenable for diagnostic and therapeutic applications of iPS cells for T1D, it would be necessary to derive multiple patient-specific iPS clones and carry out comprehensive fingerprinting of individual clones for their pluripotency and differentiation propensity. As initial screening for the integrity of derived iPS clones, the pluripotency and spontaneous differentiation propensities can be assessed by the high-throughput characterization system, recently established by Bock et al.40 We propose to include additional assessments, induction of key stage-specific markers, such as SOX17, PDX1, and NKX6.1, as well as suppression of pluripotency factors across lineage-specified iPS progeny, in the bioequivalence criteria. This would allow selection of representative iPS clones for diagnostic and disease-modeling applications based on their “central tendency” of differentiation propensity.
In summary, the presented results demonstrate reproducible generation of T1D-specific iPS cells and the requirement of appropriate regulation of pluripotency-associated genes and stage-specific pancreatic factors for successful generation of glucose-responsive INS-producing cells. Our study also highlights the intrapatient variations of patient-specific iPS clones, and the challenges for diagnostic and therapeutic applications of iPS cells. Comprehensive fingerprinting of multiple patient-specific clones and improved differentiation protocols would enable diagnostic and therapeutic applications of iPS cells for basic and translational T1D research.
Materials and Methods
Protocols were approved by Mayo Clinic Institutional Review Board and Institutional Animal Care and Use Committee. Informed consent was obtained from all patients in accordance with the Declaration of Helsinki.
Derivation of skin cells from healthy and T1D donors. Three T1D patients and one healthy volunteer were recruited for the study. Patient informations are shown in Table 1. Skin cells were isolated from 8 mm dermal biopsies as reported previously.49 Resulting fibroblasts and keratinocytes are herein referred as T1D patients #1 (T1D-1), #2 (T1D-2), #3 (T1D-3) and subject without diabetes (ND; ND-1).
Reprogramming of patient-specific fibroblasts and keratinocytes. Reprogramming of patient-derived somatic cells was performed as described.28,50 Briefly, skin cells were transduced with lentiviral vectors, pSIN-OCT4, pSIN-SOX2, pSIN-KLF4, and pSIN-cMYC50 at multiplicity of infection of 5 each. Three days after infection, cells were replated on Matrigel (#354277; BD Biosciences, San Jose, CA)-coated plates and fed with the serum-free human iPS media28 consisting of HEScGro medium (Millipore, Billerica, MA) supplemented with 20% (vol/vol) mTeSR-1 maintenance media (Stemcell Technologies, Vancouver, British Columbia, Canada). One to two weeks after vector infection, reprogrammed cells began to form iPS-like colonies and at 3–6 weeks, colonies were picked based on size and morphology. T1D and ND iPS clones were cultured under feeder-free conditions on Matrigel, and expanded using cell dissociation buffer (cat. no. 13151014; Invitrogen, Carlsbad, CA). Three clones from each individual were characterized, expanded, and iPS clones at passage 12–25 were analyzed for their biological properties, including the differentiation propensity for INS-producing cells.
Differentiation of iPS cells into INS-producing cells. In vitro differentiation of patient-specific iPS cells was carried out as described.28 Pancreatic differentiation was initiated by treating iPS clones with 100 ng/ml activin A (Peprotech, Rocky Hill, NJ) and 25 ng/ml Wnt3a (R&D Systems, Minneapolis, MN) in advanced RPMI (A-RPMI; Invitrogen) for 1 day, followed by treatment with 100 ng/ml activin A in A-RPMI supplemented with 0.2% fetal bovine serum (FBS) (Invitrogen) for 2 days. Differentiated cells were then cultured in A-RPMI medium containing 50 ng/ml FGF10 (R&D Systems), 0.25 µmol/l KAAD-cyclopamine (CYC), and 2% FBS for 2 days. Next, cells were treated with 50 ng/ml FGF10, 0.25 µmol/l CYC, and 2 µmol/l all-trans retinoic acid (Sigma-Aldrich, St Louis, MO) in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) with 1× B27 supplement (Invitrogen) for 4 days. Cells were then cultured in the presence of 50 ng/ml FGF10, 300 nmol/l IndolactamV (Axxora, San Diego, CA), and 55 nmol/l GLP-1 (Sigma-Aldrich) in DMEM with 1× B27 for 4 days. Differentiation medium including 10 µmol/l DAPT (Sigma-Aldrich) and 55 nmol/l GLP-1 in DMEM with 1× B27 was used to culture cells for the next 6 days. Finally cells were cultured in 50 ng/ml hepatocyte growth factor (R&D Systems), 50 ng/ml INS-like growth factor 1 (R&D Systems) and 55 nmol/l GLP-1 in CMRL-1066 medium (Invitrogen) with 1× B27 for 6 days. All media were supplemented with antibiotics penicillin/streptomycin. All experiments were validated three or more times.
