

Abstract
During the early years of cytochrome P450 research, a picture of conserved properties arose from studies of mammalian forms of these monooxygenases. They included the protohaem prosthetic group, the cysteine residue that coordinates to the haem iron and the reduced CO difference spectrum. Alternatively, the most variable feature of P450s was the enzymatic activities, which led to the conclusion that there are a large number of these enzymes, most of which have yet to be discovered. More recently, studies of these enzymes in other eukaryotes and in prokaryotes have led to the discovery of unexpected P450 properties. Many are variations of the original properties, whereas others are difficult to explain because of their unique nature relative to the rest of the known members of the superfamily. These novel properties expand our appreciation of the broad view of P450 structure and function, and generate curiosity concerning the evolution of P450s. In some cases, structural properties, previously not found in P450s, can lead to enzymatic activities impacting the biological function of organisms containing these enzymes; whereas, in other cases, the biological reason for the variations are not easily understood. Herein, we present particularly interesting examples in detail rather than cataloguing them all.
Keywords: cytochrome P450, evolution, biodiversity, structure/function, unusual properties
1. Introduction
Cytochrome P450 monooxygenases (P450 or CYP) constitute a superfamily of structurally diverse and functionally versatile haem-containing enzymes with more than 15 000 known genes distributed across all biological kingdoms [1]. P450 proteins have extremely diverse primary sequences and are grouped into families depending upon their amino acid sequence identity: proteins with 40 per cent identity or greater are classed in the same family (CYP1, CYP2, etc.), and members with 55 per cent identity or greater are classed in the same subfamily (CYP1A1, CYP1A2, etc.; http://drnelson.utmem.edu/CytochromeP450.html; [2]). P450 enzymes have pivotal roles in primary and secondary metabolic pathways. It has been proposed that the ancestral P450 may have been involved in the detoxification of reactive oxygen species generated in the highly volatile atmosphere of the burgeoning Earth [3]. P450s have been the subject of intense experimental interest for nearly 60 years, because of their extraordinary ability to introduce atmospheric oxygen into non-activated carbon–hydrogen bonds, showing exceptionally high degrees of regio-selectivity and stereo-selectivity according to the generalized reaction scheme:
In the resting state, the iron (Fe) atom of the P450 haem is the ferric (III) low-spin state in which the Fe is six-coordinated by four nitrogen atoms in the haem tetrapyrrole ring, the sulphur atom from cysteine and a water molecule (rather than amino acid coordination within the protein itself) in the distal position. The linkage of the haem Fe to the cysteine thiolate gives rise to the characteristic Soret absorption at 450 nm when the ferrous form of the enzyme is complexed with carbon monoxide (CO). Upon substrate binding, the water molecule is often expelled as the axial ligand to produce a five-coordinated ferric high-spin species that may have a concomitant increase of the redox potential. Next, an electron is transferred by an ancillary redox protein partner to the ferric haem, generating the ferrous five-coordinated haem. Molecular dioxygen binds to the ferrous enzyme resulting in the formation of a haem Fe producing the ferric dioxo species, which is subsequently protonated to produce the ferric peroxide complex termed compound 0. A second solvent-derived proton leads to heterolytic cleavage of the O–O bond with loss of water generating the Fe (IV) oxo-porphyrin π-radical (termed compound I). Finally, compound I extracts a proton from the substrate to form a ferryl-hydroxo species (compound II) and a substrate-centred radical, followed by rebound of oxygen from the ferryl-hydroxo intermediate, generating the ferric haem and product [4,5]. Common P450 enzymatic reactions observed include C-hydroxylation, heteroatom oxygenation, heteroatom release (dealkylation), epoxide formation and group migration. Recently, more complex (unusual) enzymatic reactions including chlorine oxygenation, dimer formation, ring coupling, ring formation, ring contraction and aromatic dehalogenation have also been attributed to P450-dependent catalysis [6]. Consequently, given the breadth and elegance of complex biotranformations that P450s can catalyse, industry and academia strive for using P450s in the development of new medicines and agrochemicals as well as using their enzymatic properties in biotechnological applications [7].
For catalytic activity, P450s require a source of electrons which are provided by reduced pyridine nucleotides (NAD(P)H or NADH). However, P450s cannot normally receive electrons directly from such cofactors and, therefore, require auxillary redox partner proteins to shuttle the electron to the P450 haem Fe. Generally these redox partners can be divided into three classes (although there are notable exceptions, some of which will be discussed later): prokaryotic P450s are reduced by Fe-containing ferredoxin (Fdx) and a flavin adenine dinucleotide (FAD)-containing ferredoxin reductase (FDR); eukaryotic P450s are reduced by a membrane-bound flavin mononucleotide (FMN)/FAD-containing NADPH cytochrome P450 oxidoreductase (CPR); and mitochrondrial P450s are reduced by adrenodoxin (Adx) and adrenodoxin reductase (ADR) [8].
2. A note on common features conserved between all P450s
Prior to the discussion of unusual properties of P450s, it is important to note that all P450s share common properties (figure 1). First, in all P450s, the central haem Fe atom is bound to the protein through the anionic, thiolate sulphur of a cysteine residue. The cysteine is the only absolutely conserved residue found in all 15 000 P450 sequences. The importance of the thiolate bond is associated with the formation of the highly reactive intermediate, Fe(IV)oxo species (compound I) that is essential in the P450 catalytic cycle to oxidize unactivated C–H bonds [5]. Site-directed mutagenesis of the cysteine residue and hence loss of the thiolate ligand invariably leads to loss of P450 function. Second, it follows that all P450s can bind and activate atmospheric dioxygen. The only known exceptions are thromboxane synthase and allene oxide synthase, where the haem Fe binds two atoms of oxygen from the substrate molecule itself [9,10]. This ability to catalyse the molecular scission of dioxygen is key to the myriad of chemical reactions that specific P450 enzymes can undertake, and is universal to all P450s. Finally, since the first P450 atomic structure was resolved in 1985 by Poulos et al. [11], the X-ray crystallographic resolution of a number of prokaryotic and eukaryotic P450s have revealed that they all share similar overall structure and shape. This reflects conserved secondary structural elements involved in protein folding, and the correct incorporation and orientation of the haem porphyrin in order to produce an active P450 enzyme [12].
Figure 1.

Examples of (a) common and (b) unusual chemical reactions catalysed by P450 monooxygenases. Many are formal oxidations but reductions and re-arrangements also occur [6].
3. Unusual P450 systems and associated redox partners
Originally P450–redox systems were divided into class I (for prokaryotic P450s consisting of the cytosolic and soluble three-component P450–Fdx–FDR electron chain) and class II (for eukaryotic P450s consisting of the endoplasmic reticulum (ER) membrane-bound P450–CPR electron chain). However, deviations from both classes began to arise during the study of mammalian adrenal P450s involved in steroid hormone biosynthesis. It was established that CYP11A1 (which converts cholesterol to pregnenolone, the first committed step in steroid hormone biosynthesis), CYP11B1 (which converts deoxycorticosterone to corticosterone and 11-deoxycortisol to cortisol) and CYP11B2 (which converts corticosterone to aldosterone) are supported by a soluble reductase partner system consisting of a 2Fe–2S protein Adx and a FAD-containing ADR, rather than a membrane-bound CPR [13]. This discovery gave clues to novel redox partner compatibility dependent upon individual P450 enzymes. Subsequently, the advent of genome sequencing and the uncovering of the multitude of P450 enzymes from organisms across biological kingdoms established the biodiversity of P450–redox systems and highlighted unusual forms. Perhaps, the first deviation from the class I/II standard described above was discovered by Narhi & Fulco [14], who isolated a naturally occurring P450 fusion protein (CYP102A1; P450BM3) consisting of a N-terminal P450 haem domain linked to a eukaryotic-like CPR containing FMN and FAD domains in the bacterium Bacillus megaterium. CYP102A1 is located in the cytosol and has enzymatic activity towards fatty acid hydroxylation with the highest turnover rate reported for any P450 monooxygenase (17 000 min−1). This is probably reflected in the rapid intra-transfer of electrons between the fused domains compared with the separate components of the vast majority of P450 and redox partner proteins [15]. Although believed to be a highly unusual P450 form, genome sequencing has revealed similar CYP102A1-like enzymes in other bacteria. Two CYP102A1 homologues (CYP102A2 and CYP102A3) are present in Bacillus subtilis and similar forms have been found in the bacteria Ralstonia metallidurans, Bradyrhizobium japonicum and various bacilli and streptomycetes [16]. Additionally, a eukaryotic counterpart of this bacterial CYP102A1 was initially discovered in the fungus Fusarium oxysporum and named CYP505A1 with homologues in various Neurospora and Aspergilli species [17]. Similar to CYP102A1, the CYP505As have fatty acid hydroxylase activity, although one member of the subfamily CYP505B1 is a hydroxylase in the production of the mycotoxin fumonisin [18]. In contrast to the CYP102 fusion proteins described above, a recently discovered CYP102 subfamily (CYP102B) was described, which exists solely as a single P450 haem domain [19]. CYP102B1 from Streptomyces coelicolor A3(2) could be reconstituted with spinach ferredoxin and ferredoxin reductase to catalyse the turnover of arachidonic acid. However, the product profile was markedly different to CYP102A1, and the turnover by CYP102B1 was approximately 1000-fold less than CYP102A1 [19] (figure 2).
