

Abstract
The discovery that somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) raised the exciting possibility of modeling diseases with patient-specific cells. Marchetto et al. (2010) now use iPSC technology to generate, characterize, and treat an in vitro model for the autism spectrum disorder, Rett syndrome.
Rett syndrome is a severe X-linked neurodevelopmental disorder that affects 1 in 10,000-20,000 girls worldwide, making it one of the most common forms of mental retardation in females (Percy & Lane 2005). A seminal discovery by Huda Zoghbi’s lab in 1999 identified a causative link between mutations in the methyl-CpG binding protein, MeCP2, and Rett syndrome (Amir et al. 1999), thus enabling mechanistic studies and providing a target for potential treatments. Importantly, restoration of MeCP2 function in a mouse model of the disease reverses the neurological symptoms in adult mice (Guy et al. 2007), raising the possibility that this disorder may be treatable in humans. Despite this progress, it remains unclear how loss of MeCP2 function leads to neurological defects, and no effective pharmacological treatments have yet been developed. A main limitation for human studies and thus drug development has been the inaccessibility of live neurons from human patients. To circumvent this shortcoming, Marchetto and colleagues (Marchetto et al. 2010, this issue of Cell) use human induced pluripotent stem cells (iPSCs) to establish a human cellular model for Rett syndrome that is amenable to mechanistic studies and drug screens.
To generate a human model of Rett syndrome, Marchetto et al. (2010) isolate fibroblasts from four female Rett patients and five healthy control individuals and then reprogram these fibroblasts into induced pluripotent stem cell (iPSC) lines (Figure 1). These cell lines express the expected pluripotency markers and give rise to cell types of all germ layers in teratomas (solid tumors derived from pluripotent cells), thus qualifying these cell lines as bona fide pluripotent cells. Given that hallmarks of Rett syndrome include changes in neuronal density and in brain size, the authors first determine whether these phenotypes are likely due to the abnormal proliferation of neural progenitor cells derived from iPSCs. Notably, they observe no overt defects in the cell cycle of neural progenitor cells, consistent with the notion that Rett is a disease of mature neurons.
Figure 1. Using iPSCs to model Rett syndrome in vitro.
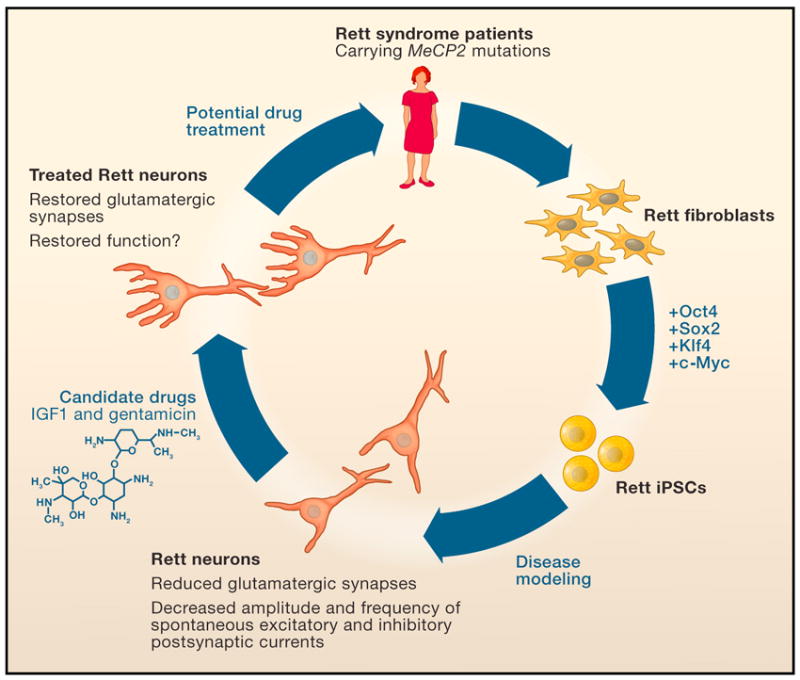
Mutations in the methyl-CpG binding protein (MeCP2) gene cause the neurodevelopmental disorder Rett syndrome. Marchetto et al. (2010) isolate fibroblasts from Rett patients with MeCP2 mutations. They then reprogram these cells into induced pluripotent stem cells (iPSCs) by the exogenous expression of the transcription factors Oct4, Sox2, Klf4 and c-Myc. These Rett-iPSCs can then differentiate into neurons in vitro, recapitulating several of the defects found in Rett syndrome patients and in animal models of the disease. These defects can be partially reversed by candidate drugs, suggesting that this disease model will facilitate large-scale drug screens and future mechanistic studies of Rett syndrome.
In contrast to neural progenitor cells, mature neurons derived from Rett iPSCs do show defects in structure and function when compared to neurons obtained from control iPSCs or embryonic stem cells (ESCs). For example, the authors detect a significant reduction in the number of synapses in glutamatergic neurons derived from Rett iPSCs when compared to neurons derived from either control iPSCs or ESCs. This reduction is likely the direct consequence of losing MeCP2 function, given that reducing the expression of MeCP2 in control ESCs produces a similar defect whereas the reintroduction of wild type MeCP2 into mutant cells rescues the phenotype.
