

Abstract
Single-molecule biology has matured in recent years, driven to greater sophistication by the development of increasingly advanced experimental techniques. A progressive appreciation for its unique strengths is attracting research that spans an exceptionally broad swath of physiological phenomena—from the function of nucleosomes to protein diffusion in the cell membrane. Newfound enthusiasm notwithstanding, the single-molecule approach is limited to an intrinsically defined set of biological questions; such limitation applies to all experimental approaches, and an explicit statement of the boundaries delineating each set offers a guide to most fruitfully orienting in vitro single-molecule research in the future. Here, we briefly describe a simple conceptual framework to categorize how submolecular, molecular and intracellular processes are studied. We highlight the domain of single-molecule biology in this scheme, with an emphasis on its ability to probe various forms of heterogeneity inherent to populations of discrete biological macromolecules. We then give a general overview of our high-throughput DNA curtain methodology for studying protein–nucleic acid interactions, and by contextualizing it within this framework, we explore what might be the most enticing avenues of future research. We anticipate that a focus on single-molecule biology's unique strengths will suggest a new generation of experiments with greater complexity and more immediately translatable physiological relevance.
Keywords: single molecule, DNA curtains, molecular heterogeneity
1. Introduction
The annals of molecular biology are written largely in the language of biochemistry and molecular genetics, with significant contributions from crystallography [1,2]. That these approaches finally grounded the gene as a physical entity and elucidated its fundamental function in the cell attests to their power in examining biological phenomena. The second half of the previous century saw cellular biology and classical genetics embrace the new understanding, such that our current conception of the cell—even in the context of whole organisms—can now be stated in molecular terms [3]. The advent of single-molecule biology has enabled research that delves still deeper into the molecular realm, engendering new conceptions of biology's elemental components.
Rotman [4] inferred the catalytic activity of individual molecules of β-d-galactosidase by measuring the accumulation of a fluorescent reaction product, and Hirschfeld [5] made the first optical observations of individual molecules. But it was not until technical advances in optics, data acquisition, microfluidics and fluorescent probes in the 1990s that single-molecule biology was established as a field in its own right [6,7] (for a review of modern single-molecule techniques, see [8–10]). However, the crucial factor was a dawning realization that observations of fluorescently labelled macromolecules could illuminate unique biological problems not accessible to other methodologies.
The anatomy of single-molecule biology emerges in useful relief with its conceptual contextualization, embedded as it is in the continuum of approaches applied to the exploration of biological macromolecules. Traditional biochemistry and molecular biology explore the chemical mechanisms and overt chemical consequences of biomolecular reactions. Single-molecule biology is a visualized and often time-resolved extension of biochemistry (figure 1), uniquely suited to detecting various forms of heterogeneity among a nominally identical sample of biological macromolecules and addressing whether any such heterogeneity is physiologically relevant. Furthermore, it explores how biological macromolecules integrate information—a complex property dictated by the set of physiological objectives that a cell must execute in order to survive and respond to its environment. A focus on these two strengths will guide in vitro single-molecule experimental design towards greater complexity and physiological interpretability.
Figure 1.

Understanding physiological complexity requires the study of molecular phenomena from multiple perspectives. The properties of biological macromolecules are often studied in bulk through structural biology, molecular biology, and biochemistry; their influence on the cell is studied through cell biology and genetics. Following this same pattern, single-molecule biology can be performed in vitro, allowing for subtle experimental manipulability, or in vivo, revealing the influence of cellular context. Uniquely, single-molecule approaches can sample various forms of heterogeneity within populations of biological macromolecules.
Information gleaned through in vitro approaches is often assumed or implied to be directly translatable to animate conditions: any such translation requires careful circumspection, and reference to in vivo conditions or results is crucial. Cell biology and genetics explore the whole-cell manifestations of a chemical or molecular property as it propagates through the complex network of physical, spatial and temporal interactions that define the living cell. In vivo single-molecule biology, the major topic of this themed issue, investigates biological macromolecules in this intricate setting: it is effectively a new form of cell biology (figure 1). We argue that a holistic grasp of the biological macromolecule will materialize from a dual single-molecule approach in which the manipulability of in vitro experiments will complement the direct physiological context of in vivo experiments.
A major limitation on real-time single-molecule approaches is sample size. The experiments are as technically challenging as ever; consequently, the acquisition of datasets large enough to push the field against new frontiers of inquiry has not yet become routine. Also, the manipulability of systems under in vitro single-molecule scrutiny—the investigator's range of options in defining and exploring the consequences of molecules’ microenvironments—has not yet reached its full potential. Our laboratory is seeking to overcome these checks on complexity in the study of protein–nucleic acid interactions by developing a robust and high-throughput system for visualizing parallel arrays of DNA molecules in real time. The platform is based on precisely nanofabricated microscope slide surfaces, a physiologically amenable passivating lipid bilayer, and signal-to-noise-ratio enhancing optics and fluorescent probes. This type of setup can be modified and expanded to encompass multi-component systems of a previously unattainable complexity, facilitating deeper examinations of the inherent heterogeneity in a population of biomolecules or set of biomolecular interactions.
2. A conceptual framework to guide the development of in vitro single-molecule biology
Briefly enunciating a conceptual framework that categorizes experimental molecular biology is useful in defining the place of single-molecule biology relative to other approaches, lending perspective to its strengths and informing future avenues of research. The following sections define conceptual categories, such that some experimental techniques prove applicable within multiple categories, whereas others fit into only one.
(a). Traditional biochemistry and molecular biology
Biological macromolecules, including proteins and nucleic acids, can in principle be defined as self-contained entities with a set of discrete chemical properties. These are general qualities such as molecular mass and isolelectric point, but also submolecular qualities dependent on structure, such as intermolecular contacts and the organization of catalytic active sites. Traditional biochemistry dominates analyses of chemical process and consequence: this type of work usually involves minimal-component systems (a purified enzyme and its substrate, for example) to reproducibly measure fundamental bulk chemical parameters and/or products. Historically, cellular extracts have also been used to great advantage in the absence of purified alternatives to study specific catalytic activities, such as DNA replication [11] and translation [12]. Biochemistry offers the most immediate and commonly used experimental toolkit in the study of biological macromolecules. For example, early work in the still-ongoing research to disentangle the nature of nucleosomes, and the roles played by their localization and compaction, characterized the components of chromatin with nuclease protection assays and spectroscopy [13]. More recently, Whitehouse and co-workers [14] studied the ATP-dependent nucleosome relocalization activity of the yeast SWI/SNF chromatin remodeller with straightforward and elegant radiolabelled-DNA gel experiments. They inferred SWI/SNF-catalysed shifts in the localization of a pre-positioned nucleosome relative to defined restriction sites by observing whether radioactivity appeared in a nucleosome-free band of DNA or a nucleosome-shifted band on a native gel. The accessibility and broad applicability of such techniques ensure their persistence.
Structural biology forms the other dominant thread in the evolution of modern molecular biology. The notion that macromolecular structure might explain chemical function was not entirely apparent at the discipline's inception, as it is today. The arduous solution of the first protein structures by X-ray crystallography in the late 1950s and early 1960s unconditionally justified the enterprise. Famously, the angstrom-scale configurations of haemoglobin and myoglobin gave satisfying physical form to their biochemically observed oxygenation curves [15–17]. Multimeric haemoglobin's highly efficient cooperative behaviour as it charges with oxygen in the lungs and discharges it in bodily tissues was revealed to involve a physical cooperativity between subunits: with oxygen binding in one precipitating structural changes that affect binding in another. Crystallography has become more routine as computing power and X-ray sources have improved, with high-resolution structures explaining how submolecular atomic contexts might determine chemical properties. The crystal structure of the nucleosome—all eight histone components in complex with DNA—revealed the set of intermolecular contacts that mediate DNA wrapping and cause perturbations to the regular helical form of DNA [18]. Such structures often prompt, and always inform, subsequent work: the discovery of precisely what mechanisms chromatin remodellers use in shifting nucleosomes will necessarily reference known structures.