Immunostaining. Immunofluorescence analysis was performed for characterization of undifferentiated and differentiated cells at various stages of differentiation. Cells were fixed for 20 minutes at room temperature in 4% paraformaldehyde in phosphate-buffered saline (PBS), washed three times in PBS and blocked for 30 minutes in PBST (PBS with 0.1% Tween-20 and 5% FBS). Cells were stained with primary antibodies overnight at 4 °C and with relevant secondary antibodies for 1 hour at room temperature. Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole) and analyzed by confocal laser-scanning microscopy (LSM 510; Zeiss, Thornwood, NY). The primary and secondary antibodies are listed in Supplementary Table S1. Alkaline phosphate staining was performed with an Alkaline Phosphatase Detection Kit (Millipore).
Spontaneous differentiation and teratoma formation assay. For spontaneous differentiation, T1D and ND iPS clones were dissociated using collagenase IV and cultured on low adhesion plates in basal HEScGRO medium (#SCM 021; Millipore) for EB formation. EBs were cultured as suspension for 10 days and then allowed to adhere in knockout DMEM with 20% FBS and further cultured for 10–14 days. Differentiated cells were analyzed for markers of three germ layers. Primary and secondary antibodies used are listed in Supplementary Table S1. Undifferentiated iPS cells dissociated with collagenase were injected subcutaneously into severe combined immunodeficiency Beige mice. Tumors were detected after 1 month of injection, and dissected out after 2–5 months. Tumor sections were analyzed by hematoxylin and eosin staining.
Karyotyping. Conventional cytogenetic analysis was performed on 20 metaphase cells of representative six iPS clones. Chromosomes were banded following standard methods for high-resolution G-banding. Cells were captured and karyotyped using a CytoVision Karyotyping System (Genetix, New Milton, UK).
Microarray. Total RNA was isolated from iPS clones using Trizol (Invitrogen). RNA probes for microarray hybridization were prepared and hybridized to HG-U133 Plus 2 (Affymetrix, Santa Clara, CA) oligonucleotide microarrays according to the manufacturer's protocols. Microarrays were scanned and data were analyzed using standard in-house MicroArray PreProcessing software. The microarray data for H9 human ES cells (GSM551202) was obtained from GEO DataSets. The fold changes were calculated for all genes in keratinocytes over the corresponding average in iPS clones. The top 100 probe sets most specifically expressed in the keratinocytes and human ES cells were selected for the heatmap generation. Partek software (Partek, St Louis, MO) was used to generate heatmap and PCA plot.
Gene expression. Total RNA was isolated with the Trizol reagent (Invitrogen) was used for semiquantitative reverse transcription-PCR and real-time PCR analysis. Reverse transcription was performed using the RNA to cDNA EcoDry TM Premix (Oligo dT) kit (cat. no. 639543; Clontech, Mountain View, CA). Platinum Taq DNA polymerase (Invitrogen) was used for PCR reactions. Primers used to determine expression of pluripotency genes (OCT4, SOX2, KLF4, NANOG, GDF3, hTERT, and c-MYC) are listed in Supplementary Table S2. Real-time PCR was performed using pancreatic genes (FOXA2, SOX17, HNF4α, HB9, HNF6, PDX1, ISL-1, NGN3, NKX 6.1, NKX2.2, GLUT-2, INS, SST, and GCG), and control GAPDH using Roche PCR probes (Roche, Indianapolis, IN). Values calculated using the comparative threshold cycle (ΔCt) method and normalized to GAPDH values. Real-time PCR primers and probes are listed in Supplementary Table S3. Quantitative real-time PCR analysis was performed for lentiviral transgenes using IDT probes, Supplementary Table S4.