Figure 2.

Overview of the different arrangements of P450 redox partners and associated fusion proteins. The schematic represents organization with the domains for P450 coloured in red; P450 reductase (CPR) in yellow; ferredoxin (fdx), adrenodoxin (adx), flavodoxin (fld) and FMN in light orange as labelled; ferredoxin reductase (FDR), adrenodoxin reductase (ADR), Fe/S reductase domain of phthalate dioxygenase (Fe/S red) and FAD in dark orange as labelled; Acyl CoA dehydrogenase (Acyl CoS-DeH) domain in blue; protein fused to Mimivirus P450 (labelled G, glycosylation site; PKC, protein kinase C phosphorylation site; C, four caesin kinase II phosphorylation sites; M, three myristoylation sites). (i) Eukaryotic class I P450 system; (ii) prokaryotic class II P450 system; (iii) mitochondrial P450 system; (iv) examples of P450s fused to a FMN/FAD containing eukaryotic-like CPR; (v) example of a naturally occurring three component soluble P450 system consisting of P450, flavodoxin and flavodoxin reductase; (vi) example of P450 fused at C-terminus to phthalate dioxygenase reductase; (vii) example of P450 fused at C-terminus to a ferredoxin; (viii) example of P450 fused at N-terminus to a flavodoxin domain; (ix) example of P450 fused at N-terminus to acyl CoA dehydrogenase; (x) example of P450 fused at C-terminus to a protein of unknown function; (xi) example of a P450 requiring no redox partners that can interact directly with NADH cofactor; and (xii) example of P450 requiring no redox partners, NADPH/NADH or O2 to function.
A different arrangement of a P450 linked to a redox partner was found in the bacterium Rhodococcus sp. NCIMB 9784. In this protein, a soluble P450 haem domain is fused at the C-terminus to a FMN- and a 2Fe–2S cluster containing a reductase that resembles phthalate dioxygenase reductase [20] and has been named CYP116B2. Furthermore, homologues of this P450 have been found in R. metallidurans (CYP116B1) and Rhodococcus ruber (CYP116B3) [21]. Although the endogenous function of these P450s are not known, it has been shown that the phthalate reductase domain can accept reducing equivalents, and drive CYP116B2 metabolism of 7-ethoxycoumarin and CYP116B3 metabolism of polycyclic aromatic hydrocarbons [22]. Another arrangement was found in the bacterium Methylococcus capsulatus. This organism is one of the few bacteria which can synthesize a sterol (initially thought to be an exclusive biochemical pathway of eukaryotes) de novo. Encoded in the genome of M. capsulatus was a soluble CYP51 (sterol 14α-demethylase) P450 haem domain fused to a ferredoxin domain [23]. The physiological role for this P450 fusion protein was established as a true CYP51, in that it catalysed the three-step oxidation of lanosterol to 4,4,-dimethyl-5α-cholesta-8,14,24-diene-3β-ol, hence confirming that an exogenous FDR can transfer electrons from NADPH to the Fdx domain, which are intramolecularly transferred to the P450 domain. Interestingly, the resulting sterol product from the CYP51 demethylation and subsequent C4 demethylation steps is the final sterol molecule in this bacterium. This finding leads to discussion and speculation on both the ancestry of CYP51, the most functionally conserved P450 in the whole superfamily, and sterol biosynthesis in general [24]. A different arrangement was found in the bacterium Rhodococcus rhodocrous consisting of a fusion between a soluble C-terminal P450 domain (XplA) and a N-terminal flavodoxin domain, a single polypeptide chain containing FMN (XplB) [25]. This P450 has been shown to catalyse the breakdown of the nitramine explosive and pollutant hexahydro-1,3,5-trinitro-1,3,5-triazine by reductive denitration [26], and when expressed in transgenic plants, hexahydro-1,3,5-trinitro-1,3,5-triazine is removed from contaminated soil, thus having application in bioremediation of contaminated military sites [27]. In another unusual mechanistic observation for a P450 enzyme, oxygen is not required for the degradation of hexahydro-1,3,5-trinitro-1,3,5-triazine, but its presence and binding is required to determine the final degradation products [28]. Finally, examples of a P450 domain fused to proteins of known and unknown function (and which have no homology to redox proteins) are now emerging. In the bacterium Pseudomonas fluorescens, a novel but uncharacterized P450 was found consisting of a P450 domain fused to acyl CoA dehydrogenase and named CYP221A1 [29]. Additionally, the recently discovered P450 found in the Mimivirus genome (CYP5253A1, the first viral P450) was expressed, spectrally characterized and shown to be a P450 domain fused at the C-terminus to a protein containing several putative post-translational modification sites, including one N glycosylation site, a protein kinase C phosphorylation site, four casein kinase II phosphorylation sites and three myristoylation sites [30]. Although the function of this P450 is unknown, this represents the first time such complex modifications are present in a P450 molecule.
Above are described novel P450 fusion proteins. However, novel arrangements of P450 with putative redox partners have now been described, which deviate from the class I and II system originally proposed. For example, the gene CYP176A1 from the bacterium Citrobacter braakii is arranged in an operon consisting of genes encoding for a flavodoxin (cindoxin) and a flavodoxin reductase, a single polypeptide chain containing FAD (cidoxin reductase) [31]. Cidoxin reductase has been resistant either to expression or isolation, but Escherichia coli flavodoxin reductase was shown to reconstitute CYP176A1 and cindoxin-mediated hydroxylation of the monoterpene cineole, allowing the bacterium to grow on this compound [31]. E. coli, which has no P450 genes encoded in its genome encodes genes for flavodoxin and flavodoxin reducatse. For many P450s, these E. coli redox proteins have been able to drive their specific catalytic activities. This has led to the hypothesis that eukaryotic CPR arose from gene fusion of the genes encoding flavodoxin and flavodoxin reductase [32].
Above, we have described eukaryotic and prokaryotic P450s and associated redox partner systems and the novel fusion forms. Two examples of P450 which function without any redox partner now follow. The first, a eukaryotic example, is CYP55A1 (P450nor) discovered in the fungus F. oxysporum. Biochemically, CYP55A1 catalyses the reduction of nitric oxide to nitrous oxide [33]. CYP55A1 is a soluble P450 and, although encoded by the same gene, is located in both the cytoplasm and mitochondria in the cell. The P450 haem prosthetic group receives electrons for nitric oxide reduction directly from NADH without participation of any NADH-linked reductase or reducing system. Kinetic experiments confirmed the direct transfer of electrons from NADH to the P450 haem and the CYP55A1 X-ray crystal structure revealed a positively charged cluster located beneath the β-helix as the likely NADH binding site [34]. CYP55A1-like soluble homologues have also been found in the yeast Trichosporon cutaneum [35] and in the fungi Cylindrocarpon tonkinense, Histoplasma capsulatum and Aspergillus oryzae (for review, [36]).
The second, a prokaryotic example, is CYP154A1 found in the bacterium S. coelicolor A3(2). Recently, CYP154A1 was shown to catalyse the intramolecular cyclization of a novel dipentaenone with a high degree of conjugation to a Paternò–Büchi-like product [37]. In an unprecendented biochemical reaction described for any P450 studied to date, CYP154A1 requires no redox partner, reducing equivalent (NADPH) or oxygen to function, although CYP154A1 retains key residues in it primary sequence seen in other P450s and produces a Soret maximum at 450 nm in the reduced CO-difference spectrum. Only a few examples of P450-catalysed rearrangement without the involvement of external reducing equivalents have been reported, e.g. the rearrangement of prostaglandin H2 and fatty acid hydroperoxides [10]. However, the substrates involved in these reactions are peroxides and the only other non-redox reactions reported for P450s are hydrolyses [38].
4. Differences in electron transport in mechanisms in P450–redox partners
In the preceding section, we have described novel combinations of different P450 and redox partners, including fusion proteins evolved to supply reducing equivalents to the P450 haem Fe for catalysis. However, the exact mechanism of electron transfer in both prokaryotic and eukaryotic P450 systems remains both elusive and controversial, despite extensive biochemical and biophysical analysis over many decades. At the forefront of these disputes in eukaryotic CPR (and CYP102A1 reductase-like component systems) are flavin interaction(s) during transfer, NADPH priming reaction requirement and the final FMN species that donates electrons to the P450 protein. The common feature of eukaryotic CPR as well as CYP102A1 reductase and its homologues and CYP505A1 is the presence of two flavin prosthetic groups, FAD and FMN, in the reductase domain that channel electrons to P450 haem Fe [39]. CPR transfers a hydride ion from NADPH to FAD and the latter transfers electrons to FMN, where they are then delivered in two one-electron transfer steps to the P450 [4,5]. However, until 2006, the model for eukaryotic CPR electron transfer mechanisms was based on the only resolved CPR crystal structure, N-terminal-truncated rat CPR, which had the disadvantage of being catalytically inactive [40]. Furthermore, the recently published structure of truncated human CPR, which shares 92.5 per cent sequence identity with rat CPR, was also inactive in driving P450 activities [41]. In contrast N-terminal-truncated yeast (Saccharomyces cerevisiae) CPR was shown to be catalytically activite in supporting different P450 activities [42], and intriguingly the resolved crystal structure of this protein suggested a new, and unusual, method of electron transfer in CPR [43]. This is still the only catalytically active CPR in driving P450 activities with a resolved three-dimensional structure. The yeast CPR structure suggested two binding sites for FMN: a FMN1 binding site that is in a similar position in rat and human CPR, and a second FMN2 binding site located at the interface of the flavin-connecting and FMN-binding domains. Hence, a new model for the CPR electron transfer pathway was proposed in which FMN shuttles between two different binding sites in the reductase molecule: FMN bound in FMN1 binding site receives an electron from FAD, is protonated and then swings to the FMN2 binding site where the electron exits to the P450 protein, as the structure revealed the FMN isoalloxazine ring is exposed to the bulk solvent in the FMN2 binding site. The FMN swings back to the FMN1 binding site, obtains a second electron and this process is repeated [43]. Such a dynamic scenario can be envisaged through protein conformational changes impacting upon flavin positioning possibly due to CPR–P450 interactions. Indeed, contentious mechanistically, it will be interesting to discover whether different CPRs have evolved unique processes to deliver electrons to their P450 complement or whether the electron transfer process is generic within the CPR family of proteins. The structural resolution of further catalytically active eukaryotic CPRs and their biochemical characterization will address this question.
