

Abstract
Objective
To compare full transcriptome expression levels of matched tumor and normal samples from patients with oropharyngeal carcinoma stratified by known tumor etiologic factors.
Patients and Methods
Full transcriptome sequencing was analyzed for 10 matched tumor and normal tissue samples from patients with previously untreated oropharyngeal carcinoma. Transcriptomes were analyzed using massively parallel messenger RNA sequencing and validated using the NanoString nCounter system. Global gene expression levels were compared in samples grouped by smoking status and human papillomavirus status. This study was completed between June 10, 2010, and June 30, 2011.
Results
Global gene expression analysis indicated tumor tissue from former smokers grouped more closely to the never smokers than the current smokers. Pathway analysis revealed alterations in the expression of genes involved in the p53 DNA damage-repair pathway, including CHEK2 and ATR, which display patterns of increased expression that is associated with human papillomavirus–negative current smokers rather than former or never smokers.
Conclusion
These findings support the application of messenger RNA sequencing technology as an important clinical tool for more accurately stratifying patients based on individual tumor biology with the goal of improving our understanding of tumor prognosis and treatment response, ultimately leading to individualized patient care strategies.
Our understanding of the molecular genetics involved with the initiation and progression of human cancers is improving our ability to create targeted therapies. Just as the characterization of important genetic alterations has transformed treatment strategies for patients with pulmonary and colorectal adenocarcinoma, malignant tumors arising in the head and neck will soon be qualified by more than simply clinical and histopathologic parameters.1-3
Head and neck cancers are the sixth most commonly observed cancers worldwide; squamous cell carcinoma (SCC) represents 95% of these cases.4 Head and neck SCCs pose a significant health risk, including a 5-year overall survival rate of only 50% in cases of advanced-stage disease.5 These cancers are clinically grouped by their anatomic site within the head and neck. Although development of SCC in the head and neck is most significantly associated with a history of tobacco and alcohol use, the incidence of oropharyngeal SCC (OPSCC) appears to be increasing in younger patients who lack these traditional risk factors. Increasing evidence supports the association of human papillomavirus (HPV), primarily types 16 and 18, in this cohort.6-9 As treatment approaches are developed to target cancers that arise from differing causes, it will be increasingly difficult to stratify patients with overlapping risk factors. This increasing difficulty further highlights the importance of developing tools that allow identification of the risk factors most significantly contributing to the development of disease in an individual patient.
To address these challenges, there are new and rapidly developing technologies in the area of high-throughput sequencing that offer a novel approach toward the clinical stratification of patients and, more importantly, successful treatment strategies. Next-generation sequencing technologies can be used to analyze the entire genome, transcriptome, and even the epigenome of matched tumor and normal tissues to yield information on transcript profiles, mutations and genomic alterations, and posttranscriptional modifications. Although all 3 approaches provide important insight into the biological mechanisms of cancer development, transcriptome sequencing will be immediately applicable to clinical patient stratification because it offers the ability to directly measure expression levels of all important clinical targets simultaneously to develop a molecular profile of specific expression levels rather than performing separate assays for each individual gene target. Because of significant advances in sequence output, the cost of full transcriptome analysis is approaching that of single pathologic tests, making this an attractive and cost-effective modality to replace the current laboratory standards on which clinically relevant measurements are based. The information obtained by sequencing will also provide the ultimate resource for retrospective patient studies to elucidate novel targets or therapeutics based on response to treatment and disease survival.