Reliant on highly ordered crystals, crystallography is static by nature. Reaction pathways can be explored to some extent by the capture of intermediate states, in the case of enzymes with non-catalysable substrates, for example. Illustratively, Wu & Beese [19] crystallized a bacterial DNA polymerase bound to a defined DNA construct, mimicking template-dependent synthesis; by supplying a nucleotide that would be mismatched to the next template base, they captured a novel ‘ajar’ polymerase conformation in between the ‘open’ and ‘closed’ states of the catalytic site. This intermediate conformation is hypothesized to ‘test’ incoming nucleotides and increase the probability of a correct selection. Nuclear magnetic resonance (NMR) is better suited to extracting the conformational space accessed by a macromolecule in response to thermal fluctuations. NMR analysis, as applied to biological macromolecules, yields an array of constraints on interatomic distances, which can then be modelled into structures, given additional information about bond angle restrictions. The result is a set of allowed conformational states that reflects both experimental error and the range of theoretically permissible fluctuations sampled in solution. One example is the NMR structure of the KH DNA-binding domain of a transcriptional regulator, notable for capturing the peptide in complex with its single-stranded DNA (ssDNA) target—an unusual substrate for transcription factors and therefore involving unique amino acid-DNA interactions [20]. Assigning specific NMR conformations to specific reaction states along a pathway can be ambiguous, though powerful advances are being made [21]. NMR yields lower-resolution structural information than crystallography, and generally cannot accommodate molecules above tens of kilodaltons. Cryogenic electron microscopy (cryo-EM) is routinely applied to large macromolecules, and it too can reveal fluctuations in conformational space by capturing—literally freezing—induced or intrinsically varying subpopulations. For example, cryo-EM has been used to study the global shifts of the ribosome's subunits as tRNAs are translocated [22]. There have recently also been advances in time-resolved EM, but this is still a developing field [23]. The usefulness of each structure-examining technique depends on the size of the molecule in question and the resolution needed to extract biological relevance.
(b). Single-molecule biology and the nature of heterogeneity among biological macromolecules
Single-molecule biology is often touted as a means to monitor molecular heterogeneities, including transient functional intermediates. Although this is true, the nature and potential significance of such heterogeneities are rarely dissected. Yet such dissection is necessary, even if in minimal terms, to inform how the field can best leverage its capabilities.
As a hypothetical illustration, consider a mole of identical organic small molecules in solution. Each molecule occupies any one of a large set of conformational states separated by small barriers so that it interconverts freely among them at room temperature. Now assume that each state endows the molecule with a unique, visible colour. A measurement of the exact colour of the entire solution, containing Avogadro's number of molecules, will yield an average colour of the ensemble. Now imagine that the colour of just one molecule could be tracked against time: intuitively, one might conclude that on some timescale—the ‘relaxation time’ of the system, dependent on the interconversion rate between states—this one molecule will sample all the thermally accessible states. Therefore, its average colour will be identical to the average colour of the solution. That this equivalence between the ensemble average and the time average of some intrinsically fluctuating variable ideally exists is termed the ‘ergodic hypothesis’, and systems that obey it are ergodic [24].
In any real system, the barriers between states and the wells defining them will not be energetically equal, and will consequently be occupied in varied proportions by the ensemble at any moment in time: such ‘static disorder’ is the minimal amount of heterogeneity in a population of molecules in solution. A biological macromolecule not only accesses a significantly larger state-space than a simple organic molecule, but the ruggedness of the associated landscape is also more pronounced: some degree of static disorder can safely be assumed. What is more, as the complexity of a system grows the energy landscape itself may fluctuate in time, giving rise to dynamic disorder [25,26]; such dynamic disorder probably applies to many systems in vivo [27]. These energy landscapes are manifestations of submacromolecular structural fluctuations. Any reasonably stable conformational states involving global rearrangements of macromolecular structure can be identified by static single-molecule techniques like cryo-EM, or atomic force microscopy (AFM), which maps gross three-dimensional structure by physically scanning over surface-immobilized macromolecules. Frequently sampled submolecular conformational states, like the thermal fluctuation of an exposed α-helix in a polypeptide, may be reflected in the disorder of a crystal structure, the density distribution of a cryo-EM image, or in the variability of an NMR reconstruction. Here too there is an averaging, or blurring, across conformational modes, and it is difficult if not impossible to tell in what proportions the macromolecules are partitioned across states or on what timescales they interconvert, if at all.
A somewhat arbitrary but convenient division can be drawn between two major classes of static disorder: (A) the energy barriers between states are easily and rapidly traversable due to thermal fluctuations, or (B) the energy barriers between states, or some subset of states, are sufficiently prominent to prevent or mitigate interconversions over the course of physiologically relevant relaxation times. In a strict sense, both (A) and (B) maintain ergodicity, but in a biological system, (B) may prove effectively non-ergodic. Only a time-resolved single-molecule approach can establish the non-ergodicity of a system because it is necessary to show that individual molecules continuously occupy distinct states. The deep energetic furrows in (B) can result from linearly identical macromolecules folding into energetically distinct conformations, generating effectively non-interconvertible subpopulations. In the cell, multiple copies of the same macromolecule may be non-equivalently altered. In any bulk assay, the different contributions of each subpopulation would be averaged out, while a static structural study might reveal their presence but tell little or nothing of their dynamics or potentially non-ergodic behaviour.
Among single-molecule methods, single-molecule fluorescence resonance energy transfer (smFRET) is particularly adept at tracking nanometre-scale conformational changes in real time. The technique relies on the highly sensitive distance-dependent efficiency of energy transfer between two fluorescent probes as their relative distance changes [10]. An smFRET study by Ditzler et al. [28] revealed the non-ergodicity of a small ribozyme. A FRET signal associated with the RNA's ‘docked’ and ‘undocked’ states demonstrated the persistence of heterogeneous distributions as the two subpopulations were found not to interconvert, even within larger and kinetically distinct subpopulations. A more tangible example of non-ergodic behaviour was uncovered by Park et al. [29] in a study of the bacterial PcrA translocase. They used a FRET signal between the two ends of a reporter ssDNA to follow the protein ‘reel in’ the reporter. The researchers were especially interested in establishing the step size taken by the molecule as it motors along ssDNA. This is a parameter that may speak to some fundamental mechanistic attributes of a given translocase, but has not been established for most. They found that earlier bulk studies averaged the differing translocation rates of distinct PcrA subpopulations, thereby overestimating step size. Their single-molecule assay distinguished individual molecules with persistently differing translocation rates, but each using the same step size (incidentally, of one nucleotide per step). Both translocation rate, obviously a function of time, and the non-ergodicity of the system could only have been discovered with a time-resolved single-molecule approach.
