

Abstract
The interaction between tumor progression and innate immune system has been well established in the last years. Indeed, several lines of clinical evidence indicate that immune cells such as tumor-associated macrophages (TAMs) interact with tumor cells, favoring growth, angiogenesis, and metastasis of a variety of cancers. In most tumors, TAMs show properties of an alternative polarization phenotype (M2) characterized by the expression of a series of chemokines, cytokines, and proteases that promote immunosuppression, tumor proliferation, and spreading of the cancer cells. Tumor suppressor genes have been traditionally linked to the regulation of cancer progression; however, a growing body of evidence indicates that these genes also play essential roles in the regulation of innate immunity pathways through molecular mechanisms that are still poorly understood. In this paper, we provide an overview of the immunobiology of TAMs as well as what is known about tumor suppressors in the context of immune responses. Recent advances regarding the role of the tumor suppressor ARF as a regulator of inflammation and macrophage polarization are also reviewed.
1. Introduction
Immune system constitutes one of the first-line defenses to prevent tumor development due to its ability to identify and destroy tumor cells. This process defined as cancer immunosurveillance was initially described by Ehrlich and subsequently revisited by Thomas and Burnet [1–3], gaining considerable attention in last years. Compelling evidence that immune system modulates cancer has emerged over the last decade from gene-targeted mice studies. Mice deficient in several immune effector cells and molecules including interferon (IFN)-γ receptor or signal transducer and activator of transcription 1 (STAT1) [4], natural killer (NK) cells, NK-T cells [5, 6], γδ T cells [7, 8], IL-12 [9], perforin [10], and granulocyte-macrophage colony stimulating factor (GM-CSF) [11] have been demonstrated to be more susceptible to tumor development. Collectively, these studies strongly support the concept that the immune response is essential in the development of tumors.
2. Macrophages: Key Immune Cells
Macrophages are one of the major widely distributed innate immune cells and present essential roles in the primary response to pathogens, maintenance of tissue homeostasis, inflammation, and immunity.
Macrophages are derived from bone marrow progenitors as immature monocytes. After circulating in the blood stream, monocytes migrate into tissues where they differentiate into resident macrophages [12].
Macrophages are dynamic cells that might modify their functional profiles in response to a variety of stimuli polarizing to functionally different phenotypes. Two distinct subsets of macrophages have been proposed, including classically activated (M1) and alternatively activated (M2) macrophages [13] (Figure 1). M1 macrophages are induced by IFN-γ either alone or cooperating with microbial stimuli such as lipopolysaccharide (LPS) or cytokines (e.g., tumor necrosis factor (TNF)-α and GM-CSF). These cells secrete high levels of classical proinflammatory cytokines such as TNF-α, interleukin (IL)-1, IL-6, IL-12, or IL-23 and increase their concentrations of nitric oxide (NO), superoxide anions, and oxygen radicals [14, 15]. Furthermore, M1 macrophages can express high levels of major histocompatibility complex (MHC) I and class II antigens and secrete complement factors that facilitate complement-mediated phagocytosis [16].
Figure 1.
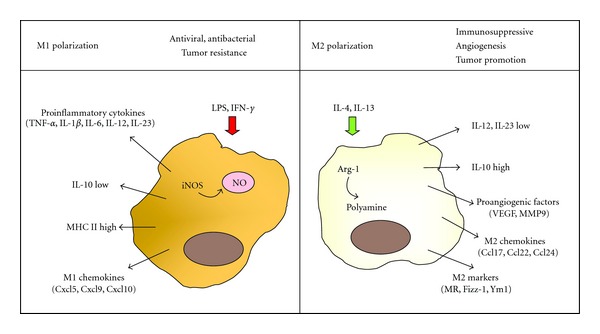
Simplified scheme for M1 and M2 activation of macrophages. M1 macrophages are induced by LPS or IFN-γ secreting high levels of classical proinflammatory cytokines such as TNF-α, IL-1, IL-6, IL-12, or IL-23, chemokines (e.g., Cxcl9, Cxcl10, and Cxcl5) and increasing their concentrations of NO. In addition, they express high levels of MHC I. IL-4/IL-13 stimulation induces M2 macrophages that downregulate IL-12 and IL-23 expression, release Ccl17, Ccl22 and Ccl24 chemokines and proangiogenic factors, and show increased expression of IL-10. Additionally, they are characterized by expression of MR, Fizz-1, and Ym1.
In contrast, IL-4/IL-13 stimulation induces M2 macrophages that reduce IL-12 and IL-23 expression while upregulate the anti-inflammatory cytokine IL-10 [17, 18]. Additionally, they are characterized by the expression of the scavenger receptors mannose receptor (MR) [13], stabilin-1 [19], CD163 [20], and some genes involved in tissue remodelling such as Found in Inflammatory Zone 1 (Fizz-1) and chitinase 3-like 3 (Ym1) [21].
An alternative metabolic pathway of L-arginine, catalyzed by arginase-1 (Arg-1) provides another feature of distinction among the two macrophage activation states. M1 macrophages upregulate iNOS to catabolize L-arginine to NO and citrulline, but M2 macrophages are characterized by high expression of Arg-1, a cytosolic enzyme which metabolizes L-arginine to ornithine and polyamines, which are precursors for collagen synthesis and cellular proliferation [22].
M1 and M2 macrophages also express a different chemokine repertoire. M1 macrophages produce proinflammatory chemokines such as (C-X-C motif) ligand 9 (Cxcl9), Cxcl10, and Cxcl5 [16], whereas M2 polarization is accompanied by production of Ccl17, Ccl22, and Ccl24 [16].
This plethora of molecules and genes leads macrophages to display distinct functions in virtue to their polarization state. Thus, classically activated macrophages are vital components in the initiation and maintenance of inflammation, as well as in host defense and priming antitumor immune response [13]. For instance, the IL-12 produced by M1 macrophages promotes the differentiation of naive T cells into Th1 cells, stimulates growth of both T and NK cells, and increases bactericidal activity of phagocytes. Moreover, IL-12 exerts an antiangiogenic activity through the increment of the chemokine inducible protein-10 (IP-10 or Cxcl10) [23]. Additionally, M1 chemokines such as Cxcl9, Cxcl10, and Cxcl5 induce the recruitment of Th1 cells, Tc1 cells, and NK cells [16].
Opposite, M2 macrophages have poor antigen-presenting capability, produce factors that suppress T-cell proliferation and activity, and mainly participate in parasite clearance, tissue remodeling, immune modulation, and tumor progression [15]. Thus, IL-10 expressed by M2 macrophages promotes the production of IL-4 and IL-13 by Th2 cells [24], inhibits the synthesis of proinflammatory cytokines such as IFN-γ, IL-2, IL-3, TNF-α and GM-CSF, and also suppresses the antigen-presentation capacity of antigen presenting cells. Furthermore, Ccl17, Ccl22, and Ccl24 production favors the attraction of immune-inhibitory cells such as regulatory T-cells (Treg) [25].
3. Macrophages and Tumor Microenvironment
Compelling evidence has emerged in recent years for macrophages playing an important function in tumor development. Although the role of macrophages in tumors is still controversial, in most human cancers such as breast, prostate, ovarian, cervical, lung carcinoma, and cutaneous melanoma, a macrophage-rich microenvironment has been correlated with a poor prognosis [26, 27]. These tumor-associated macrophages (TAMs) share many common features with the alternatively activated macrophages, showing a typical M2 marker profile with high expression of C-type lectin receptors, stabilin-1, and Arg-1 [25].