Whereas most eukaryotic organisms encode for a single CPR protein within their genome (although some fungi such as the Aspergilli species and plants encode two or more), within prokaryotes containing P450, and excluding P450–redox fusion proteins, there are also multiple numbers of possible P450–redox partner proteins encoded with the genome. It was universally thought that different Fdx and FDR were interchangeable in driving P450 reactions, with the P450 showing no preference for a particular species. Indeed, various bacterial P450s accept electrons from flavoproteins and/or Fdx and FDRs from different species including plant sources, e.g. spinach Fdx and FDR. However, this had never truly been explored definitively. Recently, the complete redox complement of six Fdx and four FDR encoded by genes in S. coelicolor A3(2) were expressed, purified and assembled in the different 24 combinations to assess whether a specific S. coelicolor P450, CYP105D5, which had been shown to hydroxylate fatty acids, had a preference for a particular redox system and hence electron transfer pathway [44]. Indeed, it was established that CYP105D5 preferred a specific electron transfer pathway (NADH → FDR1 → Fdx4 → CYP105D5) for catalytic activity raising the hypothesis that P450s have also evolved to select particular redox partners. Fdx 4 was shown to be virtually irreplaceable with the other five Fdx proteins in combining with FDR1 to drive CYP105D5 activity, thus strengthening this tenet [44]. Hence, it will be essential to apply this approach to determine optimal electron transfer pathways, particularly in bacterial systems, to fully exploit P450 applications in a biotechnological setting (figure 3).
Figure 3.
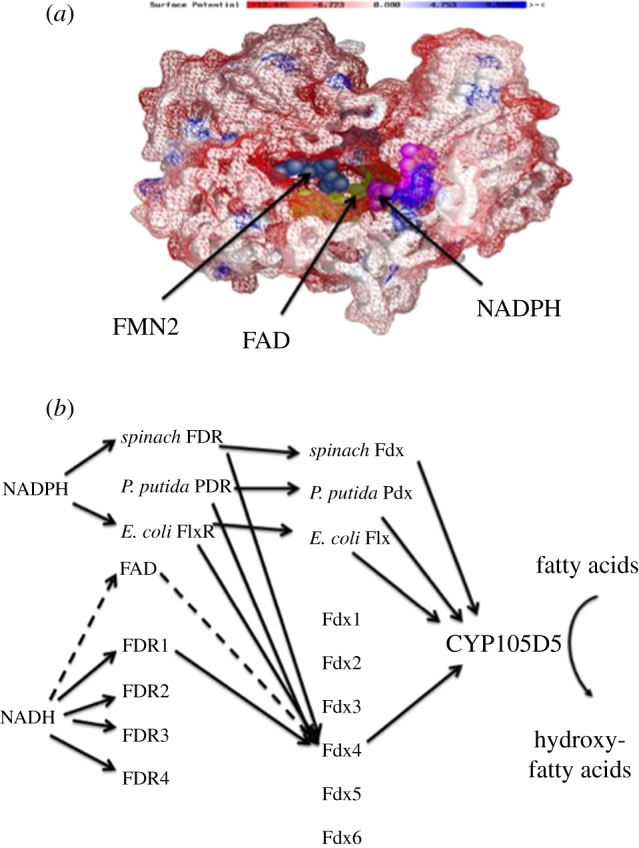
An example of a eukaryotic and a prokaryotic difference in P450 electron transport mechanisms. (a) Surface view of the overall yeast CPR structure showing three cofactors bound: FMN is blue; FAD is green; NADPH is purple. Negatively charged cluster of residues involved in cytochrome c and P450 binding are in red, distinctly clustered around the FMN2 binding site opening. The FMN cofactor in the discovered second FMN2 binding site (blue) is clearly visible through an opening with 7- and 8-CH3 groups of the flavin isoalloxazine ring exposed to the bulk solvent. (b) Schematic of the electron flow for CYP105D5-mediated fatty acid hydroxylation of the 24 Streptomyces coelicolor A3(2) redox partner combinations which could drive this P450. It was shown that the favoured flow was NADH to ferredoxin reductase 1 to ferredoxin 4 to CYP105D5. Also included are commonly used exogenous P450 redox proteins generally used in P450 turnover experiments.
5. Deviations from the accepted cellular localization of P450 enzymes
Traditionally, it is thought that all prokaryotic P450s are located in the cytoplasm and that all eukaryotic P450s are located in the ER or mitochondria of the cell. All the bacterial P450s biochemically characterized are soluble enzymes and located in the cytosol. Analysis of prokaryotic P450 primary sequences does not reveal the presence of N-terminal hydrophobic spanning regions, and a prokaryotic membrane-bound P450 has yet to be described experimentally, suggesting that indeed all prokaryotic P450s are located in the cytoplasm. Endoplasmic reticular (microsomal) P450s are integral membrane proteins with each P450 containing a single N-terminal transmembrane spanning segment. P450s are directed and retained in the ER by a signal sequence that also prevents translocation through the ER membrane [45]. Although signal sequence domains have been identified in many P450s the majority of eukaryotic P450s do not have specific identifiable signals. It is thought that overall hydrophobicity at the N-terminus of a P450 sequence directs targeting to the ER membrane [46]. Mitochondrial P450s involved in steroid biosynthesis and vitamin D metabolism have canonical and cleavable mitochondrial targeting signals at their N-termini that are distinct from the signal sequences of microsomal P450s [46]. Mitochondrial P450s are synthesized as pre-proteins containing N-terminal cleavable pre-sequences in the cytosol and post-translationally targeted to the mitochondria. After translocation into the matrix, the targeting sequence is cleaved and the mature P450 associates with the inner membrane.
Unusually, some P450 members belonging to the 1, 2 and 3 families have been shown to be bimodally targeted proteins being associated with mitochondria and plasma membrane in addition to the ER [47]. Previously, it was believed that these P450 family members where exclusively found in the ER. It has now been clearly established that the following P450s are bimodally targeted to the mitochondria: CYP1A1, CYP1A2, CYP1B1, CYP2B1, CYP2E1, CYP2D6, CYP2C11, CYP2C6, CYP3A2 and CYP4A1 [46]. That these P450s are targeted to both the mitochondria and the ER is due to unique N-terminal signals (termed chimeric signals) that carry essential elements for targeting to both organelles. These chimeric targeting signals are difficult to identify, but generally consist of an ER targeting signal at the N-terminus (which is part of the transmembrane anchor) flanked by a cryptic mitochondria targeting signal located at amino acid residues 20–36 in each of the different P450s named above. Furthermore, within the mitochondrial targeting signal are 2–5 positively charged residues that are essential for mitochondrial import [46,48]. Of course, these P450s, which are usually supported by CPR in their catalytic activities in the ER, are thus driven by mitochondrial Adx and ADR. The physiological importance of such xenobiobitic metabolizing P450s being present in the mitochondria is still not clear, but one impact lies in the activation of toxins and carcinogens resulting in mitochondrial damage and possible cell death.
6. Post-translational modification of certain P450 enzymes
It was originally believed that P450s were not modified further once they had been synthesized and folded into their mature form. However, recent work has shown that for certain eukaryotic P450 enzymes post-translational modification occurs. This includes phosphorylation (CYP2B1, CYP2B4, CYP2C6, CYP2D6, CYP2E1, CYP3A4, CYP11A1, CYP17A1, CYP19A1), ubiquitination (CYP3A4 and CYP2B1), glycosylation (CYP11A1 and CYP19A1) and nitration (CYP4A subfamily) [49]. Phosphorylation of a P450 was first shown with CYP2B4 by cAMP-dependent protein kinase (PKA) [47]. Since then a number of P450s have been shown to be phosphorylated in cell-free systems, hepatocyte incubations and in intact animals. Common features resulting in P450 phosphorylation include the presence of a cytosolically exposed PKA recognition sequence (RRXS) with the Ser residue as the kinase target [46,49]. However, other P450s do not contain this motif and other cryptic target sequences are phosphorylated by other protein kinases, e.g. protein kinase C (PKC). Much debate has ensued over why select P450s are phosphorylated. The consensus holds that P450 phosphorylation serves as a marker for P450 loss of function and/or degradation. For example, phosphorylation of CYP2E1 Ser129 and CYP2B1 Ser128 has been shown to immediately inactivate the proteins, faster than transcriptional downregulation would reduce activity through reduced protein levels [49]. Furthermore, PKA-mediated P450 phosphorylation has been shown to activate the cryptic mitochondrial targeting signal in CYP2B1, CYP2E1 and CYP2D6 [46]. It is proposed that in part, phosphorylation disrupts the ability of these P450s to be targeted to the ER (possibly through disruption of signal recognition particle binding), increasing the percentage of proteins to be imported into the mitochondria.