There are also heterogeneities associated with the chemical processes experienced or catalysed by biological macromolecules, and these can be understood on the same basic principles described earlier. Consider a chemical reaction from the substrate's point of view. Refer back to the hypothetical colour-morphing molecules and imagine that only the blue state serves as substrate to the enzyme B, which catalyses some chemical modification that shunts its substrate onto a new energy landscape (figure 2). B itself also inhabits a landscape, and the active site is competent for binding at only one point or region along it, signifying some relatively high affinity for blue. Finally, there is a third landscape associated with the process of catalysis, during which active site and substrate are in close vicinity, and then fully bound together. Importantly, B will probably have some lower affinity for the product than the initial substrate: the basis of enzymatic turnover. So, static disorder of interactants determines the occurrence of any given biochemical process. When two molecules interact in some physiologically meaningful way, a particular state of one is targeted by a particular state of the other. And at their physical and energetic intersection, the resultant complex falls onto a third landscape. (Countless variations of this unit are conceivable.) Although this illustration of the molecular perspective is oversimplified, it is still more complex than the picture taken by bulk biochemistry, in which these inherent dynamics are collapsed into one free-energy diagram as a function of the reaction coordinate. Actually characterizing energy landscapes in fine, time-resolved detail is possible to some degree with NMR-derived techniques [30,31], but remains essentially unassailable especially when biological macromolecules are involved; indeed, it is often unclear how such information should be interpreted biologically. However, these phenomena cause stochasticity, a single-molecule observable that amounts to heterogeneity in a reaction's occurrence in time.
Figure 2.
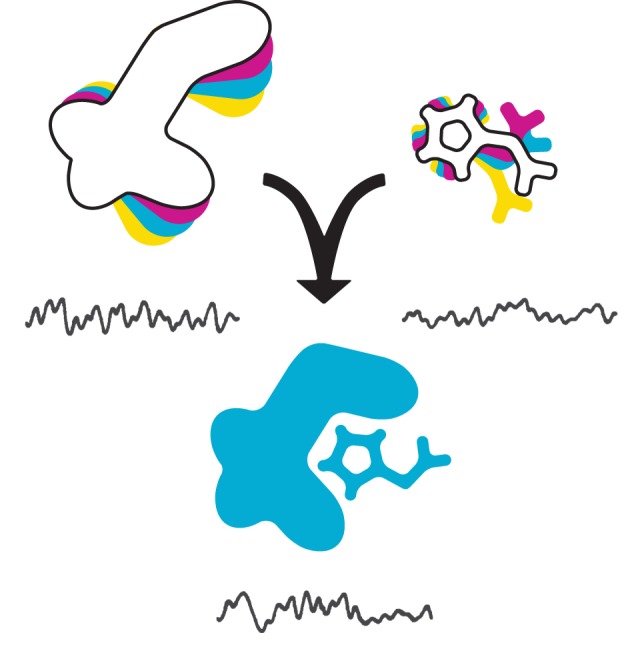
All molecules in solution inhabit an energy landscape owing to conformational fluctuations. A reaction between an enzyme (left) and a substrate molecule (right) depends on their physical interaction in space, and on their occupancy of the correct conformational states required for a physiological interaction. The reaction itself can also be described as yet another landscape. If the energy landscape of a particular species is deeply furrowed, it can give rise to static disorder. These characteristics of reaction and interaction processes also cause stochasticity, the probabilistic distribution of whether or not a reaction or interaction occurs.
Reactions are often depicted as smoothly progressive, but this is deceptive. If substrate is instantaneously added to a pool of B, then on some timescale that is short relative to the bulk reaction time, only a fraction of B enzymes will have actually engaged blue. This is an absolute requirement imposed by the energy landscapes discussed earlier, two of which must intersect for two molecules to interact productively. Consequently, whether or not a specific single molecule of B will bind blue after the bulk reaction is initiated will appear ‘random’, or stochastic, with a characteristic distribution of ‘molecules engaged’ versus time. Note that stochasticity applies to all chemical reactions—each step of each reaction in fact, even under synchronized and saturating conditions. On this front too, smFRET has made some headway [10], and for reasons addressed below, other single-molecule methods are also ideally suited to its study.
(c). Into the cell
Biochemistry, molecular biology, and in vitro single-molecule biology are all motivated by reductionism, the notion that by studying constituent elements of a system, the whole may be understood. This approach has undeniably allowed us to glimpse the inner workings of the cell, but the cell itself remains out of reach for its immense complexity. In vivo techniques preserve this living complexity and attempt to follow molecular phenomena as they propagate through incredible networks of physical, spatial and temporal interactions. In the context of our framework, these elements can be taken as forms of heterogeneity—the most elaborate kind. Classical genetics is founded on the idea that discrete genetic disruptions produce specific and overt consequences in a cell or organism, though ‘it is important to remember that mutant phenotypes provide information about the mutant, from which the behavior of the WT [wild type] is inferred’ [32, p. 928]. Molecular genetics has extended this idea to include the intervening molecular processes. Cell biology, in turn, is more difficult to unify as a field, but it is generally typified by direct or indirect observations of major cellular events, often in real time through optical microscopy.
Many single-molecule techniques rely heavily on fluorescent probes, while various in vivo disciplines built around the classical model organisms also embrace them [33]. For example, green fluorescent protein and its variants have been central to researching Drosophila development and Caenorhabditis elegans neurology, among many other problems [34]. Perhaps unsurprisingly, the two fields are being fused and an in vivo single-molecule biology has emerged. This topic is expressly and extensively explored by some of the reviews and articles in this themed issue. Here, we highlight only the work of Weigel et al. [35], as it is one of a few demonstrated cases of ergodicity breaking in any molecular biological system [36,37], all the more impressive for having been discerned in human tissue culture. The above discussion intimates that non-ergodicity arises only from pronounced static disorder, a persistent heterogeneity in a population of biological macromolecules. However, in the case of diffusion, on a cell surface for example, certain physical processes can disrupt molecular movement and cause anomalous diffusion that is either faster or slower than that expected for a strictly Brownian particle [35,38–40]. In some cases, anomalous diffusion also betrays non-ergodicity. In their study, Weigel and colleagues [35] tracked the movement of a fluorescently tagged surface transmembrane protein at the single-molecule level and observed both ergodic and non-ergodic subpopulations; they then found that the non-ergodic behaviour is abolished by the disruption of actin, a cytoskeletal component. Their results are consistent with a protein that sometimes, perhaps transiently, interacts with the cytoskeleton. The consequent pattern of movement can be modelled by ‘diffusion on a fractal’, which occasionally traps diffusing molecules in dead-ends. Only recently, any attempt at the derivation of fine single-molecule detail from an in vivo study would have seemed impracticable, but these kinds of experiments are clearly breaking new ground. In vitro single-molecule biology is poised to join this exciting new movement towards greater complexity, and bring with it technical powers beyond the reach of other methods.
3. DNA curtains: a high-throughput technique for the study of protein–DNA interactions
Chemical properties enable a biological macromolecule to carry out its biological functions, but these are not equivalent. Biological function is dictated by physiological imperative, the molecule executing commands defined by its structure, cellular localization, concentration and interactions with other molecules. For example, a particular restriction enzyme possesses a set of chemical properties that enables it to cleave a specific pair of covalent linkages along intertwined DNA backbones, but its biological function is to find and destroy DNA foreign to the bacterial cell. We conceive of biological macromolecules as hubs that actively integrate streams and packets of information to accurately and effectively accomplish their biological functions, and this principle can generally guide the time-resolved in vitro single-molecule study of protein–DNA interactions. In this section, we describe the major characteristics of our technique and illustrate how it has been used to monitor information exchange.
(a). Microscopy and curtainography
Several platforms have been developed for the single-molecule visualization of DNA-binding proteins [41], but these are generally circumscribed by a one-experiment-one-data-point limit to throughput. The most pronounced quality of the DNA curtain methodology is that each experiment offers hundreds of aligned DNA strands in defined orientation and density. The involved details of the full setup have been described elsewhere [42,43], and here we focus only on the signature elements.