Measuring glucose-stimulated C-peptide secretion from iPS-derived pancreatic hormone-expressing cells. T1D- and ND-iPS–derived islet-like cells were tested for C-peptide secretion upon glucose stimulation. Krebs-Ringer solution with bicarbonate and HEPES (KRBH; 129 mmol/l NaCl, 4.8 mmol/l KCl, 2.5 mmol/l CaCl2, 1.2 mmol/l KH2PO4, 1.2 mmol/l MgSO4, 5 mmol/l NaHCO3, 10 mmol/l HEPES, 0.1% (wt/vol) BSA) was used for the assay. Cell were initially incubated in KRBH buffer containing 3 mmol/l D-glucose for 1 hour at 37 °C, followed by glucose stimulation conditions containing 27 mmol/l D-glucose for 1 hour at 37 °C. C-peptide levels were determined using an ultrasensitive C-peptide/proinsulin ELISA kit (Alpco Diagnostics, Salem, NH).
Flow cytometry. T1D- and ND-iPS–derived cells were dissociated into single cells using TrypLE (#12605; Invitrogen) at 37 °C. Intracellular antibody staining was performed using BD Cytofix/Cytoperm and BD Perm/Wash buffer (BD, San Diego, CA). The primary and secondary antibodies are listed in Supplementary Table S1. Flow cytometry data were acquired on a Becton Dickinson FACS Calibur (Becton Dickinson, Franklin Lakes, NJ) and analyzed using Flowjo software.
SUPPLEMENTARY MATERIAL Figure S1. ND and T1D iPS clones exhibit ES-like cell morphology and express alkaline phosphatase. Figure S2. T1D-iPS clones express pluripotency markers. Figure S3. ND- and T1D-iPS clones maintain normal karyotype. Figure S4. Spontaneous differentiation and teratoma formation of T1D-iPS clones. Figure S5. Efficient induction of definitive endoderm after Activin A and Wnt 3a treatment. Figure S6. Quantification of iPS-derived definitive endoderm progeny. Figure S7. Generation of primitive gut tube cells. Figure S8. Successful generation of posterior foregut cells from iPS-derived primitive gut tube cells. Figure S9. Guided differentiation of iPS clones generated pancreatic progenitors and somatostatin- and glucagon-expressing cells. Figure S10. Pancreas development–stage-specific gene expression in differentiated T1D-specific iPS cells. Table S1. Antibodies used in this study. Table S2. Primers for characterization of human iPS cells. Table S3. Primers and probes used in this study. Table S4. Primers and probes used to detect the exogenous genes.
Acknowledgments
We thank Norman Eberhardt for providing rat insulinoma cell line. We also thank Ying Li and Christopher P Kolbert (Mayo Advanced Genomics Technology Center) for excellent technical support for the transcriptome analysis. We also thank Heather C Gilmer and Patricia T Greipp for cytogenetic analysis. This work was supported by Mayo Foundation, Marriott Individualized Medicine Award, Eisenberg Stem Cell Trust, Bernard and Edith Waterman Pilot Grants, Minnesota Partnership Grant (#10.01-P002372601) (Y.I.), Marriott Specialized Workforce Development Award in Individualized Medicine (T.T.), and National Institutes of Health (R01DK085516) (Y.C.K.) and R01HL083439 (A.T.). The authors declared no conflict of interest.
Supplementary Material
ND and T1D iPS clones exhibit ES-like cell morphology and express alkaline phosphatase.
T1D-iPS clones express pluripotency markers.
ND- and T1D-iPS clones maintain normal karyotype.
Spontaneous differentiation and teratoma formation of T1D-iPS clones.
Efficient induction of definitive endoderm after Activin A and Wnt 3a treatment.
Quantification of iPS-derived definitive endoderm progeny.
Generation of primitive gut tube cells.
Successful generation of posterior foregut cells from iPS-derived primitive gut tube cells.
Guided differentiation of iPS clones generated pancreatic progenitors and somatostatin- and glucagon-expressing cells.
Pancreas development–stage-specific gene expression in differentiated T1D-specific iPS cells.
Antibodies used in this study.
Primers for characterization of human iPS cells.
Primers and probes used in this study.
Primers and probes used to detect the exogenous genes.