Analysis of post-translational modification in the steroid hormone P450 biosynthetic enzymes reveals differences between enzymes and across species. Studies in some species have shown that CYP19As undergo glycosylation and phosphorylation. Human, bovine and equine CYP19As are glycosylated but not the porcine ovary and placenta isoforms [50]. The human CYP19A1 sequence shows potential N-glycosylation sites at Asn12 and Asn180 and it has been predicted that the glycosylation site is localized at the N-terminal membrane-spanning region [51]. Most mammalian, including human, bovine and equine CYP19As have a NXT/S (Asn12–Ile13–Thr14) motif at the N-terminal region, but porcine ovary and placenta sequences lack this motif. Interestingly, the porcine embryonic sequence contains a NXT/S motif, implying that this isoform is probably glycosylated even though it has not been experimentally tested. The NXT/S motif is present only in mammalian and amphibian species, not in birds, reptiles and both isoforms of fishes even though a few exceptions are found. Site-directed mutagenesis of murine CYP19 has demonstrated that Ser118 is a potential phosphorylation site and mutation of this site destabilized the enzyme and decreased the specific activity [52]. Unlike the less-conserved N-terminal glycosylation site, the predicted phosphorylation site Ser118 is conserved in all CYP19A sequences analysed in the present study except in the ovarian isoform of zebrafish. The differences in the levels of conservation of glycosylation and phosphorylation correlate with their effect on the function of the enzyme. While the poorly conserved N-glycosylation site does not show any effect on the activity of CYP19A1 [50], the nearly absolutely conserved phosphorylation site has a significant effect on decreasing the enzyme activity [52]. In direct contrast, phosphorylation of serines and threonines of CYP11A1 by PKC and in CYP17A1 by PKA results in an increase of both enzyme activities [53,54].
Regarding other post-translational modifications of P450s, knowledge is limited. Phosphorylation of CYP3A4 is thought to lead to the enhancement of ubiquination of this P450. This results in aggregate formation of the CYP3A4-ubiquitin form in microsomal membranes and enhanced protein degradation [55]. Clearly there is a direct link and crosstalk between P450 activity, expression, degradation and post-translational modification. It is also important to note that only certain eukaryotic P450s are directly regulated in this manner (table 1).
Table 1.
An overview of biomodally targeted (mitochondria and ER) and post-translationally modified P450 enzymes.
| phosphorylated | glycosylated | ubiquitinated | nitrated | bimodally targeted |
|---|---|---|---|---|
| CYP1A2 | CYP1A2 | CYP2B1 | CYP2B1 | CYP1A1 |
| CYP2B1 | CYP2B2 | CYP2E1 | CYP8A1 | CYP1A2 |
| CYP2B2 | CYP2B4 | CYP3A1 | CYP55A1 | CYP1B1 |
| CYP2B4 | CYP11A1 | CYP3A2 | CYP101A1 | CYP2B1 |
| CYP2C6 | CYP17A1 | CYP3A4 | CYP102A1 | CYP2E1 |
| CYP2C7 | CYP19A1 | CYP2D6 | ||
| CYP2C11 | CYP21A1 | CYP2C6 | ||
| CYP2C12 | CYP27B1 | CYP2C11 | ||
| CYP2E1 | CYP3A2 | |||
| CYP3A1 | CYP4A1 | |||
| CYP3A4 | ||||
| CYP3A6 | ||||
| CYP7A1 | ||||
| CYP11A1 | ||||
| CYP11B1 | ||||
| CYP17A1 | ||||
| CYP19A1 | ||||
| CYP51 | ||||
| CYP27A1 |
7. Selected examples of unusual biochemical properties of cytochrome P450 enzymes
(a). Variations in absolutely conserved residues in P450 superfamily
As sequencing projects advanced, it was possible to analyse and compare each P450 primary sequence and identify residues that could be considered as being essential to all P450 enzymes both functionally and structurally. Up until 2006, it was universally thought that three residues were essential in all P450 sequences: the conserved cysteine, which is the fifth ligand to the haem Fe atom, and the EXXR motif forming a charge pair in the K helix, and which is possibly involved in haem binding and overall P450 fold topology, including stability. However, the completed genome sequence of S. coelicolor revealed for the first time that three P450s—CYP156B1, CYP157A1 and CYP157C1—do not contain this EXXR motif and thus challenged this dogma [56].
Examination of the CYP157C1 amino acid sequence revealed the presence of EQSLW in place of EXXR, however, the ferrous–CO complex gave a Soret maximum at 448 nm, typical of a ‘normal’ P450 [57]. Site-directed mutagenesis to create mutant forms of CYP157C1 containing the EXXR motif were undertaken. However, all mutants produced did not result in correctly folded P450, only the incorrectly folded P420 form [57]. These experiments irrevocably proved that the EXXR motif is not required in all P450s for structural architecture. An X-ray structure of a P450 lacking EXXR is eagerly anticipated to cast new insights on P450 folding and architecture. Finally, the single residue absolutely conserved in all P450 sequences was thought to be the cysteine, which coordinates with the haem Fe. However, recent genome sequencing has revealed that members of the CYP408 family do not contain the canonical cysteine residue, but do contain the EXXR motif and have sufficient sequence identity to be classified as P450s [58]. Experimental validation to shed light on the biochemical nature of a CYP408 enzyme is still to be undertaken (figure 4).
Figure 4.

Selected examples of unusual biochemical features of P450 structure and function. (a) View of the essentially invariant EXXR motif in the crystal structure of CYP158A2. The glutamic acid and arginine residues (purple/blue) form a set of salt–bridge interactions that contribute to haem binding and overall P450 fold. In CYP157C1, replacement of these residues with glutamine and tryptophan, respectively, suggests an undertermined mechanism for haem stabilization. (b) Haem orientation in CYP154A1 is 100% in the opposite position compared with that seen in most other P450s including its subfamily member, CYP154C1, in S. coelicolor A3(2). It is not known whether this haem orientation contributes to the unique cycloaddition reaction CYP154A1 undertakes. (c) Structures of CYP158A1 and CYP158A2. Both enzymes catalyse the dimerization of flaviolin, but with differing regio-specificity in product formation. This is reflected in the different orientations the flaviolin takes when binding into each enzyme active site. (d) Structure of CYP170A1. Highlighted is the haem-binding domain and P450 active site where catalysis, consisting of two sequential allylic oxidations to convert epi-isozizaene to the antibacterial albaflavenone, occurs. Also highlighted is the moonlighting active site and FPP binding domain where catalysis of farnesyl diphosphate to farnesene occurs.
(b). Variation of P450 haem incorporation and topology
Until 2003, all P450 structures, whose X-ray crystal structure had been resolved, showed that following haem incorporation into the P450 molecule, the haem prosthetic group adopted a unique orientation based upon the positions of the two protohaem vinyl groups. However, the crystal structure of CYP121A1 from Mycobacterium tuberculosis revealed that the haem group could assume two distinct orientations, the normal orientation and a small fraction in the opposite orientation [59]. CYP121A1 catalyses the formation of an intramolecular C–C bond between two tyrosyl carbon atoms of cyclodipeptide cyclo (L-Tyr–L-Tyr) (cYY) [60]. However, it is not known whether a particular orientation of the prosthetic haem is favoured during catalysis.
Subsequently, the crystal structure of S. coelicolor CYP154A1 revealed that the haem orientation is 100 per cent opposite to that of all reported CYP structures [61]. This is not a unique characteristic of the CYP154 family, because the resolved structure of another family member, CYP154C1, contained all of its haem in the normal P450 orientation [62]. The endogenous function of CYP154C1 is unknown. However, using the substrates of Streptomyces venezuleae CYP107C1 (YC-17 and narbomycin), a P450 involved in the biosynthesis of the antibacterial agents pikromycin, methymycin and neomethymycin, CYP154C1 was shown to be active in converting both substrates to the antibacterial products [62]. Conversely CYP154A1 was shown to be inactive when these compounds were used as substrates [61]. Thus, it may be possible that the haem orientation may influence these enzymatic activities. Additionally, as described above, CYP154A1 has been shown to catalyse the unique cycloaddition of a dipentaenone in an unprecedented P450-catalysed reaction and it is not known whether the orientation of the CYP154A1 haem contributes a significant role to this reaction mechanism [37]. It will be interesting to study the process of protein folding in both P450s in order to gain further insight into haem incorporation mechanisms.