Reactions occur within a microfluidic ‘flowcell’, which consists of a thin, low volume (approx. 10–50 μl) chamber defined by a microscope slide and a coverslip fused together by double-sided tape with an excised rectangular channel (figure 3a). Nanoports allow for the introduction of buffer flow through the chamber at a defined rate with the aid of precision syringe pumps. The first challenge with any such approach is to passivate the experimental surface—in this case, the face of the microscope slide within the chamber—to prevent the non-specific deposition of molecules under investigation. Instead of applying a synthetic polymer or large quantity of protein, a lipid bilayer is generated on the slide surface by the injection of lipid micelles [45]. A lipid bilayer has two major advantages: (i) as membranes are cellularly ubiquitous, they are in principle amenable to most biological macromolecules, and, as will be discussed below, to physiological conditions, and (ii) a lipid bilayer is two-dimensionally fluid, which is absolutely necessary for the generation of ordered DNA curtains.
Figure 3.

DNA curtains consist of hundreds of individual DNA strands aligned in parallel. Single-molecule experiments are performed within microfluidic ‘flowcells’ (a) that allow buffer exchange. (b) DNA strands linked to a lipid bilayer are lined up along nanofabricated barriers by buffer flow, as illustrated in the schematic where DNA is depicted with nucleosomes. (c–e) Images of QD-labelled nucleosomes (magenta) on fluorescently stained DNA (green) curtains. Four barriers are visible in this field of view, marked B1–B4. In the presence of flow, the DNA and associated proteins are stretched into a shallow excitation field generated by TIRF (c). When flow is turned off, the DNA diffuses out of the excitation field (d); the remaining fluorescence betrays the location of the nanofabricated barriers, which become non-specifically coated with proteins and the associated fluorescent labels. Adapted with permission from Visnapuu & Greene [44].
Reproducibly manipulating multiple individual strands of DNA on a surface with any precision is a significant technical challenge, but the fluidity of the bilayer allows an elegant solution. A small subpopulation of the lipid head-groups are modified with biotin, a small organic molecule. Following lipid deposition, streptavidin is flowed into the reaction chamber: each subunit of this homotetrameric protein binds biotin tightly. Finally, DNA biotinylated on one end is added so that individual strands become linked to the bilayer through a biotin–streptavidin–biotin–lipid interaction. We prefer λDNA for our work because at approximately 48 kbp it is conveniently long, can be easily made and modified with classic phage cloning techniques, and carries complementary 12-bp overhangs so that commercially available tagged oligos can be used to differentially label the DNA ends. In the presence of buffer flow, hydrodynamic force pushes the two-dimensionally unrestricted strands of DNA, but their immobilization for visualization requires local bilayer disruption.
The fluidity of the bilayer depends on a smooth substrate, in this case the slide surface. Obstructions break this fluidity so that individual lipids, and any DNA molecule that may be attached, cannot pass [46]. Our laboratory has developed a nanofabrication procedure to pattern obstructions in the form of precisely organized nanometre-scale chrome barriers [47,48]. Briefly, the slide is coated with a polymer sensitive to electrical disruption then overlaid with a water-soluble conducting material. An electron-beam lithographer (simply a different usage of the electron microscope) ‘writes’ a design of choice into the polymer, which is then developed to reveal a negative of the desired pattern. Chrome is evaporated onto this negative, and excess polymer and chrome stripped to leave behind just the barriers. Following bilayer deposition, any lipid linked to DNA that encounters a barrier will snag, and the associated DNA will be stretched by flow into the evanescent field (see below). Each barrier can align tens or hundreds of precisely arranged individual DNA strands in the same relative linear orientation: a DNA curtain (figure 3b). A simple modification to pattern design can also be used to generate ‘double tethered’ curtains that remain aligned and properly arranged in the absence of buffer flow [48] (figure 4a).
Figure 4.

‘Double-tethered’ DNA curtains are sustained without buffer flow, as shown schematically in (a), and demonstrated in (b) where the DNA is fluorescently stained green. The lower panel in (b) shows such a curtain with QD-labelled Mlh1–Pms1 mismatch repair protein complexes. Individual complexes diffuse along DNA, and can bypass other proteins. (c) A kymogram of one unlabelled DNA strand with three nucleosomes (N) and several diffusing magenta QD-labelled Mlh1–Pms1 complexes. Note how Mlh1–Pms1 can bypass nucleosomes. Adapted with permission from Gorman et al. [49].
The near-surface localization of DNA also solves another major challenge to all fluorescent microscopy because it can be harnessed to minimize background signal. The setup is designed around total internal reflection fluorescence microscopy (TIRFM), a selective illumination method. A laser beam is directed at the interface between the slide surface and the buffer immediately beneath at or greater than the critical angle determined by the optical densities of glass and water so that the beam is totally reflected. An evanescent field penetrates the buffer beneath the interface to a shallow depth of a few hundred nanometres. Consequently, only fluorophores specifically within the small volume defined by this penetration depth are excited, whereas those in bulk solution remain dark. The selective excitation of labels bound to the surface-localized DNA significantly increases the signal-to-noise ratio. Furthermore, we generally use antibody-conjugated quantum dots (QDs) to track proteins or DNA. These fluorophores are highly photostable for many minutes, or even hours, their absorption spectra are broad but emission spectra narrow (meaning that a single illumination wavelength can be used to simultaneously excite multiple coloured QDs within a single experiment), and their quantum yields are exceptionally high and therefore well above ambient background signals [50]. The DNA itself can also be visualized with intercalating fluorescent dyes, such as YOYO-1, but this method requires the simultaneous use of an oxygen scavenging system to minimize DNA breakage by reactive oxygen species. Photons are collected with a CCD camera, and data analysed with a variety of commercially and freely available tracking software [42,43].
(b). When DNA curtain and protein meet
The DNA curtain methodology occupies a precise place within the conceptual framework. It is obviously not designed to directly access conformational changes experienced by a biological macromolecule either as it samples the static disorder landscape or undergoes a reaction, but rather tracks the wide range of functional changes that result from such fluctuations. It can follow events in real time with millisecond temporal resolution and tens-of-nanometres spatial resolution. Importantly, it is adept at detecting ‘information integration’, as described below. These and other as-yet unexploited strengths will prove pivotal in the formulation of future research.
Besides genetic information, DNA carries another type of information for targeting: sequence elements that are specifically bound by DNA-interacting proteins, which in turn may recruit additional factors. Whether or not nucleosomes respond to DNA sequences in a physiologically relevant way is highly contentious. Side-by-side in vivo and in vitro mapping of nucleosome positioning has shown that although they exhibit intrinsic local and global sequence preferences, these preferences can be disrupted by chromatin remodellers [51]. This issue was the focus of a DNA curtain study by Visnapuu & Greene [44]. First, the pattern of yeast nucleosome positioning (figure 3c) was compared with various in silico models based only on DNA sequence, with the result that nucleosomes do respond to an intrinsic energy landscape, that their complement of contacts with DNA promotes a predictable sequence-dependence. Eukaryotes harness many histone variants to mark nucleosomes as specific to certain chromosomal structures, or sites of damage, for example; as nucleosome composition can be absolutely controlled in vitro, but not in vivo, our methodology can directly assess the role of such variants in altering nucleosomal sequence preferences. Therefore, they tested the effect of a centromere-specific histone variant to find, surprisingly, that it did not alter the positioning pattern. The addition of another protein, a histone chaperone, allowed these nucleosomes to overcome an intrinsic repulsion to the AT-rich sequences that predominate in yeast centromeric DNA. By substituting a native human genetic locus for the substrate DNA, they then found that nucleosomes were highly positioned near regulatory regions without the aid of any additional factors, suggesting an evolutionary pressure on these sequences to intrinsically position nucleosomes. This work demonstrates many of the information streams supplied by the cell: DNA sequence, the subunit composition of relatively large macromolecular complexes and auxiliary protein factors. The room for insights by varying the supplied information is also apparent, illustrating how single-molecule techniques reveal the range of functional capabilities intrinsic to a biological macromolecule, and how this range can be systematically restricted by the introduction of greater and greater physiological inputs.