REFERENCES
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K.et al. (2007Induction of pluripotent stem cells from adult human fibroblasts by defined factors Cell 131861–872. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S.et al. (2007Induced pluripotent stem cell lines derived from human somatic cells Science 3181917–1920. [DOI] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA.et al. (2008Reprogramming of human somatic cells to pluripotency with defined factors Nature 451141–146. [DOI] [PubMed] [Google Scholar]
- Giorgetti A, Montserrat N, Aasen T, Gonzalez F, Rodríguez-Pizà I, Vassena R.et al. (2009Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2 Cell Stem Cell 5353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh YH, Agarwal S, Park IH, Urbach A, Huo H, Heffner GC.et al. (2009Generation of induced pluripotent stem cells from human blood Blood 1135476–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Brennand K., and, Hochedlinger K. Reprogramming of pancreatic beta cells into induced pluripotent stem cells. Curr Biol. 2008;18:890–894. doi: 10.1016/j.cub.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R.et al. (2008Generation of human induced pluripotent stem cells from dermal fibroblasts Proc Natl Acad Sci USA 1052883–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F.et al. (2008Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes Nat Biotechnol 261276–1284. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Loh YH, McLoughlin EM, Huang J, Park IH, Miller JD.et al. (2010Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients Nature 464292–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W.et al. (2008Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons Science 3211218–1221. [DOI] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A.et al. (2008Disease-specific induced pluripotent stem cells Cell 134877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raya A, Rodríguez-Pizà I, Guenechea G, Vassena R, Navarro S, Barrero MJ.et al. (2009Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells Nature 46053–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden SE, Gore A, Li Z, Fung HL, Nisler BS, Nie J.et al. (2011Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy Proc Natl Acad Sci USA 1086537–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF, Jr, Mattis VB, Lorson CL, Thomson JA.et al. (2009Induced pluripotent stem cells from a spinal muscular atrophy patient Nature 457277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Song SH, Kim KL, Yi JY, Shin GH, Kim JY.et al. (2010Functional recapitulation of smooth muscle cells via induced pluripotent stem cells from human aortic smooth muscle cells Circ Res 106120–128. [DOI] [PubMed] [Google Scholar]
- Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF.et al. (2010Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome Nature 465808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounissi-Benkalha H., and, Polychronakos C. The molecular genetics of type 1 diabetes: new genes and emerging mechanisms. Trends Mol Med. 2008;14:268–275. doi: 10.1016/j.molmed.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Yokoi N, Hayashi C, Fujiwara Y, Wang HY., and, Seino S. Genetic reconstitution of autoimmune type 1 diabetes with two major susceptibility genes in the rat. Diabetes. 2007;56:506–512. doi: 10.2337/db06-1027. [DOI] [PubMed] [Google Scholar]
- Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM.et al. (2007Derivation of pluripotent epiblast stem cells from mammalian embryos Nature 448191–195. [DOI] [PubMed] [Google Scholar]
- Bach JF., and, Mathis D. The NOD mouse. Res Immunol. 1997;148:285–286. doi: 10.1016/s0923-2494(97)87235-5. [DOI] [PubMed] [Google Scholar]
- Hanna J, Markoulaki S, Mitalipova M, Cheng AW, Cassady JP, Staerk J.et al. (2009Metastable pluripotent states in NOD-mouse-derived ESCs Cell Stem Cell 4513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG.et al. (2006Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells Nat Biotechnol 241392–1401. [DOI] [PubMed] [Google Scholar]
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S.et al. (2008Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo Nat Biotechnol 26443–452. [DOI] [PubMed] [Google Scholar]
- Jiang J, Au M, Lu K, Eshpeter A, Korbutt G, Fisk G.et al. (2007Generation of insulin-producing islet-like clusters from human embryonic stem cells Stem Cells 251940–1953. [DOI] [PubMed] [Google Scholar]
- Jiang W, Shi Y, Zhao D, Chen S, Yong J, Zhang J.et al. (2007In vitro derivation of functional insulin-producing cells from human embryonic stem cells Cell Res 17333–344. [DOI] [PubMed] [Google Scholar]
- Zhang D, Jiang W, Liu M, Sui X, Yin X, Chen S.et al. (2009Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells Cell Res 19429–438. [DOI] [PubMed] [Google Scholar]
- Tateishi K, He J, Taranova O, Liang G, D'Alessio AC., and, Zhang Y. Generation of insulin-secreting islet-like clusters from human skin fibroblasts. J Biol Chem. 2008;283:31601–31607. doi: 10.1074/jbc.M806597200. [DOI] [PubMed] [Google Scholar]