Among the cell surface molecules expressed by TAMs, several members of structurally related C-type lectin receptors such as MR and Macrophage galactose-type C-type lectin 1/2 (Mgl-1/2) are included [28]. The MR is an endocytic and phagocytic receptor that was initially described as a bridge between innate immunity and homeostasis [29] due to its ability to bind carbohydrate moieties on several pathogens such as bacteria, fungi, parasites, and viruses. Mgl-1/2 are induced on macrophages by parasitic infections or allergic asthma [28]. In the tumor microenvironment, MR and Mgl-1/2 have been documented to act as recognition molecules for glycosylated antigens on cancer cells. MR and Mgl-1/2 recognize specifically highly glycosylated molecules such as mucins present in the tumor microenvironment [30], leading to the expression of the immunoregulatory cytokine IL-10 that favors the attraction of Treg cells [25]. Indeed, Mgl-1/2 is predominantly expressed by TAMs from human ovarian carcinoma [31], on lung metastasis produced by mouse metastatic ovarian tumor cells [32], and also they have been detected after challenging tumor conditioned medium [33].
Chitinase 3-like 3 (Ym1) is a member of the mammalian chitinase family that also includes Ym2 and BRP-39 in mice, and YKL-40 in humans [34]. Increased expression of these proteins has been associated with inflammatory diseases, in particular with allergic asthma, with the induction of alternative activation of macrophages, and with progression of cancer [35–39].
Fizz-1, also known as resistin-like molecule α (RELM-α), plays a key role in the regulation of cell growth/proliferation and differentiation. Initially described in lung allergic inflammation [40], it is highly expressed in bleomycin-induced lung fibrosis and after IL-4 or IL-13 activation [41, 42]. Fizz-1 exhibits multiple functions including cell proliferation, angiogenesis, and inflammation [43], and its expression is an indicator of M2 polarization [21, 39].
Stabilin-1, a type-1 transmembrane receptor that mediates clearance of “unwanted self” components, is another marker for M2 macrophages that has been found to be expressed by TAM in several murine tumor models (e.g., B16 melanoma, pancreatic insulinoma, breast carcinoma) [19, 44]. Stabilin-1 mediates internalization of extracellular secreted protein acidic and rich in cysteine (SPARC), regulating its concentration and thereby promoting extracellular matrix remodeling, angiogenesis, and tumor progression [45].
4. Role of TAMs in Tumors
TAMs derive from circulating monocytic precursors previously recruited to the tumor region in response to the chemokines and cytokines secreted by cancer cells. In the tumor mass, TAMs exert immunosuppressive functions through the release of anti-inflammatory cytokines, modulate the tumor microenvironment producing survival/growth factors (e.g., vascular endothelial growth factor, VEGF), and facilitate the progression of tumors via proangiogenic factors release [25, 26].
4.1. TAMs and Immunosuppression
A relevant function of TAMs is to diminish the effective antitumor immune response. Several cytokines and proteases derived from TAMs, such as transforming growth factor (TGF)-β, IL-10 and Arg-1, make a significant contribution to the immunosuppressive condition [46–48].
TGF-β inhibits the antitumor response through different mechanisms including (i) inhibition of the cytolytic activity of NK cells [48, 49], (ii) differentiation of CD4+ T cells into Th2 cells [50], (iii) inhibition of the CD8+ T cells antitumoral activity [48], and (iv) maintenance of Treg cell differentiation [48].
IL-10 promotes the immune evasion impeding the production of IL-12, a cytokine known to stimulate both the proliferation and cytotoxicity of T and NK cells [51], as well as the release of the cytokine IFN-γ, which is the main factor that stimulates naive T-cell differentiation [52]. Additionally, it has been reported that IL-10 decreases the ability of epidermal antigen presenting cells (APCs) to present tumor-associated antigens, therefore interfering the induction of antitumor immune responses [51].
High Arg-1 activity has been described in TAMs from 3LL murine lung carcinoma [22], human papillomavirus E6/E7-expressing murine tumors [53], and CD11b+/CD14− myeloid cells from renal carcinoma patients [54]. Elevated Arg-1 expression might promote tumor growth via several mechanisms including downregulation of NO-mediated tumor cytotoxicity [55], increasing cellular proliferation through its participation in polyamine and proline synthesis, dysregulating the T cell receptor (TCR) signaling and subsequently inducing CD8+ T cell unresponsiveness [47] and enhancing the capacity of myeloid suppressor cells to inhibit T cell proliferation [22].
Finally, TAMs release chemokines that play fundamental roles in immunosuppression. Ccl13, Ccl18 (human only), Ccl22, and to a lesser extent Ccl17 are important chemoattractant for immune-inhibitory cells, such as Treg, which might inhibit antitumor immunity resulting in tumor growth and decreased patient survival [16]. Moreover, Ccl2 and Ccl5 suppress the T-cell responses [16].
4.2. TAMs and Angiogenesis
Accumulating evidence indicates that TAMs exert a critical function in regulating angiogenesis, the process by which new blood vessels sprout from the existing vasculature. TAMs depletion studies in mice showed reduction of blood vessel density in the tumor tissue [56] and numerous correlations between increased TAM numbers and high vascular grades have been reported for many tumor types [57–62].
TAMs support tumor cell invasion by secreting a broad repertoire of molecules, including growth factors, cytokines, proteases, and chemokines. For instance, TAMs release potent proangiogenic cytokines such as IL-8 (or Cxcl8) and growth factors as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and TGF-β, which have been reported to promote angiogenesis in tumors such as gliomas, squamous cell carcinomas of the esophagus, and breast, bladder, and prostate carcinomas [59–63]. Importantly, they also release chemokines like Ccl2, Ccl5, Cxcl9, and Cxcl16 that contribute to angiogenesis [64]. Moreover, TAM-derived proteases, such as matrix metalloproteases (MMP-1, MMP-2, MMP-3, MMP-9, and MMP-12) are also beneficial to angiogenesis [65–67].
Additionally, TAMs have been found to accumulate in hypoxic regions of human and experimental tumors (including human endometrial, breast, prostate, and ovarian carcinomas) [68]. TAMs respond to the hypoxic microenvironment by upregulating the hypoxia-inducible transcription factors HIF-1 and HIF-2 that induce the expression of proangiogenic genes, such as VEGF, Cxcl8, and Cxcl12.
4.3. TAMs and Tumor Growth
Macrophage depletion studies have proven that TAMs are essential for tumor growth [69] and TAMs infiltration has been observed in several tumors such as breast cancer, endometrial cancer, and renal cell cancer [70], demonstrating a positive correlation between proliferation of tumor cells and TAMs infiltration. Several molecules/factors secreted by TAMs such as MMP-9, IL-23, IL-10 facilitate tumor cell proliferation thereby limiting the cytotoxicity of the microenvironment.