Nucleosomes served as a physiologically relevant impediment to protein diffusion along DNA in a subsequent study [49]. The DNA landscape in vivo is crowded by hosts of proteins, and factors that travel along DNA must be able to navigate these bulky molecules as well as the DNA they presumably track. The initial steps of the yeast mismatch repair (MMR) pathway involve two protein pairs: Msh2–Msh6, which diffuses along DNA in a one-dimensional search for mismatches, and Mlh1–Pms1, which also diffuses along DNA to locate mismatch-bound Msh2–Msh6. As flow forces can disrupt diffusive processes, this work used the double-tethered curtains (figure 4a,b), and multiple colour QDs were used to simultaneously observe protein movement and relative nucleosome position. Such simultaneous traces clearly showed that Msh2–Msh6 cannot readily bypass nucleosomes, whereas Mlh1–Pms1 can (figure 4c). The former has been hypothesized to track with the replisome, which presumably reduces the load of obstructive macromolecules on nascent DNA, while the latter need only locate its target after replication. Also, it is reasonable that the component responsible for recognizing such a small error as a single mismatch should track the DNA closely, whereas the component responsible for then recognizing the first responder, which is an entire protein complex, should diffuse more freely on DNA. A small subpopulation of Msh2–Msh6 complexes was able to bypass nucleosomes (36 of 1000 molecules), though if and how this functional heterogeneity bears on MMR will be the subject of future work.
A concurrent study explored the disruption of DNA-bound proteins by the ATP-dependent bacterial translocase RecBCD [52]. Its in vivo function is to generate 3′ ssDNA overhangs to serve as an early substrate in homologous recombination during damage repair or replication fork restart. In its native cellular environment, RecBCD is likely to encounter other proteins on DNA, so the experiments challenged its movement with a variety of ‘roadblocks’. Because RecBCD is an exonuclease, the curtain assay could register its activity as the ATP-dependent shortening of individual DNA strands labelled along their length with an intercalating dye; roadblocks were tagged with QDs, and DNA-QD colocalization signalled a protein bound to DNA, while the loss of a QD signalled dissociation of a protein from DNA. RecBCD could evict RNA polymerase, EcoR1 (mutant for nuclease activity), the lactose repressor (LacI) (figure 5), and even nucleosomes. Nucleosomes are not a physiologically relevant roadblock in this collision scenario, but their eviction shows that RecBCD probably clears roadblocks by mechanical force rather than by specifically evolved protein–protein contacts, and that such force can completely disrupt the histone core proteins’ contacts with DNA. With the exception of LacI, which dissociated immediately from DNA within the resolution limit, RecBCD frequently pushed the other roadblocks some distance before causing them to dissociate. Single-molecule observations of dissociation constants in the presence and absence of RecBCD, and of RecBCD velocity (which scales with ATP concentration) enabled the proposition and testing of several models for eviction: again with the exception of LacI, the other roadblocks were cleared by undergoing repetitive steps of non-specific binding while being pushed, with an intermediate state between each step that binds relatively weakly. This model predicts the observed independence of translocation velocity and the distance a roadblock is pushed before eviction. These findings could not have been inferred without access to the behaviour of individual molecules, and without such exquisite control over the information handled by the various proteins, including DNA sequence, ATP concentration, and nature and density of RecBCD-encountered roadblocks.
Figure 5.

The ATP-dependent directional translocase/exonuclease activity of RecBCD can be followed by tracking the shortening of a DNA strand as it is digested. The upper kymogram shows RecBCD encountering a magenta QD-labelled EcoR1 that is mutant for restriction activity. EcoR1 is pushed by RecBCD all the way to the nanofabricated barrier at the leading end of the DNA. The lower kymogram shows a similar experiment with the Lac1 repressor instead of EcoR1. Lac1 repressor is evicted from the DNA, and RecBCD continues translocating. Adapted with permission from Finkelstein et al. [52].
4. Future directions
The construction of penetrating knowledge about the dynamic and intricate makeup of the cell requires both cellular level and molecular level information. In vivo studies of biological macromolecules attempt to grasp physiologically relevant conditions, but these conditions are often so abstruse in detail that fully interpreting results requires finer-scale approaches. In vitro studies revolve around the biological macromolecule itself and can more easily modulate environmental inputs, but here the drive for physiological relevance requires a shift towards greater complexity. This dichotomy is playing out on a minute scale among single-molecule methodologies.
Two veins of single-molecule biology converge on physiological complexity (figure 1). The approach can be taken to the cell, with experiments conducted in vivo. This newfound field will no doubt develop rapidly and contribute to our understanding of the biological macromolecule, but it cannot supplant its in vitro counterpart. The in vivo setting leaves target molecules under the influence of all native information streams—painting the truest picture of how the target is meant to behave—and yet for this same reason, the placement of the target molecule within the cellular network may prove difficult to tease apart without crucial contributions from other experimental approaches. In particular, the manipulability of in vitro single-molecule biology allows it to parse the information stream that a biological macromolecule might encounter in vivo. But to capitalize on this capacity, in vitro single-molecule biology must aspire as a discipline to greater information flux within experiments as a route to greater physiological complexity. The curtain approach and similar experimental configurations are poised to lead the way.
Curtain experiments with naked DNA provide an unrestricted substrate for DNA-interacting proteins, and much can be learned about the intrinsic character of such interactions, especially when they involve movement, as with the MMR proteins. The more recent work with roadblocks has demonstrated the feasibility of complicating the substrate and that interactions between proteins, each DNA-bound, can be analysed. One of the most interesting and common in vivo deviations from naked DNA is eukaryotic chromatin; so the formation of nucleosome curtains immediately suggests interesting future research. Importantly, the setup can accommodate DNA significantly longer than just the lambda substrate, which is essential because in contrast to sparse nucleosome arrays, chromatin is highly compact. One can even envision curtains composed of intact chromosomes. Also, eukaryotic DNA-binding proteins navigate chromatin, displace or shuffle nucleosomes, and/or interact directly with histones; the consequences of these processes, or the consequences of disrupting them, have been assayed in bulk and by genetics, but the mechanisms and functional intermediates remain unknown. Except for the basic unit of the nucleosome, and the canonical ‘beads-on-a-string’ found in relaxed chromatin, the higher in vivo organization of chromatin remains in question, with models ranging from 30-nm fibres to arrays of 10-nm fibres [53]. The disconnect between the excellent body of work done on nucleosomes and the uncertainty surrounding the form of in vivo chromatin highlights the space available to real-time single-molecule exploration, which may illuminate both the characteristics of chromatin and the dynamics of its formation.