(c). Different modes of substrate binding in P450s
C–C coupling reactions are key steps in the biosynthesis of many important antibiotics. For example, vancomycin biosynthesis, the last line antibiotic treatment for methicillin-resistant Staphylococcus aureus, requires three such steps catalysed by P450s [63]. Furthermore, both chloroeremomycin and balhimycin, antibiotics of the vancomycin family, contain P450-dependent biphenyl and biphenyl ether cross links [64]. S. coelicolor A3(2) contains a three-gene operon containing a type III polyketide synthase (PKSIII), CYP158A2 and a quinione-forming monooxygenase (momA). The PKSIII sequentially converts five molecules of malonyl CoA into 1,3,6,8-tetahydroxynapthalene, which is oxidized to flaviolin by momA. CYP158A2 subsquently catalyses C–C bond formation to polymerize flaviolin into the di- and trimer forms [65]. The crystal structure of CYP158A2 revealed important mechanistic features applicable to P450 coupling enzymes and P450 catalysis in general. CYP158A2 binds two molecules of flaviolin in its active site, and the crystal structure predicts that the 2-OH group of flaviolin plays a role in anchoring the substrate in the active site while the 5-OH and 7-OH stabilize water molecules important for catalysis [65]. The use of a substrate analogue, α-hydroxynapthalene (which is missing both the 5-OH and 7-OH), revealed 70-fold less activity in dimerization, thus suggesting the water molecules form a proton relay pathway to the bulk solvent [66]. During the general P450 catalytic cycle, dioxygen binding, protonation and splitting of the oxygen–oxygen bond are critical steps for product formation [4,5]. Two mechanisms have been described previously for the protonation step in catalysis. First, in CYP101A1, a hydroxyl group of a threonine residue in the P450 I helix hydrogen bonds to the dioxygen molecule in conjunction with a water molecule, with the water molecule providing the proton for catalysis [67]. This conserved threonine is found in the majority of P450 sequences. Second, in CYP107A1, the conserved threonine is not present and the crystal structure suggests that a hydroxyl group of the substrate, 6-deoxyerythronolide B, can directly donate a hydrogen bond to the Fe-linked dioxygen for the proton transfer [68]. Thus the role of active site water molecules and substrate hydroxyl groups in CYP158A2 described a third distinct mechanism of proton transfer for oxygen activation in the P450 catalytic cycle.
The S. coelicolor genome contains a second CYP158A gene, CYP158A1, where the gene product shares 61 per cent amino acid sequence identity with CYP158A2 [56]. It was found that CYP158A1 could catalyse the dimerization of flaviolin-like CYP158A2, but with differing regio-specificity [69]. The flaviolin-bound CYP158A1 crystal structure complex was different compared with that of flaviolin-bound CYP158A2, with the two molecules of flaviolin adopting different orientations in each P450 structure. One flaviolin molecule is positioned over the CYP158A1 haem similar to that found in CYP158A2, but the second flaviolin molecule is bound at the entrance to the substrate access channel in CYP158A1, thus presenting difficulties in understanding how CYP158A1 catalyses dimerization compared with CYP158A2 [69]. It has been proposed that conformational changes in CYP158A1, possibly through binding ferredoxin during the electron transfer process, allows for the second flaviolin to move closer to the first flaviolin molecule in order for the C–C bond to occur. Hence, different modes of substrate binding in two different P450 enzymes, catalysing the same biochemical reaction, is a unique observation for the P450 superfamily.
(d). Moonlighting cytochrome P450s and enzyme bifunctionality
The individual activities of P450s have been determined in only a small number of the 15 000 members of the superfamily. A moonlighting protein refers to a single protein whose normal structure has more than one function, which can arise via a number of factors including: same enzyme in a different cellular location; cofactor binding compared with the unbound form; oligomeric versus monomeric state of the enzyme; protein–protein complexes and interactions; expression of same enzyme in different cell types; and multiple enzyme active sites in a single protein molecule [70]. To date, only three P450s can be classified as being moonlighting P450 monooxygenases: eukaryotic CYP17A1 and CYP7B1, and prokaryotic CYP170A1 [71]. The first two P450s, CYP17A1 and CYP7B1, both have one enzymatic active site, but nonetheless are considered moonlighting as their catalytic reaction is changed dependent upon (i) protein interaction, as is the case for CYP17A1, or (ii) tissue location as described for CYP7B1. However, CYP170A1 is clearly a bifunctional P450 enzyme containing two enzyme active sites in a single P450 protein molecule.
Microsomal CYP17A1 plays an essential role in steroid hormone biosynthesis in animals. Specifically, in the gonads, CYP17A1 catalyses the conversion of pregnenolone to dehydroepiandrosterone and progesterone androstenedione in androgen biosynthesis, whereas in the adrenal cortex CYP17A1 hydroxylates pregnenolone at the 17α position producing 17α-hydroxypregnenolone, an intermediate in cortisol biosynthesis [72]. Unusually, this same P450 enzyme catalyses two different chemical reactions, dependent upon its cellular location. In the gonads, CYP17A1 carries out a three-step hydroxylation reaction converting the 21-carbon steroid to the 19-carbon androgen. However, in the adrenal cortex CYP17A1 only catalyses the first initial hydroxylation when the reaction stops [72]. It is believed, in the adrenal cortex, 17α-hydroxypregnenolone leaves the CYP17A1 active site and is further metabolized in the cortisol biosynthetic pathway. However, in the gonads, the product of the initial hydroxylation either remains in the CYP17A1 active site or leaves and then rebinds. The role of cytochrome b5 in the lyase reactions in the gonads appears to be pivotal and may alter CYP17A1 conformation, so that the further two hydroxylation steps (lyase) proceed [73]. CYP17A1 was the first moonlighting P450 to be discovered with two different activities dependent upon cellular location.
CYP7B1 was initially identified as a steroid 7α-hydroxylase with activity towards pregnenolone and dehydroepiandrosterone in the brain [74]. These steroid metabolites act as neurosteroids in the brain by allosterically modulating receptor function. Additionally, CYP7B1 is involved in bile acid synthesis in the liver using 25- and 27-hydroxycholesterol as substrates [74]. Furthermore, CYP7B1 metabolizes the 19-carbon steroid 5α-androstane-3β,17β-diol, which has been implicated in prostate shrinkage and ovarian failure [75]. Finally, CYP7B1 catalyses the conversion of dehydroepiandrosterone to 7α-hydroxy-dehydroepiandrosterone, a steroid that has been associated with causing chronic inflammation leading to rheumatoid arthritis [76]. Clearly the moonlighting activity of this P450 is dependent upon its tissue localization.
CYP170A1 is the first bifunctional P450 discovered, which contains two different active sites in the same P450 protein molecule. The sco5223 gene encoding CYP170A1 in S. coelicolor A3(2) is part of a two-gene cluster with a sesquiterpene cyclase gene (sco5222) with which it shares a 4-nt ATGA transcriptional overlap at its 5′-end. The sco5222 gene product has been shown to catalyse the synthesis of a novel sesquiterpene previously unseen in nature, epi-isozizaene, by cyclization of the universal sesquiterpene synthase substrate, farnesyl diphosphate (FPP). Using an in vitro assay, it was established that purified CYP170A1 carries out two sequential allylic oxidations to convert epi-isozizaene to an epimeric mixture of albaflavenols and ultimately to the single ketone sesquiterpene, the antibiotic albaflavenone [77]. Both epimers of albaflavenol as well as albaflavenone are produced by the wild-type strain, while none of these three metabolites can be detected in the extracts of the CYP170A1 knock-out strain. Hence, the in vivo observations verified the results of the in vitro experiments. Although there are numerous examples described of three-step oxidations of methyl groups to carboxylic acids that are catalysed by a single P450, the two-step oxidation of an allylic methylene to a conjugated ketone by a single P450 is unusual. Notably, in the biosynthesis of oxidized monoterpenes P450-catalysed allylic oxidation is normally followed by a conventional NAD(P)+-dependent dehydrogenation, as for example in the conversion of (–)-limonene to (–)-isopiperitenone by way of (–)-trans-isopiperitenol in peppermint catalysed by successive P450 and dehydrogenase enzymes [78].
During initial studies of the monooxygenase activity of CYP170A1, an unexpected additional catalytic activity was observed. Surprisingly, gas chromatography/mass spectrometry analysis of the products of the SCO5222, CYP170A1 and redox partners incubation mixture revealed the formation not only of both epimers of albaflavenol and the fully oxidized sesquiterpene antibiotic albaflavenone, but also the formation of (E)-β-farnesene (61%), (3E,6E)-α-farnesene (26%), (3Z,6E)-α-farnesene (6.8%), nerolidol (4.9%) and farnesol (1.8%). Subsequently it was shown that purified CYP170A1 alone can convert FPP to the farnesene mixture without the need for redox partners and NADPH but Mg2+ was essential for the reaction to proceed. Consequently, the crystal structure of CYP170A1 revealed the presence of a novel terpene synthase active site which is moonlighting on the normal P450 structure [79]. This includes signature sequences for divalent cation binding and an α-helical barrel. This barrel is unusual because it consists of only four helices rather than the six found in all other terpene synthases. Mutagenesis established that this barrel is essential for the terpene synthase activity of CYP170A1 but not for the monooxygenase activity [79]. The presence of two such distinct and unrelated biochemical activities in a single protein molecule is unprecedented within the P450 superfamily.