4.4. TAMs and Metastasis
Another process in which TAMs have been involved is in the regulation of metastasis. Indeed, a correspondence between the number of macrophages in metastatic sites and the metastatic potential of the tumor has been observed [71], and systemic depletion of macrophages results in reduced formation of lung metastases [72]. These findings are in line with clinical studies showing that increased numbers of macrophages in regional lymph node metastases correlates with poor patient survival [73].
TAMs appear to influence the microenvironment to facilitate migration of tumor cells [74, 75] by the release of MMPs, for example, MMP-2, MMP-7, and MMP-9. Those MMPs contribute to transform the proteins of the extracellular matrix and induce the expression of lymphatic endothelial growth factor (VEGF-C), events that promote dissemination of tumor cells by stimulating the formation of lymphatic vessels in tumors [76].
5. Tumor Suppressors and Immune System
Tumor suppressor genes act as sensors of multiple forms of cellular stress, being regulated to induce cell-cycle arrest, senescence, or apoptosis. Nevertheless, in last decades, a growing body of evidence indicates that tumor suppressors play a key role in the modulation of innate immune system, a function more relevant even than their activity as cancer inhibitors.
For instance, innate immune response in the metazoan Caenorhabditis elegans has been described to be dependent on p53 function [77]. A clear function in antiviral defense has been reported for the tumor suppressors p53, the promyelocytic leukemia (PML) protein, and the alternative reading frame (ARF) [78–82]. Activation of PML, p53, and ARF has been described after IFN treatment, expression of viral proteins and viral infection [79, 82–87].
In addition, a molecular link between p53, ARF, retinoblastoma protein (Rb), and Toll-like receptors (TLRs) has been shown [87–89] and recent studies have demonstrated that ARF regulates inflammatory response [87].
5.1. The Retinoblastoma Protein Rb
Rb was identified as the protein responsible for the congenital tumor retinoblastoma [90] and plays pivotal roles in cell cycle control, differentiation, and inhibition of oncogenic transformation. Rb regulates cellular proliferation by directly binding to E2F transcription factors [91, 92], a family of transcription factors that regulates cellular proliferation, growth, and differentiation.
Furthermore, additional functions of Rb in the control of immune response have been described including a novel role in viral infection surveillance. Thus Rb is required for the activation of the NF-κB pathway in response to virus infection [93]. In addition, a recent report has shown that Rb positively regulates expression of TLR3, the sensing receptor for viral double-stranded RNA [89]. The mechanism involves modulation of the transcription factor E2F1, which directly binds to the proximal promoter of TLR3.
5.2. The Promyelocytic Leukemia (PML) Protein
The PML gene was originally identified in acute promyelocytic leukemia (APL), being implicated in numerous cellular functions including oncogenesis, DNA damage, senescence, apoptosis, and protein degradation. In addition, accumulating reports have also demonstrated the role of PML in host antiviral defense [78]. PML functions as the organizer of PML nuclear bodies (NBs) that contains some proteins recruited in a transient manner and two permanent NB-associated proteins, the IFN-stimulated gene product Speckled protein of 100 kDa (Sp100) and death-associated dead protein (Daxx) [94]. PML is induced by IFN leading to a marked increase in the expression of several PML isoforms (PMLI-PMLVII) and NBs. PML confers resistance to numerous virus including foamy virus (HFV), vesicular stomatitis virus (VSV), influenza virus, poliovirus, rabies virus, lymphocytic choriomeningitis virus (LCMV), and encephalomyocarditis virus [78, 95–99]. Interestingly, viruses inhibited by PML have developed various strategies to counteract the antiviral defense mechanisms by altering PML expression and/or localization on nuclear bodies [100].
5.3. Tumor Suppressor p53
The tumor suppressor p53 also known as “the guardian of genome” is activated in response to several types of cellular stress, including DNA damage and oncogene expression. Under normal conditions, p53 is maintained at very low levels through regulation by murine double minute 2 (Mdm2) protein. Mdm2 inhibits p53 transactivation and prompts p53 for proteasomal degradation by promoting its ubiquitination [101, 102]. However, in response to cellular stress, such as DNA damage, heat shock, or hypoxia, p53 levels rise as a consequence of activation of the tumor suppressor ARF that binds to Mdm2 and inhibit the ubiquitination, nuclear export, and subsequent degradation of p53 [103].
p53 has been implicated in multiple functions that play key roles in health and disease, including ribosome biogenesis, control of aging, cell cycle arrest, and apoptosis, having a clear importance in tumor suppression [104]. Interestingly, several lines of evidence also indicate that p53 may have a broader function in antiviral defense. Activation of p53 by IFN has been reported [80, 81] and p53-deficient mice are more permissive to viral infection [82]. This p53-mediated protection against viral infection is related with an induction of apoptosis, which is associated with reduced viral replication [82]. Moreover, in addition to activation of p53 by IFN, several genes involved in innate immunity have been described to be p53 direct transcriptional targets. IFN regulatory factors (IRFs) such as IRF-9 and IRF-5 have been described to be modulated by p53 [80]. Several mechanisms have been proposed for regulation of IRF-9 by p53 including upregulation at the transcriptional level [80], transactivation in response to influenza virus infection [105], and direct p53-IRF9 protein interaction upon Hepatitis C virus (HCV) infection [106]. Regarding IRF-5, an increase in IRF-5 levels in cancer cell lines has been shown through p53 binding and transactivation of the IRF-5 promoter [107].
Pattern recognition receptors such as TLR3 have also been reported to be regulated by p53 [88]. TLR3 plays a major role in the recognition of virus infection leading to the induction of the IFN pathway [108]. p53 activates TLR3 transcription by binding to the p53 consensus site in the TLR3 promoter. Moreover, TLR3 expression was downregulated in liver and intestine of p53−/− mice and HCT116 p53−/− cells, leading to a dysfunction in both NF-κB and IRF-3 signaling pathways [88]. Upregulation of TLR3 activity by p53 may also be responsible for the activation of interferon-stimulated gene 15 (ISG15). ISG15 is strongly induced by type I interferons and displays antiviral activity. Although a functional p53 binding site adjacent to the core ISRE site of ISG15 has been reported, upregulation of ISG15 has been observed after dsRNA stimulation rather than in response to IFN treatment or virus infection, suggesting that the observed effects on ISG15 could be mediated through p53-dependent upregulation of TLR3 activity [109].
Finally, important proinflammatory chemokines such as monocyte-chemoattractant protein (MCP)-1 have also been reported to be transcriptionally regulated by p53 [110]. MCP-1, also known as Ccl2, triggers the infiltration and activation of cells of the monocyte-macrophage lineage and has been linked with antitumor immunity [111] and cervical cancer [112].
Thus, in addition to the well-established function of p53 as a tumor suppressor through regulation of apoptosis or cell cycle, p53 exerts essential roles in the expression of key molecules of the innate immune response. Indeed, the loss of p53 function during carcinogenesis might affect the recognition of tumor cells by the immune system through interfering with inflammatory mediators expression.