- Thatava T, Nelson TJ, Edukulla R, Sakuma T, Ohmine S, Tonne JM.et al. (2011Indolactam V/GLP-1-mediated differentiation of human iPS cells into glucose-responsive insulin-secreting progeny Gene Ther 18283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nostro MC, Sarangi F, Ogawa S, Holtzinger A, Corneo B, Li X.et al. (2011Stage-specific signaling through TGFß family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells Development 138861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R.et al. (2009Generation of pluripotent stem cells from patients with type 1 diabetes Proc Natl Acad Sci USA 10615768–15773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taapken SM, Nisler BS, Newton MA, Sampsell-Barron TL, Leonhard KA, McIntire EM.et al. (2011Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells Nat Biotechnol 29313–314. [DOI] [PubMed] [Google Scholar]
- Kudva YC, Ohmine S, Greder LV, Dutton JR, Armstrong A, De Lamo JG.et al. (2012Transgene-free disease-specific induced pluripotent stem cells from patients with type 1 and type 2 diabetes Stem Cells Trans Med 1451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R.et al. (2011Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations Cell 146318–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Närvä E.et al. (2011Copy number variation and selection during reprogramming to pluripotency Nature 47158–62. [DOI] [PubMed] [Google Scholar]
- Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J.et al. (2011Somatic coding mutations in human induced pluripotent stem cells Nature 47163–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G.et al. (2011Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells Nature 47168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT.et al. (2010Identification and classification of chromosomal aberrations in human induced pluripotent stem cells Cell Stem Cell 7521–531. [DOI] [PubMed] [Google Scholar]
- Zhao T, Zhang ZN, Rong Z., and, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–215. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz-Gerald CS, Sato Y.et al. (2008Marked differences in differentiation propensity among human embryonic stem cell lines Nat Biotechnol 26313–315. [DOI] [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD.et al. (2011Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines Cell 144439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY.et al. (2010Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells Nat Biotechnol 28848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta A., and, Ellis J. Retroviral vector silencing during iPS cell induction: an epigenetic beacon that signals distinct pluripotent states. J Cell Biochem. 2008;105:940–948. doi: 10.1002/jcb.21912. [DOI] [PubMed] [Google Scholar]
- Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P.et al. (2011Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells Nat Biotechnol 291117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry AB., and, Daley GQ. Reprogramming cellular identity for regenerative medicine. Cell. 2012;148:1110–1122. doi: 10.1016/j.cell.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II.et al. (2009Human induced pluripotent stem cells free of vector and transgene sequences Science 324797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VandenDriessche T, Ivics Z, Izsvák Z., and, Chuah MK. Emerging potential of transposons for gene therapy and generation of induced pluripotent stem cells. Blood. 2009;114:1461–1468. doi: 10.1182/blood-2009-04-210427. [DOI] [PubMed] [Google Scholar]
- Seki T, Yuasa S, Oda M, Egashira T, Yae K, Kusumoto D.et al. (2010Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells Cell Stem Cell 711–14. [DOI] [PubMed] [Google Scholar]
- Anokye-Danso F, Trivedi CM, Juhr D, Gupta M, Cui Z, Tian Y.et al. (2011Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency Cell Stem Cell 8376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatava T, Armstrong AS, De Lamo JG, Edukulla R, Khan YK, Sakuma T.et al. (2011Successful disease-specific induced pluripotent stem cell generation from patients with kidney transplantation Stem Cell Res Ther 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson TJ, Martinez-Fernandez A, Yamada S, Mael AA, Terzic A., and, Ikeda Y. Induced pluripotent reprogramming from promiscuous human stemness related factors. Clin Transl Sci. 2009;2:118–126. doi: 10.1111/j.1752-8062.2009.00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ND and T1D iPS clones exhibit ES-like cell morphology and express alkaline phosphatase.
T1D-iPS clones express pluripotency markers.
ND- and T1D-iPS clones maintain normal karyotype.
Spontaneous differentiation and teratoma formation of T1D-iPS clones.
Efficient induction of definitive endoderm after Activin A and Wnt 3a treatment.
Quantification of iPS-derived definitive endoderm progeny.
Generation of primitive gut tube cells.
Successful generation of posterior foregut cells from iPS-derived primitive gut tube cells.
Guided differentiation of iPS clones generated pancreatic progenitors and somatostatin- and glucagon-expressing cells.
Pancreas development–stage-specific gene expression in differentiated T1D-specific iPS cells.
Antibodies used in this study.
Primers for characterization of human iPS cells.
Primers and probes used in this study.
Primers and probes used to detect the exogenous genes.