Certain aspects of DNA metabolism, and certain structures in eukaryotic chromosomes involve large assemblages of many different proteins. DNA replication, double-strand break repair, telomeres and centromeres are outstanding examples. The constituents have been identified piecemeal by genetics and associations in bulk biochemical assays, but temporal and organizational relationships are incredibly difficult to formulate this way. These ultrastructures can be considered functional units and may not simply amount to the ‘sum of their parts’. A whole kinetochore complex, for example, is absolutely intransigent to fine structural analysis, though AFM and cryo-EM are powerful on this front [54]. Understanding the many functional transitions and informational exchanges necessary to manage such complexes as they form or operate is a major goal on the horizon of single-molecule biology.
The upper limit of informational complexity achievable by any in vitro methodology is set by the use of cell extracts, an approach that laid the early foundations for our current molecular-level understanding of many biochemical systems. Vast informational streams, especially those related to spatial organization, are dammed or confounded by the destruction of a cell, which is why in vivo single-molecule biology holds such promise, but what remains in a crude extract is macromolecular context: the complement of molecules usually surrounding a particular component of interest is often retained. Although extracts are generally used to catalyse some specific effect when the participants are unknown or inseparable, they also offer some distinct benefits. Crude cellular extracts made possible many seminal discoveries in biology. Moreover, from a modern perspective, they can now be more easily manipulated to modify and/or label specific protein components. Bacterial and yeast genetics are so advanced and accessible that mutants or variants of many proteins are already available, and purification of a target is unnecessary. With the Xenopus system lacking such genetics, specific components can be immunodepleted, and this method is useful for essential factors in all cases. Bulk biochemical assays with extracts ‘screen’ for a very specific catalytic effect, such as radiolabelled amino acid incorporation, if an extract is active for translation [12]. In other cases, the activity of interest is more difficult to characterize, as in early work with extract-mediated chromatin formation. For example, Wood & Earnshaw [55] observed chromosome-like compaction of DNA when nuclei were exposed to mitotic extracts in several cross-species combinations. The assay allowed them to explore topoisomerase requirements for compaction, but otherwise interpreting the process of chromatin formation was not possible. Single-molecule assays can also be tuned to selectively target a specific activity in an extract, but because extracts are rich in components, we anticipate that picking out the desired function will not prove easy. Some studies have already affirmed the usefulness of extracts for single-molecule analysis, including work by Hoskins et al. [56] on the spliceosome. Although the splicing reaction pathway is known, its malleability, especially for alternative splicing, is poorly understood. The researchers visualized spliceosome components by expressing them with orthogonal tags, then labelling the products in whole-cell yeast extract with fluorescent dyes. Spliceosome component associations and dissociations on mRNA were observed with TIRFM. They were able to confirm the order of the established pathway, but also showed that pre-association of the various components is not required for splicing (which they could also track using a fluorescent tag on the substrate RNA), that no one step dominates in rate-limiting the reaction, and that no one step commits the complex to execute a splicing event (i.e. the steps are reversible). The findings will inform future work into the role of step reversibility in alternative splicing, and the experiments demonstrated the compatibility of whole-cell extracts as sources of catalytic activity and in vitro single-molecule biology.
The earlier-mentioned ideas all intensify information flux, but the curtain methodology is also open to broadening substrate range beyond double-stranded DNA (dsDNA) and dsDNA-interacting proteins. dsDNA has a reasonably high persistence length (a measure of inflexibility). By contrast, ssDNA does not extend in our system owing to intrastrand base-pair interactions in the absence of a complement. The flow-rates attainable in our flowcells cannot exert the approximately 10 pN required to extend ssDNA [57]. If this limitation could be somehow overcome and ssDNA curtains could be easily generated, then an interesting new set of questions could be addressed because ssDNA is an intermediate in DNA replication, repair and telomere maintenance [58,59]. RNA poses the same extension challenge, but here too a solution would make possible single-molecule studies of eukaryotic mRNA processing and even ribosome movement on a curtain.
If DNA is considered simply as a polymer, then even transiently stable long and linear biological macromolecules can in principle be linked to a lipid bilayer and aligned in parallel arrays. Microfilaments and microtubules are both highly dynamic, and could be assayed similarly to DNA length changes provided the actin and tubulin subunits can be stably labelled for TIRFM. There are also many cytoskeleton-interaction proteins and protein complexes of particular interest to single-molecule biophysicists because they are often molecular motors. These few suggestive examples show that in vitro single-molecule biology is yet in its infancy: an exciting time for any science.
5. Concluding remarks
Although biomolecular heterogeneity remains largely unexplored, what makes it truly enticing is its unknown physiological relevance. This dark line of questioning is open to single-molecule biology. Static disorder, as manifested in functional heterogeneity, is particularly remarkable because it produces a definite effect at the level of individual molecules. Its corollary–ergodicity breaking–can only be assessed by single-molecule techniques with the stipulation that datasets are large enough to distinguish a distinction between individual trajectories and the population average. Ergodicity breaking in diffusive processes is also of great interest, and can now be more readily identified and quantified. The central physiological issue is whether or not the cell harnesses heterogeneity. Because heterogeneity is a consequence of chemical and physical processes, exaggerated by the complexity of living systems, it is possible that evolution has simply incorporated it as an intrinsic quality of macromolecular behaviour (the same way it incorporates the diffusive properties of small organic molecules). But static disorder and functional heterogeneity, ergodicity breaking and stochasticity are all linked to macromolecular structural properties and may very well be generally modulated by evolution. If this is true, then it predicts wide variations in the extent of heterogeneity within systems, and each will be correlated with the specific function of the given system.
There has been significant interest recently in one particular manifestation of molecular heterogeneity: the stochasticity of gene expression—also termed ‘noise’ within the field [60]. Researchers have long observed that transcription and translation occur in bursts; even when a gene is signalled to be ‘on’ by the cell, mRNA and protein production follow a time series of on/off fluctuations resulting in heterogeneous expression in an otherwise isogenic population. The molecular causes of such noise remain unknown, but are thought to include static disorder in the transcriptional machinery as well as external environmental fluctuations [27,61]. Interestingly, noise is subject to evolutionary and cellular tuning [62–64]. Illustratively, So et al. [65] counted mRNA copy numbers of specific bacterial genes in vivo after cells responded to stimuli effecting each gene. They found the same correlation between ‘burstiness’ and average expression level, irrespective of the gene in question, suggesting that noise results from some fundamental transcriptional process. They further calculated that the cells tune ‘burstiness’ to extract as much information about the external environment as possible. If stochasticity in gene expression is in fact a consequence of molecular heterogeneities, then results like these hint at a host of entirely new insights by single-molecule biology as the layers of physiological complexity are slowly resolved.
The biological macromolecule is the cell in microcosm: it lives in a complicated and fluctuating environment, channelling information through its structure, localization and interactions. Single-molecule biology is an attempt to capture the moments when information streams cross, and endow the molecule with physiological meaning.
Acknowledgements
We thank Sam Sternberg and members of our laboratory for reviewing the manuscript, and especially Sy Redding, Ilya Finkelstein, Tim Silverstein, Bryan Gibb and Feng Wang for insightful discussions, and Myles Marshall for assistance with the figures. Research in the Greene laboratory is supported by the National Institutes of Health, the National Science Foundation and the Howard Hughes Medical Institute. D.D. is supported in part by an award from the Paul and Daisy Soros Fellowships for New Americans.