8. Conclusions and perspectives
While the identification of P450s by genome sequencing projects is exhilarating given the wonderful diversity of life found on Earth, it is also daunting because in the majority of cases the endogenous function and contribution to the biology of an individual organism is unknown. While this is not unexpected, it is surprising to find that more and more unusual properties of P450 enzymes are being found. This overview increases our expectation that many more novel properties of P450s will be discovered. Hence, we can expect to find additional novel properties of P450s as the number of different P450 gene families are studied in greater detail. However, it should be noted that only a small number of the approximately 15 000 P450 enzymes have been biochemically characterized, to date, and, in some instances, the unusual P450 properties described herein may actually be rather common within the superfamily.
Acknowledgements
This work was supported by a National Institutes of Health grant to M.R.W. (grant no. R01 GM69970) and by a Wellcome Trust Sabbatical Award (to D.C.L.).
References
- 1.Ortiz de Montellano PR. 2005. Cytochrome P450, structure, mechanism and biochemistry, 3rd edn New York, NY: Kluwer Academic/Plenum Publishers [Google Scholar]
- 2.Nelson DR, et al. 1996. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6, 1–42 10.1097/00008571-199602000-00002 (doi:10.1097/00008571-199602000-00002) [DOI] [PubMed] [Google Scholar]
- 3.Wickramasinghe RH, Ville CA. 1975. Early role during chemical evolution for cytochrome P450 in oxygen detoxification. Nature 256, 509–511 10.1038/256509a0 (doi:10.1038/256509a0) [DOI] [PubMed] [Google Scholar]
- 4.Schlichting I, et al. 2000. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287, 1615–1622 10.1126/science.287.5458.1615 (doi:10.1126/science.287.5458.1615) [DOI] [PubMed] [Google Scholar]
- 5.Rittle J, Green MT. 2010. Cytochrome P450 compound. I. Capture, characterization and C–H bond activation kinetics. Science 330, 933–937 10.1126/science.1193478 (doi:10.1126/science.1193478) [DOI] [PubMed] [Google Scholar]
- 6.Guengerich FP. 2001. Common and uncommon cytochrome P450 reactions related to metabolism and toxicity. Chem. Res. Toxicol. 14, 611–650 10.1021/tx0002583 (doi:10.1021/tx0002583) [DOI] [PubMed] [Google Scholar]
- 7.Lamb DC, Waterman MR, Kelly SL, Guengerich FP. 2007. Cytochrome P450 and drug discovery. Curr. Opin. Biotechnol. 18, 504–512 10.1016/j.copbio.2007.09.010 (doi:10.1016/j.copbio.2007.09.010) [DOI] [PubMed] [Google Scholar]
- 8.Paine MJI, Scrutton NS, Munro AW, Guiterrez A, Roberts GCK, Wolf CR. 2005. Electron transfer partners of cytochrome P450. In Cytochrome P450, structure, mechanism and biochemistry (ed. PR Ortiz de Montellano) pp. 115–148 New York, NY: Kluwer Academic/Plenum Publishers [Google Scholar]
- 9.Watanabe T, Narumiya S, Shimizu T, Hayaishi O. 1982. Characterization of the biosynthetic pathway of prostaglandin D2 in human platelet-rich plasma. J. Biol. Chem. 257, 14 847–14 853 [PubMed] [Google Scholar]
- 10.Song WC, Brash AR. 1991. Purification of an allene oxide synthase and identification of the enzyme as a cytochrome P450. Science 253, 781–784 10.1126/science.1876834 (doi:10.1126/science.1876834) [DOI] [PubMed] [Google Scholar]
- 11.Poulos TL, Finzel BC, Gunsalus IC, Wagner GC, Kraut J. 1985. The 2.6 Å crystal structure of Pseudomonas putida cytochrome P450. J. Biol. Chem. 260, 16 122–16 130 [PubMed] [Google Scholar]
- 12.Johnson EF, Stout CD. 2005. Structural diversity of human xenobiotic-metabolising cytochrome P450 monooxygenases. Biochem. Biophys. Res. Commun. 338, 331–336 10.1016/j.bbrc.2005.08.190 (doi:10.1016/j.bbrc.2005.08.190) [DOI] [PubMed] [Google Scholar]
- 13.Seybert DW, Lancaster JR, Lambeth JD, Kamin H. 1979. Participation of the membrane in the side chain cleavage of cholesterol. Reconstitution of cytochrome P450scc into phospholipid vesicles. J. Biol. Chem. 254, 12 088–12 098 [PubMed] [Google Scholar]
- 14.Narhi LO, Fulco AJ. 1986. Characterization of a catalytically self-sufficient 119 000-dalton cytochrome P450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 261, 7160–7169 [PubMed] [Google Scholar]
- 15.Miles JS, Munro AW, Rospendowski BN, Smith WE, McKnight J, Thomson AJ. 1992. Domains of the catalytically self-sufficient cytochrome P450BM-3. Genetic construction, overexpression, purification and spectroscopic characterization. Biochem. J. 288, 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafsson MC, et al. 2004. Expression, purification, and characterization of Bacillus subtilis cytochromes P450 CYP102A2 and CYP102A3: flavocytochrome homologues of P450 BM3 from Bacillus megaterium. Biochemistry 43, 5474–5487 10.1021/bi035904m (doi:10.1021/bi035904m) [DOI] [PubMed] [Google Scholar]
- 17.Nakayama N, Takemae A, Shoun H. 1996. Cytochrome P450foxy, a catalytically self-sufficient fatty acid hydroxylase of the fungus Fusarium oxysporum. J. Biochem. 119, 435–440 10.1093/oxfordjournals.jbchem.a021260 (doi:10.1093/oxfordjournals.jbchem.a021260) [DOI] [PubMed] [Google Scholar]
- 18.Seo JA, Proctor RH, Plattner RD. 2001. Characterization of four clustered and coregulated genes associated with fumonisin biosynthesis in Fusarium verticillioides. Fungal Genet. Biol. 34, 155–165 10.1006/fgbi.2001.1299 (doi:10.1006/fgbi.2001.1299) [DOI] [PubMed] [Google Scholar]
- 19.Lamb DC, et al. 2010. Streptomyces coelicolor A3(2) CYP102 protein, a novel fatty acid hydroxylase encoded as a heme domain without an N-terminal redox partner. Appl. Environ. Microbiol. 76, 1975–1980 10.1128/AEM.03000-09 (doi:10.1128/AEM.03000-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Correll CC, Batie CJ, Ballou DP, Ludwig ML. 1992. Phthalate dioxygenase reductase: a modular structure for electron transfer from pyridine nucleotides to [2Fe–2S]. Science 258, 1604–1610 10.1126/science.1280857 (doi:10.1126/science.1280857) [DOI] [PubMed] [Google Scholar]
- 21.De Mot R, Parret AHA. 2002. A novel class of self-sufficient cytochrome P450 monooxygenases in prokaryotes. Trends Microbiol. 10, 502–508 10.1016/S0966-842X(02)02458-7 (doi:10.1016/S0966-842X(02)02458-7) [DOI] [PubMed] [Google Scholar]
- 22.Hunter DJ, Roberts GA, Ost TW, White JH, Muller S, Turner NJ, Flitsch SL, Chapman SK. 2005. Analysis of the domain properties of the novel cytochrome P450 RhF. FEBS Lett. 579, 2215–2220 10.1016/j.febslet.2005.03.016 (doi:10.1016/j.febslet.2005.03.016) [DOI] [PubMed] [Google Scholar]
- 23.Jackson CJ, Lamb DC, Marczylo TH, Warrilow AG, Manning NJ, Lowe DJ, Kelly DE, Kelly SL. 2002. A novel sterol 14alpha-demethylase/ferredoxin fusion protein (MCCYP51FX) from Methylococcus capsulatus represents a new class of the cytochrome P450 superfamily. J. Biol. Chem. 277, 46 959–46 965 10.1074/jbc.M203523200 (doi:10.1074/jbc.M203523200) [DOI] [PubMed] [Google Scholar]
- 24.Lamb DC, Jackson CJ, Warrilow AG, Manning NJ, Kelly DE, Kelly SL. 2007. Lanosterol biosynthesis in the prokaryote Methylococcus capsulatus: insight into the evolution of sterol biosynthesis. Mol. Biol. Evol. 24, 1714–1721 10.1093/molbev/msm090 (doi:10.1093/molbev/msm090) [DOI] [PubMed] [Google Scholar]
- 25.Rylott EL, Jackson RG, Edwards J, Womack GL, Seth-Smith HM, Rathbone DA, Strand SE, Bruce NC. 2006. An explosive-degrading cytochrome P450 activity and its targeted application for the phytoremediation of RDX. Nat. Biotechnol. 24, 216–219 10.1038/nbt1184 (doi:10.1038/nbt1184) [DOI] [PubMed] [Google Scholar]