5.4. The Tumor Suppressor ARF
Tumor suppressor ARF (p14ARF in human, p19ARF in mouse) is among the most frequent genes mutated in human cancer [113]. ARF is encoded by the INK4a/ARF locus (Cdkn2a) that generates two unrelated proteins, the cyclin-dependent kinase inhibitor p16INK4a and ARF, which, respectively, regulate the activity of Rb and the p53 transcription factor [114, 115]. ARF activates p53 by sequestering Mdm2, an E3 ubiquitin ligase, to the nucleolus, thereby inhibiting the Mdm2-mediated proteasomal degradation of p53. p53 subsequently activates p21 (CIP1/WAF1), which inhibits the cell cycle [103, 116]. Although tumor suppressor activity of ARF was initially attributed to p53 regulation, several p53-independent actions for ARF have been described [117]. Thus, ARF inhibits ribosomal RNA processing [118] and transcriptional factors that induce proliferation such as E2F1, Myc, and Forkhead box M1 (Foxm1b) [119–121]. In addition, ARF interacts with the protein related with cell proliferation nucleophosmin (NPM) [122].
Recent studies have shown that the tumor suppressor ARF is more than a simple tumor suppressor and acts a general sensor for different situation of cellular stress. In this context, a regulatory network between Heat shock protein 70 (Hsp70), ARF, and β-catenin has been shown after oxidative stress that leads to the induction of apoptosis [123]. ARF deficiency has been reported to aggravate atherosclerosis through the reduction of macrophage and vascular smooth muscle cell apoptosis [124]. ARF is also expressed transiently during mouse male germ cell and eye development and its inactivation compromises spermatogenesis as mice age and leads to aberrant postnatal proliferation of cells in the vitreous of the eye, resulting in blindness [125].
Furthermore, ARF plays an important role in the regulation of innate immunity and inflammatory processes. Several reports have described an antiviral action of ARF as well as its activation after the expression of viral proteins, viral infection, or type I IFN treatment [79, 85–87]. Indeed, the analysis of the ARF promoter revealed the presence of IFN response elements such as IRF-3 and interferon-sensitive response element (ISRE). The protective effect of ARF against viral infection seems to be a general feature for IFN-sensitive viruses as demonstrated the studies using VSV and Sindbis virus or vaccinia virus (VV) [79]. Mechanism involved in this protection is due, at least in part, to interaction with NPM and activation of the double-stranded RNA-dependent protein kinase, PKR [79].
Interestingly, it has been recently described that ARF is a critical modulator of the inflammatory response and macrophage activation. It has also been reported a molecular link between ARF and TLRs [87]. Additionally, a manuscript by Herranz et al. [126] suggests that ARF might modulate the M1/M2 polarization and functional plasticity of macrophages (Figure 2).
Figure 2.
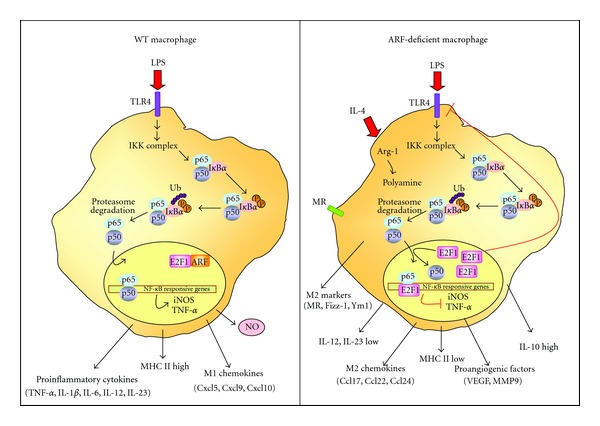
Alternative activation in ARF-deficient macrophages. In WT macrophages, a balance between M1 and M2 phenotype is established, depending on stimuli. Inflammatory stimuli induce NF-κB signaling pathways through the phosphorylation and subsequent ubiquitin-dependent degradation of IκBα by the 26S proteasome. Then, NF-κB translocates to the nucleus inducing target gene expression. ARF present in the nucleus displays physical and functional interaction with E2F1 resulting in destabilization of E2F1 protein and activation of NF-κB. However, ARF-deficient macrophages establish an immunosuppressive and tolerant microenvironment via impairment of M1 signals. When NF-κB translocates to the nucleus, excessive E2F1 inhibits NF-κB by binding to its subunit p65 in competition with the heterodimeric partner p50. Moreover, excessive E2F1 may inhibit transcriptional expression of TLRs. This leads to secretion of M2 chemokines Ccl17 and Ccl22, release of the anti-inflammatory cytokine IL-10, and stimulation of angiogenesis through expression of VEGF and MMP-9.
Mice lacking the ARF gene are resistant to LPS-endotoxic shock, and a significant reduction of leukocyte recruitment in a model of thioglicollate-induced peritonitis was also reported [87]. Moreover, ARF-deficient macrophages present an impaired ability to develop proinflammatory properties showing a relevant downregulation of genes involved in M1 macrophage phenotype and inhibition of the antimicrobial and antitumoral responses, including expression of proinflammatory cytokines (TNF-α, IL-1β), chemokines (Cxcl10, Cxcl1, Ccl4), and inflammatory mediators (iNOS/PGE2). Mechanisms involved in this inhibitory effect have not been fully explored, although a decrease in NF-κB and Mitogen-activated protein kinases (MAPK) activation have been described in ARF−/− macrophages after stimulation with LPS [87], as well as inhibition of IκB degradation in ARF-deficient macrophages stimulated with VSV [79]. Furthermore, an increase of the transcription factor E2F1 at basal state and after LPS-stimulation has been shown in absence of ARF [87]. E2F1 has been proposed to be an anti-inflammatory and immunosuppressive transcription factor since it represses NF-κB-dependent inflammatory signaling [127, 128]. Therefore, it has been proposed that in normal cells, ARF interacts with E2F1, resulting in destabilization of E2F1 protein and activation of NF-κB. In contrast, in the absence of ARF, E2F1 is overexpressed, and although NF-κB translocates to the nucleus, excessive E2F1 inhibits its activity by binding to p65 and thereby suppressing NF-κB-dependent genes (iNOS, COX-2, chemokines, etc.).
Loss of ARF gene can abrogate tumor surveillance mechanisms and increase cancer susceptibility. Indeed, mice lacking p19ARF are highly prone to tumor development [129, 130] and deletion of ARF has been described in a variety of malignancies, including glioblastoma, melanoma, pancreatic adenocarcinoma, and non-small-cell lung cancer. Interestingly, M2-polarized TAMs have been demonstrated to be associated with poor prognosis and progression in many of these tumors.
However, the link between ARF and immune response in the tumoral context remains an open question. A manuscript by Herranz et al. [126] goes deep into the immune role of ARF, evaluating its possible contribution to the M1/M2 polarization of macrophages. In this study, authors demonstrate that ARF deficiency switches macrophages to a M2-like phenotype. Thus, in addition to downregulation of proinflammatory mediators, typical hallmarks of an anti-inflammatory M2-activation state were increased in both resting and IL-4-treated ARF−/− macrophages, as exemplified by the upregulation of Arg-1, Fizz-1 and Ym1. Moreover, the cytokine/chemokine pattern induced by IL-4 stimulation appeared upregulated in macrophages isolated from ARF−/− mice showing an M2-phenotype with higher levels of IL-10, Ccl22, Ccl5, Ccr3, or Ccr5. Together with these M2 markers, important proangiogenic factors such as VEGF and MMP-9 were also increased, suggesting the potential protumoral action of ARF−/− macrophages. Consistent with this notion, recent studies have demonstrated a role for ARF in suppressing tumor angiogenesis via modulation of VEGF expression [131] and the activity of its transactivator HIF [132]. Notably, ARF has also been described to inhibit angiogenesis by up-regulating the expression of TIMP3, an inhibitor of MMPs activity [133].