References
- 1.Cairns J. (ed.) 2007. Phage and the origins of molecular biology, the centennial edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- 2.Judson HF. 1996. The eighth day of creation: the makers of the revolution in biology (25th anniversary edition). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- 3.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. 2007. Molecular biology of the cell, 5th edn New York, NY: Garland Science [Google Scholar]
- 4.Rotman B. 1961. Measurement of activity of single molecules of β-d-galactosidase. Proc. Natl Acad. Sci. USA 47, 1981–1991 10.1073/pnas.47.12.1981 (doi:10.1073/pnas.47.12.1981) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirschfeld T. 1976. Optical microscopic observation of single small molecules. Appl. Opt. 15, 2965–2966 10.1364/AO.15.002965 (doi:10.1364/AO.15.002965) [DOI] [PubMed] [Google Scholar]
- 6.Shera EB, Seitzinger NK, Davis LM, Keller RA, Soper SA. 1990. Detection of single fluorescent molecules. Chem. Phys. Lett. 174, 553–557 10.1016/0009-2614(90)85485-U (doi:10.1016/0009-2614(90)85485-U) [DOI] [Google Scholar]
- 7.Betzig E, Chichester RJ. 1993. Single molecules observed by near-field scanning optical microscopy. Science 262, 1422–1425 10.1126/science.262.5138.1422 (doi:10.1126/science.262.5138.1422) [DOI] [PubMed] [Google Scholar]
- 8.Greenleaf WJ, Woodside MT, Block SM. 2007. High-resolution single-molecule measurements of biomolecular motion. Annu. Rev. Biophys. Biomol. Struct. 36, 171–190 10.1146/annurev.biophys.36.101106.101452 (doi:10.1146/annurev.biophys.36.101106.101452) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wennmalm S, Simon SM. 2007. Studying individual events in biology. Annu. Rev. Biochem. 76, 419–446 10.1146/annurev.biochem.76.062305.094225 (doi:10.1146/annurev.biochem.76.062305.094225) [DOI] [PubMed] [Google Scholar]
- 10.Tinoco I, Jr, Gonzalez RL., Jr 2011. Biological mechanisms, one molecule at a time. Genes Dev. 25, 1205–1231 10.1101/gad.2050011 (doi:10.1101/gad.2050011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kornberg A, Lehman IR, Bessman MJ, Simms ES. 1956. Enzymatic synthesis of deoxyribonucleic acid. Biochim. Biophys. Acta 21, 197–198 10.1016/0006-3002(56)90127-5 (doi:10.1016/0006-3002(56)90127-5) [DOI] [PubMed] [Google Scholar]
- 12.Lamborg MR, Zamecnik PC. 1960. Amino acid incorporation into protein by extracts of E. coli. Biochim. Biophys. Acta 42, 206–211 10.1016/0006-3002(60)90782-4 (doi:10.1016/0006-3002(60)90782-4) [DOI] [PubMed] [Google Scholar]
- 13.McGhee JD, Felsenfeld G. 1980. Nucleosome structure. Annu. Rev. Biochem. 49, 1115–1156 10.1146/annurev.bi.49.070180.005343 (doi:10.1146/annurev.bi.49.070180.005343) [DOI] [PubMed] [Google Scholar]
- 14.Whitehouse I, Flaus A, Cairns BR, White MF, Workman JL, Owen-Hughes T. 1999. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature 400, 784–787 10.1038/23506 (doi:10.1038/23506) [DOI] [PubMed] [Google Scholar]
- 15.Perutz MF, Rossmann MG, Cullins AF, Muirhead H, Will G, North ACT. 1960. Structure of haemoglobin: a three-dimensional Fourier synthesis at 5.5-Å. resolution obtained by X-ray analysis. Nature 185, 416–422 10.1038/185416a0 (doi:10.1038/185416a0) [DOI] [PubMed] [Google Scholar]
- 16.Kendrew JC, Dickerson RE, Strandberg BE, Hart RG, Davies DR, Phillips DC, Shore VC. 1960. Structure of myoglobin: a three-dimensional Fourier synthesis at 2Å resolution. Nature 185, 422–427 10.1038/185422a0 (doi:10.1038/185422a0) [DOI] [PubMed] [Google Scholar]
- 17.Muirhead H, Perutz MF. 1963. Structure of haemoglobin: a three-dimensional Fourier synthesis of reduced human haemoglobin at 5.5 Å resolution. Nature 199, 633–638 10.1038/199633a0 (doi:10.1038/199633a0) [DOI] [PubMed] [Google Scholar]
- 18.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 10.1038/38444 (doi:10.1038/38444) [DOI] [PubMed] [Google Scholar]
- 19.Wu E, Beese LS. 2011. The structure of a high fidelity DNA polymerase bound to a mismatched nucleotide reveals an ‘ajar’ intermediate conformation in the nucleotide selection mechanism. J. Biol. Chem. 22, 19 758–19 767 10.1074/jbc.M110.191130 (doi:10.1074/jbc.M110.191130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braddock DT, Baber JL, Levens D, Clore GM. 2002. Molecular basis of sequence-specific single-stranded DNA recognition by KH domains: solution structure of a complex between hnRNP K KH3 and single-stranded DNA. EMBO J. 21, 3476–3485 10.1093/emboj/cdf352 (doi:10.1093/emboj/cdf352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mittermaier A, Kay LE. 2006. New tools provide new insights in NMR studies of protein dynamics. Science 312, 224–228 10.1126/science.1124964 (doi:10.1126/science.1124964) [DOI] [PubMed] [Google Scholar]
- 22.Fu J, Munro JB, Blanchard SC, Frank J. 2011. Cryo-EM structures of the ribosome complex in intermediate states during tRNA translocation. Proc. Natl Acad. Sci. USA 12, 4817–4821 10.1073/pnas.1101503108 (doi:10.1073/pnas.1101503108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zewail AH, Thomas JM. 2010. 4D electron microsocpy: imaging in space and time. London, UK: Imperial College Press [Google Scholar]
- 24.Chandler R. 1987. Introduction to modern statistical mechanics. New York, NY: Oxford University Press [Google Scholar]
- 25.Zwanzig R. 1990. Rate processes with dynamical disorder. Acc. Chem. Res. 23, 148–152 10.1021/ar00173a005 (doi:10.1021/ar00173a005) [DOI] [Google Scholar]
- 26.Weber SC, Spakowitz AJ, Theriot JA. 2012. Nonthermal ATP-dependent fluctuations contribute to the in vivo motion of chromosomal loci. Proc. Natl Acad. Sci. USA 19, 7338–7343 10.1073/pnas.1119505109 (doi:10.1073/pnas.1119505109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hilfinger A, Paulsson J. 2011. Separating intrinsic from extrinsic fluctuations in dynamic biological systems. Proc. Natl Acad. Sci USA 108, 12 167–12 172 10.1073/pnas.1018832108 (doi:10.1073/pnas.1018832108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ditzler MA, Rueda D, Mo J, Håkansson K, Walter NG. 2008. A rugged free energy landscape separates multiple functional RNA folds throughout denaturation. Nucleic Acids Res. 36, 7088–7099 10.1093/nar/gkn871 (doi:10.1093/nar/gkn871) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J, Myong S, Niedziela-Majka A, Lee KS, Yu J, Lohman TM, Ha T. 2010. PcrA helicase dismantles RecA filaments by reeling in DNA in uniform steps. Cell 142, 544–555 10.1016/j.cell.2010.07.016 (doi:10.1016/j.cell.2010.07.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clore GM. 2008. Visualizing lowly-populated regions of the free-energy landscape of macromolecular complexes by paramagnetic relaxation enhancement. Mol. BioSyst. 4, 1058–1069 10.1039/b810232e (doi:10.1039/b810232e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clore GM. 2011. Exploring translocation of proteins on DNA by NMR. J. Biomol. NMR 51, 209–219 10.1007/s10858-011-9555-8 (doi:10.1007/s10858-011-9555-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfingstein JS, Goodrich KJ, Taabazuing C, Ouenzar F, Chartrand P, Cech TR. 2012. Mutually exclusive binding of telomerase RNA and DNA by Ku alters telomerase recruitment model. Cell 148, 922–932 10.1016/j.cell.2012.01033 (doi:10.1016/j.cell.2012.01033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. 2006. The fluorescent toolbox for assessing protein location and function. Science 312, 217–224 10.1126/science.1124618 (doi:10.1126/science.1124618) [DOI] [PubMed] [Google Scholar]