- 26.Jackson RG, Rylott EL, Fournier D, Hawari J, Bruce NC. 2007. Exploring the biochemical properties and remediation applications of the unusual explosive-degrading P450 system XplA/B. Proc. Natl Acad. Sci. USA 104, 16 822–16 827 10.1073/pnas.0705110104 (doi:10.1073/pnas.0705110104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rylott EL, Budarina MV, Barker A, Lorenz A, Strand SE, Bruce NC. 2011. Engineering plants for the phytoremediation of RDX in the presence of the co-contaminating explosive TNT. New Phytol. 192, 405–413 10.1111/j.1469–8137.2011.03807.x (doi:10.1111/j.1469–8137.2011.03807.x) [DOI] [PubMed] [Google Scholar]
- 28.Sabbadin F, Jackson R, Haider K, Tampi G, Turkenburg JP, Hart S, Bruce NC, Grogan G. 2009. The 1.5-Å structure of XplA-heme, an unusual cytochrome P450 heme domain that catalyzes reductive biotransformation of royal demolition explosive. J. Biol. Chem. 284, 28 467–28 475 10.1074/jbc.M109.031559 (doi:10.1074/jbc.M109.031559) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghisla S, Thorpe C. 2004. Acyl-CoA dehydrogenases. A mechanistic overview. Eur. J. Biochem. 271, 494–508 10.1046/j.1432-1033.2003.03946.x (doi:10.1046/j.1432-1033.2003.03946.x) [DOI] [PubMed] [Google Scholar]
- 30.Lamb DC, Lei L, Warrilow AG, Lepesheva GI, Mullins JG, Waterman MR, Kelly SL. 2009. The first virally encoded cytochrome P450. J. Virol. 83, 8266–8269 10.1128/JVI.00289-09 (doi:10.1128/JVI.00289-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawkes DB, Adams GW, Burlingame AL, Ortiz de Montellano PR, De Voss JJ. 2002. Cytochrome P450(cin) (CYP176A), isolation, expression, and characterization. J. Biol. Chem. 277, 27 725–27 732 10.1074/jbc.M203382200 (doi:10.1074/jbc.M203382200) [DOI] [PubMed] [Google Scholar]
- 32.Jenkins CM, Waterman MR. 1994. Flavodoxin and NADPH-flavodoxin reductase from Escherichia coli support bovine cytochrome P450c17 hydroxylase activities. J. Biol. Chem. 269, 27 401–27 408 [PubMed] [Google Scholar]
- 33.Nakahara K, Tanimoto T, Hatano K, Usuda K, Shoun H. 1993. Cytochrome P-450 55A1 (P-450dNIR) acts as nitric oxide reductase employing NADH as the direct electron donor. J. Biol. Chem. 268, 8350–8355 [PubMed] [Google Scholar]
- 34.Oshima R, Fushinobu S, Su F, Zhang L, Takaya N, Shoun H. 2004. Structural evidence for direct hydride transfer from NADH to cytochrome P450nor. J. Mol. Biol. 342, 207–217 10.1016/j.jmb.2004.07.009 (doi:10.1016/j.jmb.2004.07.009) [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Takaya N, Kitazume T, Kondo T, Shoun H. 2001. Purification and cDNA cloning of nitric oxide reductase cytochrome P450nor (CYP55A4) from Trichosporon cutaneum. Eur. J. Biochem. 268, 3198–3204 10.1046/j.1432–1327.2001.02206.x (doi:10.1046/j.1432–1327.2001.02206.x) [DOI] [PubMed] [Google Scholar]
- 36.Omura T. 2010. Structural diversity of cytochrome P450 enzyme system. J. Biochem. 147, 297–306 10.1093/jb/mvq001 (doi:10.1093/jb/mvq001) [DOI] [PubMed] [Google Scholar]
- 37.Cheng Q, Lamb DC, Kelly SL, Lei L, Guengerich FP. 2010. Cyclization of a cellular dipentaenone by Streptomyces coelicolor cytochrome P450 154A1 without oxidation/reduction. J. Am. Chem. Soc. 132, 15 173–15 175 10.1021/ja107801v (doi:10.1021/ja107801v) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yun CH, Ahn T, Guengerich FP, Yamazaki H, Shimada T. 1999. Phospholipase D activity of cytochrome P450 in human liver endoplasmic reticulum. Arch. Biochem. Biophys. 367, 81–88 10.1006/abbi.1999.1254 (doi:10.1006/abbi.1999.1254) [DOI] [PubMed] [Google Scholar]
- 39.Munro AW, Girvan HM, Munro AW. 2007. Variations on a (t)heme—novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 24, 585–609 10.1039/b604190f (doi:10.1039/b604190f) [DOI] [PubMed] [Google Scholar]
- 40.Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. 1997. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl Acad. Sci. USA 94, 8411–8416 10.1073/pnas.94.16.8411 (doi:10.1073/pnas.94.16.8411) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia C, Panda SP, Marohnic CC, Martásek P, Masters BS, Kim JJ. 2011. Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc. Natl Acad. Sci. USA 108, 13 486–13 491 10.1073/pnas.1106632108 (doi:10.1073/pnas.1106632108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venkateswarlu K, Lamb DC, Kelly DE, Manning NJ, Kelly SL. 1998. The N-terminal membrane domain of yeast NADPH-cytochrome P450 (CYP) oxidoreductase is not required for catalytic activity in sterol biosynthesis or in reconstitution of CYP activity. J. Biol. Chem. 273, 4492–4496 10.1074/jbc.273.8.4492 (doi:10.1074/jbc.273.8.4492) [DOI] [PubMed] [Google Scholar]
- 43.Lamb DC, Kim Y, Yermalitskaya LV, Yermalitsky VN, Lepesheva GI, Kelly SL, Waterman MR, Podust LM. 2006. A second FMN binding site in yeast NADPH-cytochrome P450 reductase suggests a mechanism of electron transfer by diflavin reductases. Structure 14, 51–61 10.1016/j.str.2005.09.015 (doi:10.1016/j.str.2005.09.015) [DOI] [PubMed] [Google Scholar]
- 44.Chun YJ, et al. 2007. Electron transport pathway for a Streptomyces cytochrome P450: cytochrome P450 105D5-catalyzed fatty acid hydroxylation in Streptomyces coelicolor A3(2). J. Biol. Chem. 282, 17 486–17 500 10.1074/jbc.M700863200 (doi:10.1074/jbc.M700863200) [DOI] [PubMed] [Google Scholar]
- 45.Sakaguchi M, Mihara K, Sato R. 1984. Signal recognition particle is required for co-translational insertion of cytochrome P450 into microsomal membranes. Proc. Natl Acad. Sci. USA 81, 3361–3364 10.1073/pnas.81.11.3361 (doi:10.1073/pnas.81.11.3361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Avadhani NG, Sangar MC, Bansal S, Bajpai P. 2011. Bimodal targeting of cytochrome P450s to endoplasmic reticulum and mitochondria: the concept of chimeric signals. FEBS J. 278, 4218–4229 10.1111/j.1742-4658.2011.08356.x (doi:10.1111/j.1742-4658.2011.08356.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anandatheerthavarada HK, Biswas G, Mullick J, Sepuri NB, Otvos L, Pain D, Avadhani NG. 1999. Dual targeting of cytochrome P4502B1 to endoplasmic reticulum and mitochondria involves a novel signal activation by cyclic AMP-dependent phosphorylation at ser128. EMBO J. 18, 5494–5504 10.1093/emboj/18.20.5494 (doi:10.1093/emboj/18.20.5494) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robin MA, Anandatheerthavarada HK, Biswas G, Sepuri NB, Gordon DM, Pain D, Avadhani NG. 2002. Bimodal targeting of microsomal CYP2E1 to mitochondria through activation of an N-terminal chimeric signal by cAMP-mediated phosphorylation. J. Biol. Chem. 277, 40 583–40 593 10.1074/jbc.M203292200 (doi:10.1074/jbc.M203292200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oesch-Bartlomowicz B, Oesch F. 2003. Cytochrome-P450 phosphorylation as a functional switch. Arch. Biochem. Biophys. 409, 228–234 10.1016/S0003-9861(02)00558-1 (doi:10.1016/S0003-9861(02)00558-1) [DOI] [PubMed] [Google Scholar]
- 50.Jo Corbin C, Mapes SM, Lee YM, Conley AJ. 2003. Structural and functional differences among purified recombinant mammalian aromatases: glycosylation, N-terminal sequence and kinetic analysis of human, bovine and the porcine placental and gonadal isozymes. Mol. Cell Endocrinol. 206, 147–157 10.1016/S0303-7207(02)00422-7 (doi:10.1016/S0303-7207(02)00422-7) [DOI] [PubMed] [Google Scholar]
- 51.Shimozawa O, Sakaguchi M, Ogawa H, Harada N, Mihara K, Omura T. 1993. Core glycosylation of cytochrome P-450(arom). Evidence for localization of N terminus of microsomal cytochrome P-450 in the lumen. J. Biol. Chem. 268, 21 399–21 402 [PubMed] [Google Scholar]
- 52.Miller TW, Shin I, Kagawa N, Evans DB, Waterman MR, Arteaga CL. 2008. Aromatase is phosphorylated in situ at serine-118. J. Steroid Biochem. Mol. Biol. 112, 95–101 10.1016/j.jsbmb.2008.09.001 (doi:10.1016/j.jsbmb.2008.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vilgrain I, Defaye G, Chambaz EM. 1984. Adrenocortical cytochrome P-450 responsible for cholesterol side chain cleavage (P-450scc) is phosphorylated by the calcium-activated, phospholipid-sensitive protein kinase (protein kinase C). Biochem. Biophys. Res. Commun. 125, 554–561 10.1016/0006-291X(84)90575-8 (doi:10.1016/0006-291X(84)90575-8) [DOI] [PubMed] [Google Scholar]