Taken together, it is tempting to speculate that ARF has a profound influence in regulating the polarization of macrophages. Thus, ARF deficiency might modify the immune tumor microenvironment as a result of the induction of multiple activities including (a) immune suppression, through the production of the anti-inflammatory cytokine IL-10 and the secretion of Ccl22 and Ccl2 that attracts Tregs, (b) stimulation of angiogenesis through expression of VEGF and MMP-9, and (c) induction of matrix remodeling through the production of MMP-9, Fizz-1, and Ym1.
6. Conclusions
More and more evidence indicates that tumor suppressors play an essential role in host immunity, placing them as general sensors and modulators of innate immune response. Mechanisms involved in this process remain unclear, although transcriptional regulation of several inflammatory mediators has been reported. In addition, M1/M2 polarization and functional plasticity of macrophages have been shown to be modulated by tumor suppressors. These studies reveal positive effects of tumor suppressors on cancer immunosurveillance that will pave the way for new targeted therapies.
Therefore, identification of the role of tumor suppressors in the pathways responsible for the skewing of macrophage function as well as in the regulation of inflammatory mediators will remain an important area of investigation in the years to come.
Acknowledgment
This study was supported by Grants PI11.0036 from the FIS and MPY 1410/09 from ISCIII to S. Hortelano.
References
- 1.Ehrlich P. Ueber den jetzigen Stand der Karzinomforschung. Nederlands Tijdschrift voor Geneeskunde. 1909;5:273–290. [Google Scholar]
- 2.Burnet FM. Immunological surveillance in neoplasia. Transplantation reviews. 1971;7:3–25. doi: 10.1111/j.1600-065x.1971.tb00461.x. [DOI] [PubMed] [Google Scholar]
- 3.Thomas L. Discussion of medawar: reactions to homologous tissue antigens in relation to hypersensitivity. In: Lawrence HS, editor. Cellular and Humoral Aspects of the Hypersensitivity States. New York, NY, USA: Paul Hoeber; 1959. pp. 529–532. [Google Scholar]
- 4.Shankaran V, Ikeda H, Bruce AT, et al. IFNγ, and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 5.Smyth MJ, Thia KYT, Street SEA, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. Journal of Experimental Medicine. 2000;191(4):661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. International Immunology. 2001;13(4):459–463. doi: 10.1093/intimm/13.4.459. [DOI] [PubMed] [Google Scholar]
- 7.Gao Y, Yang W, Pan M, et al. γδ T cells provide an early source of interferon γ in tumor immunity. Journal of Experimental Medicine. 2003;198(3):433–442. doi: 10.1084/jem.20030584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girardi M, Oppenheim DE, Steele CR, et al. Regulation of cutaneous malignancy by γδ T cells. Science. 2001;294(5542):605–609. doi: 10.1126/science.1063916. [DOI] [PubMed] [Google Scholar]
- 9.Noguchi Y, Jungbluth A, Richards EC, Old LJ. Effect of interleukin 12 on tumor induction by 3-methylcholanthrene. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(21):11798–11801. doi: 10.1073/pnas.93.21.11798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van den Broek MF, Kägi D, Ossendorp F, et al. Decreased tumor surveillance in perforin-deficient mice. Journal of Experimental Medicine. 1996;184(5):1781–1790. doi: 10.1084/jem.184.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enzler T, Gillessen S, Manis JP, et al. Deficiencies of GM-CSF and interferon γ link inflammation and cancer. Journal of Experimental Medicine. 2003;197(9):1213–1219. doi: 10.1084/jem.20021258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annual Review of Immunology. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 13.Gordon S. Alternative activation of macrophages. Nature Reviews Immunology. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 14.Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase-arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. European The Journal of Immunology. 1995;25(4):1101–1104. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
- 15.Fairweather D, Cihakova D. Alternatively activated macrophages in infection and autoimmunity. Journal of Autoimmunity. 2009;33(3-4):222–230. doi: 10.1016/j.jaut.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. Journal of Leukocyte Biology. 2009;86(5):1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 18.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Kzhyshkowska J, Mamidi S, Gratchev A, et al. Novel stabilin-1 interacting chitinase-like protein (SI-CLP) is up-regulated in alternatively activated macrophages and secreted via lysosomal pathway. Blood. 2006;107(8):3221–3228. doi: 10.1182/blood-2005-07-2843. [DOI] [PubMed] [Google Scholar]
- 20.Högger P, Dreier J, Droste A, Buck F, Sorg C. Identification of the integral membrane protein RM3/1 on human monocytes as a glucocorticoid-inducible member of the scavenger receptor cysteine-rich family (CD163) The Journal of Immunology. 1998;161(4):1883–1890. [PubMed] [Google Scholar]
- 21.Raes G, de Baetselier P, Noël W, Beschin A, Brombacher F, Hassanzadeh GG. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. Journal of Leukocyte Biology. 2002;71(4):597–602. [PubMed] [Google Scholar]
- 22.Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunological Reviews. 2008;222(1):180–191. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sgadari C, Angiolillo AL, Tosato G. Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood. 1996;87(9):3877–3882. [PubMed] [Google Scholar]
- 24.Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Human Immunology. 2009;70(5):325–330. doi: 10.1016/j.humimm.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in Immunology. 2002;23(11):549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 26.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nature Reviews Cancer. 2004;4(1):71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 27.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. European Journal of Cancer. 2006;42(6):717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Raes G, Brys L, Dahal BK, et al. Macrophage galactose-type C-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. Journal of Leukocyte Biology. 2005;77(3):321–327. doi: 10.1189/jlb.0304212. [DOI] [PubMed] [Google Scholar]
- 29.Geijtenbeek TBH, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nature Reviews Immunology. 2009;9(7):465–479. doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nature Reviews Cancer. 2009;9(12):874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allavena P, Chieppa M, Bianchi G, et al. Engagement of the mannose receptor by tumoral mucins activates an immune suppressive phenotype in human tumor-associated macrophages. Clinical and Developmental Immunology. 2010;2010:10 pages. doi: 10.1155/2010/547179.547179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imai Y, Akimoto Y, Mizuochi S, Kimura T, Hirano H, Irimura T. Restricted expression of galactose/N-acetylgalactosamine-specific macrophage C-type lectin to connective tissue and to metastatic lesions in mouse lung. Immunology. 1995;86(4):591–598. [PMC free article] [PubMed] [Google Scholar]