In this report, we describe our initial efforts in characterizing the transcriptome of OPSCC, demonstrating both the clinical and research utility of this technology as applied to this cancer. We obtained full transcriptome data for 10 OPSCC tumors and patient-matched normal oropharyngeal tissue. These patients represented the full clinical spectrum observed with this cancer, including those with current, former, or no history of tobacco use, patients with positive or negative HPV status, and those with a range of tumor stage and grade at the time of surgery. To our knowledge, this is the first reported use of massively parallel messenger RNA sequencing (mRNA-seq) technology to examine transcriptomic changes in oropharyngeal SCC with simultaneous comparison to matched normal tissue. In this work we compared global patterns of dysregulated gene expression when grouping patients based on smoking history and discovered that former smokers (with smoking cessation >10 years) display gene regulation patterns that are more similar to those who never smoked (never smokers) than current smokers. In addition, we identified dysregulated gene pathways that parallel these trends in smoking history, particularly DNA damage–responsive targets within the p53 signaling pathway. Our analysis of specific gene targets in the p53 pathway identified that ATR and CHEK2 were correlated with both current smoking and negative HPV activity.
Patients and Methods
Collection and Processing of Tissues
After institutional review board approval, oropharyngeal tumor samples were obtained from patients undergoing surgical treatment of OPSCC at Mayo Clinic, Rochester, MN. After achieving negative surgical margins, adjacent normal mucosal tissue was collected from the oropharynx. All tissue was snap-frozen in liquid nitrogen for storage. Samples were evaluated by frozen sectioning, with a fresh blade between samples, and preparation of hematoxylin-eosin slides. The slides for each sample were evaluated by a pathologist to confirm the presence or absence of tumor. In the former case, the representation of overall tumor composition was also assessed, with more than 80% of cells being neoplastic considered optimal. The HPV status of each patient was determined by polymerase chain reaction quantification of HPV-16 E6 and E7 viral oncogenes (Supplemental Table 1, available online at http://www.mayoclinicproceedings.org).
RNA Extraction
Preparations of total RNA were extracted from frozen tissue samples using the PureLink RNA Mini Kit (Life Technologies, Carlsbad, CA) according to the manufacturer's protocol. Extracted RNA was quantified by NanoDrop ND1000 (Thermo Fisher Scientific, Waltham, MA), and the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) was used to assess RNA quality.
RNA Sequencing
Messenger RNA-seq complementary DNA (cDNA) libraries were prepared from 1 μg of total RNA using the standard Illumina mRNA-seq protocol (Supplemental Table 1, available at http://www.mayoclinicproceedings.org). These fragments were amplified by polymerase chain reaction and sequenced using the Illumina Cluster Station and Genome Analyzer (Illumina, Inc, San Diego, CA). Paired-end sequence analysis (51 cycles per end) was conducted with primers specific to the ends of the bridge-amplified cDNA fragments to obtain 51 nucleotides of sequence from each end of all cDNA fragments.
mRNA-seq Data Analysis
A whole transcriptome analysis approach was applied for the purpose of assessing gene expression, exon/intron usage, and splicing across the entire transcriptome. Raw reads from normal (control) and tumor cell lines were aligned to the human genome reference, build 36, using the Geospiza GeneSifter Analysis Edition pipeline (Supplemental Table 1, available at http://www.mayoclinicproceedings.org). From the aligned data, expression values for annotated genes were calculated by adding the number of reads mapping to all exons and splicing events for a given gene and dividing that value by the total number of mapped million reads in a sample. Similarly, exon expression values, used to detect differential splicing, were calculated by counting the number of reads mapped to an exon. Exon count values were then normalized by the total number of reads mapped to the gene.
Validation of mRNA-seq With nCounter
Gene targets of interest identified using mRNA-seq analysis were validated using a custom code set and the nCounter counting system by NanoString Technologies (Seattle, WA) according to manufacturer's protocol (Supplemental Table 1, available at http://www.mayoclinicproceedings.org). Wilcoxon rank sum test was applied to the nCounter data to identify differentially expressed genes between samples grouped by smoking history (never smokers, former smokers, and current smokers).