- 54.Zhang LH, Rodriguez H, Ohno S, Miller WL. 1995. Serine phosphorylation of human P450c17 increases 17,20-lyase activity: implications for adrenarche and the polycystic ovary syndrome. Proc. Natl Acad. Sci. USA 92, 10 619–10 623 10.1073/pnas.92.23.10619 (doi:10.1073/pnas.92.23.10619) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y, Liao M, Hoe N, Acharya P, Deng C, Krutchinsky AN, Correia MA. 2009. A role for protein phosphorylation in cytochrome P450 3A4 ubiquitin-dependent proteasomal degradation. J. Biol. Chem. 284, 5671–5684 10.1074/jbc.M806104200 (doi:10.1074/jbc.M806104200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamb DC, Skaug T, Song HL, Jackson CJ, Podust LM, Waterman MR, Kell DB, Kelly DE, Kelly SL. 2002. The cytochrome P450 complement (CYPome) of Streptomyces coelicolor A3(2). J. Biol. Chem. 277, 24 000–24 005 10.1074/jbc.M111109200 (doi:10.1074/jbc.M111109200) [DOI] [PubMed] [Google Scholar]
- 57.Rupasinghe S, Schuler MA, Kagawa N, Yuan H, Lei L, Zhao B, Kelly SL, Waterman MR, Lamb DC. 2006. The cytochrome P450 gene family CYP157 does not contain EXXR in the K-helix reducing the absolute conserved P450 residues to a single cysteine. FEBS Lett. 580, 6338–6342 10.1016/j.febslet.2006.10.043 (doi:10.1016/j.febslet.2006.10.043) [DOI] [PubMed] [Google Scholar]
- 58.Nelson DR. 2009. The cytochrome P450 homepage. Hum. Genomics 4, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leys D, Mowat CG, McLean KJ, Richmond A, Chapman SK, Walkinshaw MD, Munro AW. 2003. Atomic structure of Mycobacterium tuberculosis CYP121 to 1.06 Å reveals novel features of cytochrome P450. J. Biol. Chem. 278, 5141–5147 10.1074/jbc.M209928200 (doi:10.1074/jbc.M209928200) [DOI] [PubMed] [Google Scholar]
- 60.Belin P, et al. 2009. Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA 106, 7426–7431 10.1073/pnas.0812191106 (doi:10.1073/pnas.0812191106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Podust LM, Bach H, Kim Y, Lamb DC, Arase M, Sherman DH, Kelly SL, Waterman MR. 2004. Comparison of the 1.85 Å structure of CYP154A1 from Streptomyces coelicolor A3(2) with the closely related CYP154C1 and CYPs from antibiotic biosynthetic pathways. Protein Sci. 13, 255–268 10.1110/ps.03384804 (doi:10.1110/ps.03384804) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Podust LM, et al. 2003. The 1.92-Å structure of Streptomyces coelicolor A3(2) CYP154C1. A new monooxygenase that functionalizes macrolide ring systems. J. Biol. Chem. 278, 12 214–12 221 10.1074/jbc.M212210200 (doi:10.1074/jbc.M212210200) [DOI] [PubMed] [Google Scholar]
- 63.Zerbe K, et al. 2002. Crystal structure of OxyB, a cytochrome P450 implicated in an oxidative phenol coupling reaction during vancomycin biosynthesis. J. Biol. Chem. 277, 47 476–47 485 10.1074/jbc.M206342200 (doi:10.1074/jbc.M206342200) [DOI] [PubMed] [Google Scholar]
- 64.Süssmuth RD, Pelzer S, Nicholson G, Walk T, Wohlleben W, Ung G. 1999. New advances in the biosynthesis of glycopeptide antibiotics of the vancomycin type from Amycolatopsis mediterranei. Angew. Chem. Int. Ed. 38, 1976–1979 10.1002/(SICI)1521–3773(19990712)38:13/14<1976::AID-ANIE1976>3.0.CO;2-3 (doi:10.1002/(SICI)1521–3773(19990712)38:13/14<1976::AID-ANIE1976>3.0.CO;2-3) [DOI] [PubMed] [Google Scholar]
- 65.Zhao B, et al. 2005. Bindings of two flaviolin substrate molecules, oxidative coupling, and crystal structure of Streptomyces coelicolor A3(2) cytochrome P450 158A2. J. Biol. Chem. 280, 11 599–11 607 10.1074/jbc.M4109332000 (doi:10.1074/jbc.M4109332000) [DOI] [PubMed] [Google Scholar]
- 66.Zhao B, Guengerich FP, Voehler M, Waterman MR. 2005. Role of active site water molecules and substrate hydroxyl groups in oxygen activation by cytochrome P450 158A2. A new mechanism of proton transfer. J. Biol. Chem. 280, 42 188–42 197 10.1074/jbc.M509220200 (doi:10.1074/jbc.M509220200) [DOI] [PubMed] [Google Scholar]
- 67.Raag R, Martinis SA, Sligar SG, Poulos TL. 1991. Crystal structure of cytochrome P450CAM active site mutant Thr252Ala. Biochemistry 20, 9252–9253 10.1021/bi00112a008 (doi:10.1021/bi00112a008) [DOI] [PubMed] [Google Scholar]
- 68.Nagano S, Cupp-Vickery JR, Poulos TL. 2005. Crystal structures of the ferrous dioxygen complex of wild-type cytochrome P450eryF and its mutants, A245S and A245T: investigation of the proton transfer system in P450eryF. J. Biol. Chem. 280, 22 101–22 107 10.1074/jbc.M501732200 (doi:10.1074/jbc.M501732200) [DOI] [PubMed] [Google Scholar]
- 69.Zhao B, Lamb DC, Lei L, Kelly SL, Yuan H, Hachey DL, Waterman MR. 2007. Different binding modes of two flaviolin substrate molecules in cytochrome P450 158A1 (CYP158A1) compared to CYP158A2. Biochemistry 46, 8725–8733 10.1021/bi7006959 (doi:10.1021/bi7006959) [DOI] [PubMed] [Google Scholar]
- 70.Jeffery CJ. 1999. Moonlighting proteins. Trends Biochem. Sci. 24, 8–11 10.1016/S0968-0004(98)01335-8 (doi:10.1016/S0968-0004(98)01335-8) [DOI] [PubMed] [Google Scholar]
- 71.Zhao B, Waterman MR. 2011. Moonlighting cytochrome P450 monooxygenases. IUBMB Life 63, 473–477 10.1002/iub.501 (doi:10.1002/iub.501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yanase T, Simpson ER, Waterman MR. 1991. 17α-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocrine Rev. 12, 91–108 10.1210/edrv-12-1-91 (doi:10.1210/edrv-12-1-91) [DOI] [PubMed] [Google Scholar]
- 73.Katagiri M, Kagawa N, Waterman MR. 1995. The role of cytochrome b(5) in the biosynthesis of androgens by human P450c 17. Arch. Biochem. Biophys. 317, 343–347 10.1006/abbi.1995.1173 (doi:10.1006/abbi.1995.1173) [DOI] [PubMed] [Google Scholar]
- 74.Stiles AR, McDonald JG, Bauman DR, Russell DW. 2009. CYP7B1: one cytochrome P450, two human genetic diseases and multiple physiological functions. J. Biol. Chem. 284, 28 485–28 489 10.1074/jbc.R109.042168 (doi:10.1074/jbc.R109.042168). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weihua Z, Lathe R, Warner M, Gustafsson JA. 2002. An endocrine pathway in the prostrate, ERbeta, AR, 5α-androstane-3β,17β-diol and CYP7B1, regulates prostate growth. Proc. Natl Acad. Sci. USA 99, 13 589–13 594 10.1073/pnas.162477299 (doi:10.1073/pnas.162477299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dulos J, van der Vleuten MAJ, Kavelaars A, Heijnen CJ, Boots AM. 2005. CYP7B expression and activity in fibroblast synoviocytes from patients with rheumatoid arthritis: regulation by proinflammatory cytokines. Arthritis Rheum. 52, 770–778 10.1002/art.20950 (doi:10.1002/art.20950) [DOI] [PubMed] [Google Scholar]
- 77.Zhao B, Lin X, Lei L, Lamb DC, Kelly SL, Waterman MR, Cane DE. 2008. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor A3(2). J. Biol. Chem. 283, 8183–8189 10.1074/jbc.M710421200 (doi:10.1074/jbc.M710421200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Karp F, Mihaliak CA, Harris JL, Croteau R. 1990. Monoterpene biosynthesis: specificity of the hydroxylations of (−)-limonene by enzyme preparations from peppermint (Mentha piperita), spearmint (Mentha spicata), and perilla (Perilla frutescens) leaves. Arch. Biochem. Biophys. 276, 219–226 10.1016/0003-9861(90)90029-X (doi:10.1016/0003-9861(90)90029-X) [DOI] [PubMed] [Google Scholar]
- 79.Zhao B, Lei L, Vassylyev DG, Lin X, Cane DE, Kelly SL, Yuan H, Lamb DC, Waterman MR. 2009. Crystal structure of albaflavenone monooxygenase containing a moonlighting terpene synthase active site . J. Biol. Chem. 284, 36 711–36 719 10.1074/jbc.M109.064683 (doi:10.1074/jbc.M109.064683) [DOI] [PMC free article] [PubMed] [Google Scholar]