- 33.Solinas G, Schiarea S, Liguori M, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. The Journal of Immunology. 2010;185(1):642–652. doi: 10.4049/jimmunol.1000413. [DOI] [PubMed] [Google Scholar]
- 34.Bussink AP, Speijer D, Aerts JMFG, Boot RG. Evolution of mammalian chitinase(-like) members of family 18 glycosyl hydrolases. Genetics. 2007;177(2):959–970. doi: 10.1534/genetics.107.075846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shuhui L, Mok YK, Wong WSF. Role of mammalian chitinases in asthma. International Archives of Allergy and Immunology. 2009;149(4):369–377. doi: 10.1159/000205583. [DOI] [PubMed] [Google Scholar]
- 36.Cai Y, Kumar RK, Zhou J, Foster PS, Webb DC. Ym1/2 promotes Th2 cytokine expression by inhibiting 12/15(S)-lipoxygenase: identification of a novel pathway for regulating allergic inflammation. The Journal of Immunology. 2009;182(9):5393–5399. doi: 10.4049/jimmunol.0803874. [DOI] [PubMed] [Google Scholar]
- 37.Chang NCA, Hung SI, Hwa KY, et al. A macrophage protein, Ym1, transiently expressed during inflammation is a novel mammalian lectin. The Journal of Biological Chemistry. 2001;276(20):17497–17506. doi: 10.1074/jbc.M010417200. [DOI] [PubMed] [Google Scholar]
- 38.Lee CG, da Silva CA, dela Cruz CS, et al. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annual Review of Physiology. 2011;73:479–501. doi: 10.1146/annurev-physiol-012110-142250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nair MG, Cochrane DW, Allen JE. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunology Letters. 2003;85(2):173–180. doi: 10.1016/s0165-2478(02)00225-0. [DOI] [PubMed] [Google Scholar]
- 40.Holcomb IN, Kabakoff RC, Chan B, et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. The EMBO Journal. 2000;19(15):4046–4055. doi: 10.1093/emboj/19.15.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu T, Dhanasekaran SM, Jin H, et al. FIZZ1 stimulation of myofibroblast differentiation. American Journal of Pathology. 2004;164(4):1315–1326. doi: 10.1016/S0002-9440(10)63218-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu T, Jin H, Ullenbruch M, et al. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. The Journal of Immunology. 2004;173(5):3425–3431. doi: 10.4049/jimmunol.173.5.3425. [DOI] [PubMed] [Google Scholar]
- 43.Teng X, Li D, Champion HC, Johns RA. FIZZ1/RELMα, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circulation Research. 2003;92(10):1065–1067. doi: 10.1161/01.RES.0000073999.07698.33. [DOI] [PubMed] [Google Scholar]
- 44.Schledzewski K, Falkowski M, Moldenhauer G, et al. Lympathic endothelium-specific hyaluronan receptor LYVE-1 is expressed by stabilin-1+, F4/80+, CD11b+ macropahages in malignant tumours and wound healing tissue in vivo and in bone marrow cultures in vitro: implications for the assessment of lymphangiogenesis. Journal of Pathology. 2006;209(1):67–77. doi: 10.1002/path.1942. [DOI] [PubMed] [Google Scholar]
- 45.Kzhyshkowska J, Workman G, Cardó-Vila M, et al. Novel function of alternatively activated macrophages: stabilin-1-mediated clearance of SPARC. The Journal of Immunology. 2006;176(10):5825–5832. doi: 10.4049/jimmunol.176.10.5825. [DOI] [PubMed] [Google Scholar]
- 46.Kurte M, López M, Aguirre A, et al. A synthetic peptide homologous to functional domain of human IL-10 down-regulates expression of MHC class I and transporter associated with antigen processing 1/2 in human melanoma cells. The Journal of Immunology. 2004;173(3):1731–1737. doi: 10.4049/jimmunol.173.3.1731. [DOI] [PubMed] [Google Scholar]
- 47.Bak SP, Alonso A, Turk MJ, Berwin B. Murine ovarian cancer vascular leukocytes require arginase-1 activity for T cell suppression. Molecular Immunology. 2008;46(2):258–268. doi: 10.1016/j.molimm.2008.08.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFβ . Nature Reviews Immunology. 2010;10(8):554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castriconi R, Cantoni C, Chiesa MD, et al. Transforming growth factor β1 inhibits expression of NKP30 and NKG2d receptors: consequences for the NK-mediated killing of dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(7):4120–4125. doi: 10.1073/pnas.0730640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maeda H, Shiraishi A. TGF-β contributes to the shift toward Th2-type responses through direct and IL-10-mediated pathways in tumor-bearing mice. The Journal of Immunology. 1996;156(1):73–78. [PubMed] [Google Scholar]
- 51.Beissert S, Hosoi J, Grabbe S, Asahina A, Granstein RD. IL-10 inhibits tumor antigen presentation by epidermal antigen-presenting cells. The Journal of Immunology. 1995;154(3):1280–1286. [PubMed] [Google Scholar]
- 52.Sica A, Saccani A, Bottazzi B, et al. Autocrine production of IL-10 mediates defective IL-12 production and NF-κB activation in tumor-associated macrophages. The Journal of Immunology. 2000;164(2):762–767. doi: 10.4049/jimmunol.164.2.762. [DOI] [PubMed] [Google Scholar]
- 53.Lepique AP, Daghastanli KRP, Cuccovia I, Villa LL. HPV16 tumor associated macrophages suppress antitumor T cell responses. Clinical Cancer Research. 2009;15(13):4391–4400. doi: 10.1158/1078-0432.CCR-09-0489. [DOI] [PubMed] [Google Scholar]
- 54.Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clinical Cancer Research. 2007;13(2):721s–726s. doi: 10.1158/1078-0432.CCR-06-2197. [DOI] [PubMed] [Google Scholar]
- 55.Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Research. 2001;61(3):1100–1106. [PubMed] [Google Scholar]
- 56.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjällman AHM, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. British Journal of Cancer. 2006;95(3):272–281. doi: 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Research. 1996;56(20):4625–4629. [PubMed] [Google Scholar]
- 58.Mäkitie T, Summanen P, Tarkkanen A, Kivelä T. Tumor-infiltrating macrophages (CD68+ cells) and prognosis in malignant uveal melanoma. Investigative Ophthalmology and Visual Science. 2001;42(7):1414–1421. [PubMed] [Google Scholar]
- 59.Nishie A, Ono M, Shono T, et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clinical Cancer Research. 1999;5(5):1107–1113. [PubMed] [Google Scholar]
- 60.Koide N, Nishio A, Sato T, Sugiyama A, Miyagawa SI. Significance of macrophage chemoattractant protein-1 expression and macrophage infiltration in squamous cell carcinoma of the esophagus. American Journal of Gastroenterology. 2004;99(9):1667–1674. doi: 10.1111/j.1572-0241.2004.30733.x. [DOI] [PubMed] [Google Scholar]
- 61.Hanada T, Nakagawa M, Emoto A, Nomura T, Nasu N, Nomura Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. International Journal of Urology. 2000;7(7):263–269. doi: 10.1046/j.1442-2042.2000.00190.x. [DOI] [PubMed] [Google Scholar]
- 62.Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. International Journal of Oncology. 2000;17(3):445–451. doi: 10.3892/ijo.17.3.445. [DOI] [PubMed] [Google Scholar]
- 63.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. Journal of Pathology. 2002;196(3):254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 64.Balkwill F. Cancer and the chemokine network. Nature Reviews Cancer. 2004;4(7):540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 65.Hagemann T, Wilson J, Burke F, et al. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. The Journal of Immunology. 2006;176(8):5023–5032. doi: 10.4049/jimmunol.176.8.5023. [DOI] [PubMed] [Google Scholar]