Results
Measurement of Gene Expression in OPSCC and Patient-Matched Normal Tissue by RNA Transcriptional Sequencing
We constructed mRNA-Seq libraries from poly A+ selected transcripts, and each library was sequenced on a single lane of the Illumina GAIIx machine. Using paired-end, 51-bp sequencing, we were able to obtain a mean of 63 million reads per transcriptome. We then compared mRNA-seq analysis for 10 patient-matched samples representing primary SCC and normal tissue (Table 1). One of the sequenced samples was excluded because of incomplete smoking history. The remaining 9 tumor and normal paired samples were grouped by smoking status based on patient-reported smoking history and include 3 never smokers (no reported smoking history), 3 past smokers (smoking cessation history >10 years), and 3 current smokers with a reported use of at least 2 packs per week (Table 2). Additional clinical information was collected, including age, sex, HPV status, location of the primary tumor, and tumor stage and grade but was not used in grouping the patients for data analysis.
TABLE 1.
Alignment Statistics for Transcriptome Reads
| Patient No. | No. of reads (in millions) (%) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Total |
Aligned to filter |
Aligned to genome |
Aligned to RefSeq exon |
|||||
| Tumor | Normal | Tumor | Normal | Tumor | Normal | Tumor | Normal | |
| 1 | 53 | 67 | 8 (16) | 9 (14) | 42 (80) | 45 (68) | 29 (55) | 32 (48) |
| 2 | 71 | 72 | 15 (21) | 16 (22) | 52 (73) | 63 (72) | 33 (47) | 32 (45) |
| 3 | 58 | 62 | 15 (21) | 16 (22) | 52 (73) | 52 (72) | 33 (47) | 32 (45) |
| 4 | 67 | 59 | 15 (21) | 16 (22) | 52 (73) | 52 (72) | 33 (47) | 32 (45) |
| 5 | 61 | 62 | 15 (21) | 16 (22) | 52 (73) | 52 (72) | 33 (47) | 32 (45) |
| 6 | 75 | 71 | 15 (21) | 16 (22) | 52 (73) | 52 (72) | 33 (47) | 32 (45) |
| 7 | 61 | 57 | 15 (21) | 16 (22) | 52 (73) | 52 (72) | 33 (47) | 32 (45) |
| 8 | 61 | 53 | 15 (21) | 16 (22) | 52 (73) | 52 (72) | 33 (47) | 32 (45) |
| 9 | 64 | 65 | 15 (21) | 16 (22) | 52 (73) | 52 (72) | 33 (47) | 32 (45) |
| 10 | 65 | 61 | 3 (5) | 5 (8) | 53 (82) | 48 (79) | 35 (59) | 32 (53) |
Read counts are expressed in millions (percentage of the total reads processed) for each patient-matched tumor and normal tissue sample. RefSeq = National Center for Biotechnology Information (NCBI) Reference Sequence Database.
TABLE 2.
Patient Demographics Grouped by Smoking Status
| Patient No. | Age (y) | Sex | Subsite | HPV-16 test result | T stage | N stage | M stage | Grade |
|---|---|---|---|---|---|---|---|---|
| Current smokers | ||||||||
| 1 | 56 | M | Tonsil | Positive | 1 | 0 | 0 | 4 |
| 2 | 73 | M | Tonsil | Negative | 4a | 2c | 0 | 3 |
| 3 | 54 | M | Base of tongue | Negative | 4 | 2b | 0 | 3 |
| Former smokers | ||||||||
| 4 | 48 | M | Tonsil | Positive | 4a | 2c | 0 | 3 |
| 5 | 64 | F | Tonsil | Positive | 1 | 2a | 0 | 4 |
| 6 | 66 | M | Base of tongue | Positive | 2 | 2b | 0 | 3 |
| Never smokers | ||||||||
| 7 | 46 | M | Tonsil | Positive | 3 | 2b | 1 | 3 |
| 8 | 49 | F | Tonsil | Positive | 2 | 0 | 0 | 4 |
| 9 | 73 | M | Tonsil | Negative | 2 | 2b | 0 | 3 |
Messenger RNA sequencing transcriptome data were analyzed for 9 patient-matched tumor and normal tissue samples. These patients were divided into analysis groups based on smoking status, including current smokers, former smokers with greater than 10 years of cessation, and never smokers. The human papillomavirus (HPV) status of each patient was determined by polymerase chain reaction (PCR) detection of HPV DNA and PCR quantification of E6 and E7 viral oncogenes. Additional clinical information collected was not used in grouping the patients for data analysis.