The authors further confirm MeCP2’s role in regulating synapse formation by showing that overexpression of MeCP2 in control iPSCs leads to an increase in glutamatergic synapse numbers. These results are in accordance with observations from mouse models in which the loss or overexpression of MeCP2 leads to a respective decrease or increase of glutamatergic synapses (Chao et al. 2007). In further agreement with research performed with mouse cells (Chen et al. 2001, Guy et al., 2001) and on human autopsies, neurons derived from Rett iPSCs are smaller in size and have fewer dendritic spines when compared to control iPSC and ESC neurons.
Importantly, neurons derived from Rett iPSCs are also functionally impaired when compared to neurons derived from control iPSCs. Specifically, neurons derived from Rett iPSCs exhibit a reduction in the transient rise of intracellular calcium levels typical of active synapses. They also show a decrease in the frequency and amplitude of spontaneous excitatory and inhibitory post-synaptic currents when compared to control cells. Together, these findings provide compelling evidence that molecular and functional defects found in Rett syndrome patients can be recapitulated in iPSC-derived neurons.
Given the reversibility of the Rett phenotype in mouse, Marchetto et al. (2010) then ask whether they could rescue their in vitro phenotype using candidate drugs. First, they examine the effects of insulin-like growth factor 1 (IGF-1), which had previously been shown to partially rescue Rett symptoms in Mecp2-defficient mice (Tropea et al. 2009). Indeed, treating neurons derived from human Rett iPSCs with this growth factor increases the number of glutamatergic synapses.
Because the majority of MeCP2 mutations create premature stop codons in the gene (i.e., nonsense mutations), the authors also test the effect of the drug gentamicin, which facilitates ribosomal read-through of stop codons. Following treatment with low doses of gentamicin, Rett neurons harboring nonsense mutations in the MeCP2 gene express elevated levels of MeCP2 protein and display a striking increase of glutamatergic synapses, reaching levels similar to those seen in control neurons. Whether IGF-1 or gentamicin treatment leads to a functional recovery of neurons, however, remains unexplored in this study. In summary, these results show that previously identified drugs are effective on neurons derived from Rett iPSCs and thus validate the use of large-scale drug screens to identify new and more effective compounds that could ameliorate the symptoms of Rett.
Given the recent recognition that mouse iPSCs can exhibit molecular and functional differences compared with mouse ESCs (Stadtfeld and Hochedlinger, 2010), it will be important to ensure that any cell culture disease model is an accurate representation of its in vivo counterpart rather than an artifact of the reprogramming procedure. In that regard, data presented by Marchetto and colleagues for their Rett syndrome model agree well with what is known from mouse models of Rett syndrome and post-mortem analyses in humans. However, the observation by the authors that differentiated neurons derived from Rett iPSCs exhibit a severe skewing of X-chromosome inactivation – meaning that one copy of the X chromosome is more likely to undergo inactivation than the other - remains unexplained. This observation is of particular relevance because MeCP2 is an X-linked gene and patients’ cells in vivo are mosaic for the MeCP2 mutation due to random X-chromosome inactivation. One possible explanation for the skewing of X-inactivation in vitro is that the previously inactive X chromosome in Rett fibroblasts does not fully reactivate in iPSCs and remains inactive in iPSC-derived neurons. Indeed, a recent study on skewed X-chromosome inactivation in human iPSCs by Kathrin Plath’s lab is consistent with this interpretation (Tchieu et al. 2010). If the skewing reflects incomplete reactivation, then the studies performed by Marchetto and colleagues likely relied exclusively on iPSC clones derived from Rett-fibroblasts that had inactivated the wild-type MeCP2 allele. Thus, to harness the power of cell culture disease models, future work will need to further explore the differences between iPSCs and ESCs.
The ability to produce disease-specific differentiated cells is one of the major promises of iPSC technology because it holds the potential for disease modeling, drug development, and ultimately cell therapy. The model of Rett syndrome presented by Marchetto and colleagues is not only an additional proof of principle that human iPSCs may be useful in drug development, but also a promising opportunity to gain insights into the pathology of Rett syndrome in live human neurons.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Nat Genet. 2001;27:327–31. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chao H, Zoghbi HY, Rosenmund C. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Science. 2007;315:1143–7. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. Nat Genet. 2001;27(3):322–6. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo G, Yangling M, Chen G, Gage FH, Muotri AR. Cell. 2010 doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy AK, Lane JB. J Child Neurol. 2005;20(9):718–21. doi: 10.1177/08830738050200090301. [DOI] [PubMed] [Google Scholar]
- Tchieu J, Kuoy E, Chin MH, Trinh H, Patterson M, Sherman SP, Aimiuwu O, Lindgren A, Hakimian S, Zack JA, Clark AT, Pyle AD, Lowry WE, Plath K. Cell Stem Cell. 2010;7(3):329–42. doi: 10.1016/j.stem.2010.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Proc Natl Acad Sci U S A. 2009;106(6):2029–34. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Hochedlinger K. Genes Dev. 2010;24:2239–63. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]