- 66.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9—expressing macrophages and angiogenesis to impair cervical carcinogenesis. Journal of Clinical Investigation. 2004;114(5):623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krecicki T, Zalesska-Krecicka M, Jelen M, Szkudlarek T, Horobiowska M. Expression of type IV collagen and matrix metalloproteinase-2 (type IV collagenase) in relation to nodal status in laryngeal cancer. Clinical Otolaryngology and Allied Sciences. 2001;26(6):469–472. doi: 10.1046/j.0307-7772.2001.00503.x. [DOI] [PubMed] [Google Scholar]
- 68.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Research. 2006;66(2):605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 69.Polverini PJ, Leibovich SJ. Effect of macrophage depletion on growth and neovascularization of hamster buccal pouch carcinomas. Journal of Oral Pathology. 1987;16(9):436–441. doi: 10.1111/j.1600-0714.1987.tb00714.x. [DOI] [PubMed] [Google Scholar]
- 70.Tsutsui S, Yasuda K, Suzuki K, Tahara K, Higashi H, Era S. Macrophage infiltration and its prognostic implications in breast cancer: the relationship with VEGF expression and microvessel density. Oncology Reports. 2005;14(2):425–431. [PubMed] [Google Scholar]
- 71.Ohno S, Ohno Y, Suzuki N, et al. Correlation of histological localization of tumor-associated macrophages with clinicopathological features in endometrial cancer. Anticancer Research. 2004;24(5 C):3335–3342. [PubMed] [Google Scholar]
- 72.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. Journal of Experimental Medicine. 2001;193(6):727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Öberg A, Samii S, Stenling R, Lindmark G. Different occurrence of CD8+, CD45R0+, and CD68 + immune cells in regional lymph node metastases from colorectal cancer as potential prognostic predictors. International Journal of Colorectal Disease. 2002;17(1):25–29. doi: 10.1007/s003840100337. [DOI] [PubMed] [Google Scholar]
- 74.Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochimica et Biophysica Acta. 2009;1796(1):11–18. doi: 10.1016/j.bbcan.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 75.Wyckoff J, Wang W, Lin EY, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Research. 2004;64(19):7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 76.Schoppmann SF. Lymphangiogenesis, inflammation and metastasis. Anticancer Research. 2005;25(6):4503–4511. [PubMed] [Google Scholar]
- 77.Fuhrman LE, Goel AK, Smith J, Shianna KV, Aballay A. Nucleolar proteins suppress Caenorhabditis elegans innate immunity by inhibiting p53/CEP-1. PLoS Genetics. 2009;5(9) doi: 10.1371/journal.pgen.1000657.e1000657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chelbi-Alix MK, Quignon F, Pelicano L, Koken MHM, de Thé H. Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. Journal of Virology. 1998;72(2):1043–1051. doi: 10.1128/jvi.72.2.1043-1051.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.García MA, Collado M, Mũoz-Fontela C, et al. Antiviral action of the tumor suppressor ARF. The EMBO Journal. 2006;25(18):4284–4292. doi: 10.1038/sj.emboj.7601302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muñoz-Fontela C, Macip S, Martínez-Sobrido L, et al. Transcriptional role of p53 in interferon-mediated antiviral immunity. Journal of Experimental Medicine. 2008;205(8):1929–1938. doi: 10.1084/jem.20080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muñoz-Fontela C, Garcia MA, Garcia-Cao I, et al. Resistance to viral infection of super p53 mice. Oncogene. 2005;24(18):3059–3062. doi: 10.1038/sj.onc.1208477. [DOI] [PubMed] [Google Scholar]
- 82.Takaoka A, Hayakawa S, Yanai H, et al. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 83.Lavau C, Marchio A, Fagioli M, et al. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene. 1995;11(5):871–876. [PubMed] [Google Scholar]
- 84.Grötzinger T, Sternsdorf T, Jensen K, Will H. Interferon-modulated expression of genes encoding the nuclear-dot-associated proteins Sp100 and promyelocytic leukemia protein (PML) European Journal of Biochemistry. 1996;238(2):554–560. doi: 10.1111/j.1432-1033.1996.0554z.x. [DOI] [PubMed] [Google Scholar]
- 85.Yang CT, You L, Uematsu K, Yeh CC, McCormick F, Jablons DM. p14ARF modulates the cytolytic effect of ONYX-015 in mesothelioma cells with wild-type p53. Cancer Research. 2001;61(16):5959–5963. [PubMed] [Google Scholar]
- 86.Pollice A, Nasti V, Ronca R, et al. Functional and physical interaction of the human ARF tumor suppressor with tat-binding protein-1. The Journal of Biological Chemistry. 2004;279(8):6345–6353. doi: 10.1074/jbc.M310957200. [DOI] [PubMed] [Google Scholar]
- 87.Traves PG, Lopez-Fontal R, Luque A, Hortelano S. The tumor suppressor ARF regulates innate immune responses in mice. The Journal of Immunology. 2011;187(12):6527–6538. doi: 10.4049/jimmunol.1004070. [DOI] [PubMed] [Google Scholar]
- 88.Taura M, Eguma A, Suico MA, et al. p53 regulates toll-like receptor 3 expression and function in human epithelial cell lines. Molecular and Cellular Biology. 2008;28(21):6557–6567. doi: 10.1128/MCB.01202-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taura M, Suico MA, Koyama K, et al. Rb/E2F1 regulates the Iinnate immune receptor toll-like receptor 3 in epithelial cells. Molecular and Cellular Biology. 2012;32(8):1581–1590. doi: 10.1128/MCB.06454-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cavenee WK, Dryja TP, Phillips RA, et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305(5937):779–784. doi: 10.1038/305779a0. [DOI] [PubMed] [Google Scholar]
- 91.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65(6):1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 92.Shirodkar S, Ewen M, DeCaprio JA, Morgan J, Livingston DM, Chittenden T. The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell. 1992;68(1):157–166. doi: 10.1016/0092-8674(92)90214-w. [DOI] [PubMed] [Google Scholar]
- 93.Garcia MA, Gallego P, Campagna M, et al. Activation of NF-κB pathway by virus infection requires Rb expression. PLoS ONE. 2009;4(7) doi: 10.1371/journal.pone.0006422.e6422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Regad T, Chelbi-Alix MK. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene. 2001;20(49):7274–7286. doi: 10.1038/sj.onc.1204854. [DOI] [PubMed] [Google Scholar]
- 95.Pampin M, Simonin Y, Blondel B, Percherancier Y, Chelbi-Alix MK. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. Journal of Virology. 2006;80(17):8582–8592. doi: 10.1128/JVI.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Regad T, Saib A, Lallemand-Breitenbach V, Pandolfi PP, de Thé H, Chelbi-Alix MK. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. The EMBO Journal. 2001;20(13):3495–3505. doi: 10.1093/emboj/20.13.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blondel D, Kheddache S, Lahaye X, Dianoux L, Chelbi-Alix MK. Resistance to rabies virus infection conferred by the PMLIV isoform. Journal of Virology. 2010;84(20):10719–10726. doi: 10.1128/JVI.01286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bonilla WV, Pinschewer DD, Klenerman P, et al. Effects of promyelocytic leukemia protein on virus-host balance. Journal of Virology. 2002;76(8):3810–3818. doi: 10.1128/JVI.76.8.3810-3818.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.El Mchichi B, Regad T, Maroui MA, et al. SUMOylation promotes PML degradation during encephalomyocarditis virus infection. Journal of Virology. 2010;84(22):11634–11645. doi: 10.1128/JVI.01321-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Everett RD, Chelbi-Alix MK. PML and PML nuclear bodies: implications in antiviral defence. Biochimie. 2007;89(6-7):819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 101.Momand J, Wu HH, Dasgupta G. MDM2—master regulator of the p53 tumor suppressor protein. Gene. 2000;242(1-2):15–29. doi: 10.1016/s0378-1119(99)00487-4. [DOI] [PubMed] [Google Scholar]