Global Patterns of Gene Expression From Transcriptome Analysis of Patients Grouped by Smoking Status
We were interested in characterizing the global expression patterns of genes differentially expressed in patient-matched tumor and normal tissues when patients were grouped according to their smoking history. Using hierarchical clustering and principal component analysis, we observed the expected observation that tumor tissues from never smokers are dissimilar to current smokers. More interestingly, we also discovered that when comparing overall expression patterns of all dysregulated genes, tumor tissue from former smokers grouped more closely to the never smokers than the current smokers (Figure 1). Global expression of genes from normal tissues is similar in all patients, regardless of smoking status. Further study is under way to determine the full spectrum of alterations associated with each smoking group to determine whether the former smokers represent a lesser degree of genomic change and altered translocation patterns than current smokers or whether significant periods of smoking cessation result in transcriptional patterns reverting to those seen with the never smokers. Regardless of the mechanism, this transcriptome analysis indicates that patients with a smoking history should be investigated as an individual group when stratifying patients by risk factor exposure.
FIGURE 1.
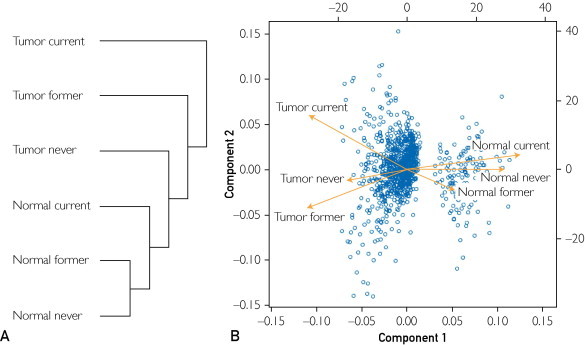
Global gene expression patterns in samples grouped by smoking status. A, Hierarchical clustering shows that tumors from current smokers show a distinct pattern of expressed genes when compared with tumors from individuals who have stopped smoking or never smoked. B, Similarly, a principal component analysis shows differential gene expression between normal and tumor samples and that the tumor from current smokers is different from the other tumor samples.
Identification of Differentially Regulated Targets Related to Smoking Status
To identify the most significantly differential targets when grouping patients by smoking status, we used the GeneSifter program to conduct 2-way analysis of variance. This analysis identified targets that were differential among tumor and normal tissue types and smoking risk factor groupings. We identified a list of transcript targets representing the most differentially up-regulated and down-regulated targets within the 9 samples analyzed (data not shown). In addition, we used the GeneSifter pathway analysis feature, which allowed us to identify a number of differentially expressed pathways when grouping samples by smoking status. One pathway that stood out with interesting trends of expression was the p53 signaling pathway. We next chose to investigate several members of the p53 signaling pathway, representing DNA damage–responsive genes, based on patterns of expression that differed among the smoking risk factor groups (Supplemental Figure, available at http://www.mayoclinicproceedings.org). However, although analysis of the transcriptome sequencing data identified these intriguing patterns of regulation in important pathways, the minimal sample size of 3 patient pairs per group resulted in a lack of statistical power, requiring the use of validation experiments on larger numbers of samples.
Validation of Differentially Expressed DNA Damage–Responsive Genes in the p53 Signaling Pathway Among Samples Grouped by Smoking and HPV Status
To perform a larger-scale validation of targets of interest and gain statistical power, we applied the NanoString nCounter technology to measure the transcript expression of the custom set of targets involved in DNA damage response and p53 signaling in 38 oropharyngeal patient-matched tumor and normal tissue pairs (Supplemental Table 2, available at http://www.mayoclinicproceedings.org).
We identified a significant expression pattern between smoking groups for 2 specific genes in the p53 signaling pathway, ATR and CHEK2, both of which have kinase activity and are involved in p53 regulation with a demonstrated response to DNA damage. The expression of ATR is significantly different when using a Wilcoxon rank sum test to compare either never smokers with current smokers (P=.05) or former smokers with current smokers (P=.04) but not between never and former smokers (Figure 2A). When ATR expression is examined using a Wilcoxon rank sum test in patients grouped both by current or never smoking status and HPV status, HPV-negative patients with a current smoking history are found to express significantly higher levels of ATR than HPV-positive (P=.008) or HPV-negative (P=.002) patients who never smoked (Figure 2B). A second gene, CHEK2, a cell cycle checkpoint regulator and putative tumor suppressor that is both responsive to DNA damage and involved in stabilization of the tumor suppressor p53, was evaluated by Wilcoxon rank sum test in patients grouped by HPV and smoking status. We found that CHEK2 has significantly higher expression in HPV-negative patients with a current smoking history compared with HPV-positive never smokers (P=.02) and current smokers (P=.002) and with HPV-negative never smokers (P=.04) (Figure 3). These findings demonstrate that the patterns of increased expression of ATR and CHEK2 DNA damage–responsive genes clearly are associated specifically with those patients who have a current smoking history and lack an active HPV infection.
FIGURE 2.

Expression of ATR in patient samples grouped by smoking and human papillomavirus (HPV) status. A, RNA transcripts corresponding to ATR were quantified using nCounter technology for patients grouped by smoking status and identified significant differences between current smokers and either never or former smokers but no difference between never and former smokers using a Wilcoxon rank sum test. B, The groups were further divided by adding the additional factor of HPV status, and significant differences were identified between HPV-negative current smokers and all never smokers irrespective of their HPV status but not HPV-positive current smokers.
FIGURE 3.
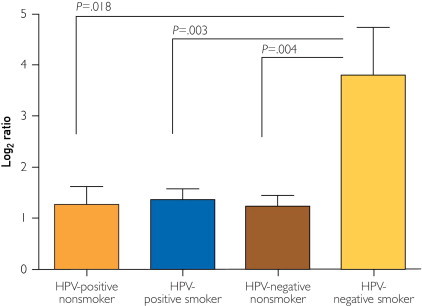
Expression of CHEK2 in patient samples grouped by smoking and human papillomavirus (HPV) status. RNA transcripts corresponding to CHEK2 were quantified using nCounter technology for patients grouped by smoking and HPV status. Significant differences were identified when comparing HPV-negative current smokers with all other groups using a Wilcoxon rank sum test.
Discussion
In this report we used total transcriptome sequencing analysis to characterize a group of oropharyngeal tumors and patient-matched normal tissues. Oropharyngeal tumors display a wide range of behavior, including differences in patterns of invasion, tendency for metastasis, and widely varying treatment responses. Clinicians' inability to accurately predict tumor behavior leads to widely varying treatment strategies, often resulting in significant morbidity and even mortality. Increasing evidence suggests that patient prognosis and treatment should be stratified on the basis of exposure to tobacco and HPV infection and activity.10-12 These classifications are complicated by the overlap of factors in many patients and the unreliable nature of self-reporting smoking history and the estimation of pack-year exposure (eg, HPV-positive patients who also have a smoking history, HPV-negative patients who lack a smoking history, patients with a past smoking history but significant periods of cessation, and patients with a history of secondhand tobacco exposure). In the future, molecular profiling of target genes identified as markers of smoking exposure will play an important role in sorting out this intricate collection of risk factors in individual patients. Naturally, this will assist clinicians in applying the most effective treatments based on individual tumor and patient characteristics.