- 102.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 103.Pomerantz J, Schreiber-Agus N, Liégeois NJ, et al. The Ink4a tumor suppressor gene product, p19(Arf), interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92(6):713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 104.Fuster JJ, Sanz-González SM, Moll UM, Andrés V. Classic and novel roles of p53: prospects for anticancer therapy. Trends in Molecular Medicine. 2007;13(5):192–199. doi: 10.1016/j.molmed.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 105.Shen Y, Wang X, Guo L, et al. Influenza A virus induces p53 accumulation in a biphasic pattern. Biochemical and Biophysical Research Communications. 2009;382(2):331–335. doi: 10.1016/j.bbrc.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 106.Dharel N, Kato N, Muroyama R, et al. Potential contribution of tumor suppressor p53 in the host defense against hepatitis C virus. Hepatology. 2008;47(4):1136–1149. doi: 10.1002/hep.22176. [DOI] [PubMed] [Google Scholar]
- 107.Mori T, Anazawa Y, Iiizumi M, Fukuda S, Nakamura Y, Arakawa H. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene. 2002;21(18):2914–2918. doi: 10.1038/sj.onc.1205459. [DOI] [PubMed] [Google Scholar]
- 108.Uematsu S, Akira S. Toll-like receptors and innate immunity. Journal of Molecular Medicine. 2006;84(9):712–725. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- 109.Hummer BT, Li XL, Hassel BA. Role for p53 in gene induction by double-stranded RNA. Journal of Virology. 2001;75(16):7774–7777. doi: 10.1128/JVI.75.16.7774-7777.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hacke K, Rincon-Orozco B, Buchwalter G, et al. Regulation of MCP-1 chemokine transcription by p53. Molecular Cancer. 2010;9, article 82 doi: 10.1186/1476-4598-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang S, Singh RK, Xie K, et al. Expression of the JE/MCP-1 gene suppresses metastatic potential in murine colon carcinoma cells. Cancer Immunology Immunotherapy. 1994;39(4):231–238. doi: 10.1007/BF01525986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kleine-Lowinski K, Rheinwald JG, Fichorova RN, et al. Selective suppression of monocyte chemoattractant protein-1 expression by human papillomavirus E6 and E7 oncoproteins in human cervical epithelial and epidermal cells. International Journal of Cancer. 2003;107(3):407–415. doi: 10.1002/ijc.11411. [DOI] [PubMed] [Google Scholar]
- 113.Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutation Research. 2005;576(1-2):22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 114.Serrano M. The tumor suppressor protein p16(INK4a) Experimental Cell Research. 1997;237(1):7–13. doi: 10.1006/excr.1997.3824. [DOI] [PubMed] [Google Scholar]
- 115.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83(6):993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 116.Stott FJ, Bates S, James MC, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. The EMBO Journal. 1998;17(17):5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Weber JD, Jeffers JR, Rehg JE, et al. p53-independent functions of the p19(ARF) tumor suppressor. Genes and Development. 2000;14(18):2358–2365. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sugimoto M, Kuo ML, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Molecular Cell. 2003;11(2):415–424. doi: 10.1016/s1097-2765(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 119.Martelli F, Hamilton T, Silver DP, et al. p19ARF targets certain E2F species for degradation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(8):4455–4460. doi: 10.1073/pnas.081061398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature. 2004;431(7009):712–717. doi: 10.1038/nature02958. [DOI] [PubMed] [Google Scholar]
- 121.Kuo ML, Duncavage EJ, Mathew R, et al. Arf induces p53-dependent and -independent antiproliferative genes. Cancer Research. 2003;63(5):1046–1053. [PubMed] [Google Scholar]
- 122.Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Molecular and Cellular Biology. 2004;24(3):985–996. doi: 10.1128/MCB.24.3.985-996.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Damalas A, Velimezi G, Kalaitzakis A, et al. Loss of p14ARF confers resistance to heat shock- and oxidative stress-mediated cell death by upregulating β-catenin. International Journal of Cancer. 2011;128(8):1989–1995. doi: 10.1002/ijc.25510. [DOI] [PubMed] [Google Scholar]
- 124.González-Navarro H, Abu Nabah YN, Vinué A, et al. p19ARF deficiency reduces macrophage and vascular smooth muscle cell apoptosis and aggravates atherosclerosis. Journal of the American College of Cardiology. 2010;55(20):2258–2268. doi: 10.1016/j.jacc.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 125.Gromley A, Churchman ML, Zindy F, Sherr CJ. Transient expression of the Arf tumor suppressor during male germ cell and eye development in Arf-Cre reporter mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(15):6285–6290. doi: 10.1073/pnas.0902310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herranz S, Traves PG, Luque A, Hortelano S. Role of the tumor suppressor ARF in macrophage polarization. Enhancement of the M2 phenotype in ARF-deficient mice. OncoImmunology. 2012;1(8):1227–1238. doi: 10.4161/onci.21207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen M, Capps C, Willerson JT, Zoldhelyi P. E2F-1 regulates nuclear factor-κB activity and cell adhesion: potential antiinflammatory activity of the transcription factor E2F-1. Circulation. 2002;106(21):2707–2713. doi: 10.1161/01.cir.0000038706.30661.86. [DOI] [PubMed] [Google Scholar]
- 128.Fang F, Wang Y, Li R, et al. Transcription factor E2F1 suppresses dendritic cell maturation. The Journal of Immunology. 2010;184(11):6084–6091. doi: 10.4049/jimmunol.0902561. [DOI] [PubMed] [Google Scholar]
- 129.Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19(ARF) Cell. 1997;91(5):649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 130.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Research. 1999;59(9):2217–2222. [PubMed] [Google Scholar]
- 131.Kawagishi H, Nakamura H, Maruyama M, et al. ARF suppresses tumor angiogenesis through translational control of VEGFA mRNA. Cancer Research. 2010;70(11):4749–4758. doi: 10.1158/0008-5472.CAN-10-0368. [DOI] [PubMed] [Google Scholar]
- 132.Fatyol K, Szalay AA. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1α (HIF-1α) and inhibits HIF-1-mediated transcription. The Journal of Biological Chemistry. 2001;276(30):28421–28429. doi: 10.1074/jbc.M102847200. [DOI] [PubMed] [Google Scholar]
- 133.Zerrouqi A, Pyrzynska B, Febbraio M, Brat DJ, Van Meir EG. P14ARF inhibits human glioblastoma-induced angiogenesis by upregulating the expression of TIMP3. Journal of Clinical Investigation. 2012;122(4):1283–1295. doi: 10.1172/JCI38596. [DOI] [PMC free article] [PubMed] [Google Scholar]