Although the work presented focused exclusively on transcriptome expression analysis, we recognize the need to integrate DNA sequence analysis in future studies. From our data, it is clear, however, that transcriptome analysis is an effective and perhaps more powerful way to initiate a deep understanding of cancer biology from the perspective of defining clinical biomarkers and pharmacologic targets. Ongoing work by others clearly demonstrates how DNA sequencing can be used to identify mutations associated with cancer development to help explain mechanisms of carcinogenesis. However, DNA analysis shows only what changes have occurred to the DNA sequence, whereas transcriptome analysis demonstrates the effect of those changes, which is critically important in identifying which mutations and rearrangements could be the best diagnostic and prognostic indicators.
In our transcriptome analysis, a global comparison of dysregulated genes from patients representing all smoking risk factor groups, including never smokers, former smokers with more than 10 years of cessation, and current smokers, revealed patterns that suggest that tumor tissues of former smokers are transcriptionally more similar to never smokers than current smokers. These patterns may suggest that a significant period of cessation allows the regulation of many transcripts to return to patterns more closely resembling patients who lack such a history. The next important step will involve a more comprehensive analysis of genomic changes occurring in these patients to determine whether former smokers have fewer genomic alterations than patients with a current smoking history. One must also consider the potential effects these findings could have on both prognostic and therapeutic classification.
Full characterization of the transcriptome for the 9 samples analyzed resulted in the identification of important trends and gene targets of interest, but the small sample size reduced statistical power. A current limitation in using mRNA-seq technology to analyze large groups of patient samples is the current cost of preparing samples and sequencing and analyzing the resulting data. However, each of these steps is rapidly progressing toward more automated platforms with drastically lower costs. These improvements in workflow will rapidly allow for large sample sets to be analyzed directly by mRNA-seq and eliminate the need for secondary technology for validation experiments. To validate the targets identified by this work, we applied NanoString nCounter technology on a larger number of samples. One of the advantages of this technology is that only 100 ng of total RNA is sufficient to accurately measure the expression of up to several hundred targets per sample. When mRNA-seq is coupled with high-throughput quantitative assays, like nCounter, trends observed in lower-sample, high-coverage assays can be verified quickly by high-sample, targeted assays that increase statistical power.
We therefore chose genes from pathways of interest that were differentially expressed in the small number of oropharyngeal samples whose transcriptomes were sequenced. We then validated these patterns of expression in 38 oropharyngeal tumor-normal pairs, which demonstrated similar patterns of regulation for 2 genes related to the DNA damage and the p53 signaling pathway (ATR and CHEK2). We examined the expression of these genes in patient samples grouped by smoking status and demonstrated increased expression in patients with a current smoking history compared with those with a former history and those who lack exposure. This same pattern was evident even when incorporating the confounding clinical factor of active HPV infections in these same patient groups.
Patient stratification in OPSCC is complex because of the potential interactions of overlapping risk factors, such as smoking status and HPV infection, and activity that can affect tumor behavior, prognosis, and treatment selection. Transcriptome sequencing will aid clinicians in determining which risk factors most significantly influence the development of an individual patient's cancer. The accumulation of these data will also provide an invaluable retrospective research data set that will lead to the identification of patient and tumor markers most strongly associated with treatment outcomes and selection. As our understanding of this technology improves, it will become an invaluable prospective tool for clinicians, allowing them to select individualized treatment strategies based on objective patient and tumor biology.
Conclusion
Transcriptional profiling by mRNA-seq allowed us to identify unique patterns of global gene regulation that correlated with patient exposure to known oncogenic risk factors. Specific gene targets involved in the p53 DNA damage-repair pathway, including CHEK2 and ATR, were analyzed and displayed patterns of increased expression associated with HPV-negative current smokers when compared with past smokers or nonsmokers. Identification and validation of these and related gene targets will serve as valuable molecular tools for more accurately stratifying patients based on individual tumor biology and behavior.
Acknowledgments
We thank Charles Beatty, MD, chair of otorhinolaryngology, Mayo Clinic, Rochester, MN, for his ongoing support of this work, and the Stoll Foundation for postdoctoral fellowship support.
Footnotes
For editorial comment, see page 211
Grant Support: This work was supported by internal funding from the Mayo Foundation for Medical Education and Research. The efforts of Todd M. Smith and N. Eric Olson were supported by award 2R44HG005297 from the National Human Genome Research Institute.
Potential Competing Interests: Todd M. Smith and N. Eric Olson are employed by Geospiza, a PerkinElmer company. The authors have no financial interests to disclose.
Supplemental Online Material
Analysis of messenger RNA sequencing data corresponding to 9 patient-matched tumor and normal pairs grouped based on smoking status. The GeneSifter pathway analysis feature was used to identify dysregulated genes within pathways relevant to cancer development. Targets involved in p53 signaling and DNA damage response were chosen for validation experiments based on patterns of expression when comparing patients grouped by smoking status (A) and patient-matched tumor and normal ratios of reads per million.values (B).
References
- 1.Pao W., Miller V., Zakowski M. EGF receptor gene mutations are common in lung cancers from ”never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Acad Sci U S A. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogino S., Meyerhardt J.A., Cantor M. Molecular alterations in tumors and response to combination chemotherapy with gefitinib for advanced colorectal cancer. Clin Cancer Res. 2005;11(18):6650–6656. doi: 10.1158/1078-0432.CCR-05-0738. [DOI] [PubMed] [Google Scholar]
- 3.Tsuchihashi Z., Khambata-Ford S., Hanna N., Jänne P.A. Responsiveness to cetuximab without mutations in EGFR. N Engl J Med. 2005;353(2):208–209. doi: 10.1056/NEJM200507143530218. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 5.Jemal A., Center M.M., DeSantis C., Ward E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- 6.Attner P., Du J., Nasman A. The role of human papillomavirus in the increased incidence of base of tongue cancer. Int J Cancer. 2010;126(12):2879–2884. doi: 10.1002/ijc.24994. [DOI] [PubMed] [Google Scholar]
- 7.Chung C.H., Gillison M.L. Human papillomavirus in head and neck cancer: its role in pathogenesis and clinical implications. Clin Cancer Res. 2009;15(22):6758–6762. doi: 10.1158/1078-0432.CCR-09-0784. [DOI] [PubMed] [Google Scholar]
- 8.D'Souza G., Kreimer A.R., Viscidi R. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356(19):1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 9.Gillison M. HPV and its effect on head and neck cancer prognosis. Clin Adv Hematol Oncol. 2010;8(10):680–682. [PubMed] [Google Scholar]
- 10.Hoffmann M., Ihloff A.S., Gorogh T. p16(INK4a) overexpression predicts translational active human papillomavirus infection in tonsillar cancer. Int J Cancer. 2010;127(7):1595–1602. doi: 10.1002/ijc.25174. [DOI] [PubMed] [Google Scholar]
- 11.Smith E.M., Pawlita M., Rubenstein L.M., Haugen T.H., Hamsikova E., Turek L.P. Risk factors and survival by HPV-16 E6 and E7 antibody status in human papillomavirus positive head and neck cancer. Int J Cancer. 2010;127(1):111–117. doi: 10.1002/ijc.25015. [DOI] [PubMed] [Google Scholar]
- 12.Rubenstein L.M., Smith E.M., Pawlita M., Haugen T.H., Hamsikova E., Turek L.P. Human papillomavirus serologic follow-up response and relationship to survival in head and neck cancer: a case-comparison study. Infect Agent Cancer. 2011;6:9. doi: 10.1186/1750-9378-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of messenger RNA sequencing data corresponding to 9 patient-matched tumor and normal pairs grouped based on smoking status. The GeneSifter pathway analysis feature was used to identify dysregulated genes within pathways relevant to cancer development. Targets involved in p53 signaling and DNA damage response were chosen for validation experiments based on patterns of expression when comparing patients grouped by smoking status (A) and patient-matched tumor and normal ratios of reads per million.values (B).