- 34.Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. 2010. Fluorescent proteins and their applications in imaging live cells and tissues. Physiol. Rev. 90, 1103–1163 10.1152/physrev.00038.2009 (doi:10.1152/physrev.00038.2009) [DOI] [PubMed] [Google Scholar]
- 35.Weigel AV, Simon B, Tamkun MM, Krapf D. 2011. Ergodic and nonergodic processes coexist in the plasma membrane as observed by single-molecule tracking. Proc. Natl Acad. Sci USA 108, 6438–6443 10.1073/pnas.1016325108 (doi:10.1073/pnas.1016325108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He Y, Burov S, Metzler R, Barkai E. 2008. Random time-scale invariant diffusion and transport coefficients. Phys. Rev. Lett. 101, 058101. 10.1103/physrevlett.101.058101 (doi:10.1103/physrevlett.101.058101) [DOI] [PubMed] [Google Scholar]
- 37.Jeon J-H, Tejedor V, Burov S, Barkai E, Selhuber-Unkel C, Berg-Sorensen K, Oddershede L, Metzler R. 2011. In vivo anomalous diffusion and weak ergodicity breaking of lipid granules. Phys. Rev. Lett. 106, 048103. 10.1103/PhysRevLett.106.048103 (doi:10.1103/PhysRevLett.106.048103) [DOI] [PubMed] [Google Scholar]
- 38.Sokolov IM. 2008. Statistics and the single molecule. Physics 1, 8–10 10.1103/physics.1.8 (doi:10.1103/physics.1.8) [DOI] [Google Scholar]
- 39.Kusumi A, Nakada C, Ritchie K, Murase K, Suzuki K, Murakoshi H, Kasai RS, Kodo J, Fujiwara T. 2005. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34, 351–378 10.1146/annurev.biophys.34.040204.144637 (doi:10.1146/annurev.biophys.34.040204.144637) [DOI] [PubMed] [Google Scholar]
- 40.Robson A, Burrage K, Leake MC. 2013. Inferring diffusion in single live cells at the single-molecule level. Phil. Trans. R. Soc. B 368, 20120029. 10.1098/rstb.2012.0029 (doi:10.1098/rstb.2012.0029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visnapuu M-L, Duzdevich D, Greene EC. 2008. The importance of surfaces in single-molecule bioscience. Mol. BioSyst. 4, 394–403 10.1039/B800444G (doi:10.1039/B800444G) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greene EC, Wind S, Fazio T, Gorman J, Visnapuu M-L. 2010. DNA curtains for high-throughput single-molecule optical imaging. Methods Enzymol. 472, 293–315 10.1016/S0076-6879(10)72006-1 (doi:10.1016/S0076-6879(10)72006-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finkelstein IJ, Greene EC. 2011. Supported lipid bilayers and DNA curtains for high-throughput single-molecule studies. Methods Mol. Biol. 745, 447–461 10.1007/978-1-61779-129-1-26 (doi:10.1007/978-1-61779-129-1-26) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Visnapuu M-L, Greene EC. 2009. Single-molecule imaging of DNA curtains reveals intrinsic energy landscapes for nucleosome deposition. Nat. Struct. Mol. Biol. 16, 1056–1063 10.1038/nsmb.1655 (doi:10.1038/nsmb.1655) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Granéli A, Yeykal C, Prasad TK, Greene EC. 2006. Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir 22, 292–299 10.1021/la051944a (doi:10.1021/la051944a) [DOI] [PubMed] [Google Scholar]
- 46.Fazio T, Visnapuu M-L, Wind S, Greene EC. 2008. DNA curtains and nanoscale curtain rods: high-throughput tools for single molecule imaging. Langmuir 24, 10 524–10 531 10.1021/la801762h (doi:10.1021/la801762h) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Visnapuu M-L, Fazio T, Wind S, Greene EC. 2008. Parallel arrays of geometric nanowells for assembling curtains of DNA with controlled lateral dispersion. Langmuir 24, 11 293–11 299 10.1021/la8017634 (doi:10.1021/la8017634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gorman J, Fazio T, Wang F, Wind S, Greene EC. 2009. Nanofabricated racks of aligned and anchored DNA substrates for single-molecule imaging. Langmuir 26, 1372–1379 10.1021/la902443e (doi:10.1021/la902443e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gorman J, Plys AJ, Visnapuu M-L, Alani E, Greene EC. 2010. Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat. Struct. Mol. Biol. 17, 932–938 10.1038/nsmb.1858 (doi:10.1038/nsmb.1858) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bruchez MP. 2011. Quantum dots find their stride in single molecule tracking. Curr. Opin. Chem. Biol. 15, 775–780 10.1016/j.cbpa.2011.10.011 (doi:10.1016/j.cbpa.2011.10.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, Struhl K. 2009. Intrinsic histone–DNA interactions are not the major determinant of nucleosome positions in vivo. Nat. Struct. Mol. Biol. 16, 847–853 10.1038/nsmb.1636 (doi:10.1038/nsmb.1636) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Finkelstein IJ, Visnapuu M-L, Greene EC. 2010. Single-molecule imaging reveals mechanisms of protein disruption by a DNA translocase. Nature 468, 983–987 10.1038/nature09561 (doi:10.1038/nature09561) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maeshima K, Hihara S, Eltsov M. 2010. Chromatin structure: does the 30-nm fibre exist in vivo? Curr. Opin. Cell Biol. 22, 291–297 10.1016/j.ceb.2010.03.001 (doi:10.1016/j.ceb.2010.03.001) [DOI] [PubMed] [Google Scholar]
- 54.Alushin G, Nogales E. 2011. Visualizing kinetochore architecture. Curr. Opin. Struct. Biol. 21, 661–669 10.1016/j.sbi.2011.07.009 (doi:10.1016/j.sbi.2011.07.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wood ER, Earnshaw WC. 1990. Mitotic chromatin condensation in vitro using somatic cell extracts and nuclei with variable levels of endogenous topoisomerase II. J. Cell Biol. 111, 2839–2850 10.1083/jcb.111.6.2839 (doi:10.1083/jcb.111.6.2839) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoskins AA, et al. 2011. Ordered and dynamic assembly of single spliceosomes. Science 331, 1289–1295 10.1126/science.1198830 (doi:10.1126/science.1198830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bustamante C, Smith SB, Liphardt J, Smith D. 2000. Single-molecule studies of DNA mechanics. Curr. Opin. Struct. Biol. 10, 279–285 10.1016/S0959-440X(00)00085-3 (doi:10.1016/S0959-440X(00)00085-3) [DOI] [PubMed] [Google Scholar]
- 58.Baumann P, Price C. 2010. Pot1 and telomere maintenance. FEBS Lett. 584, 3779–3784 10.1016/j.febslet.2010.05.024 (doi:10.1016/j.febslet.2010.05.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dewar JM, Lydall D. 2011. Similarities and differences between ‘uncapped’ telomeres and DNA double-strand breaks. Chromosoma 121, 117–130 10.1007/s00412-011-0357-2 (doi:10.1007/s00412-011-0357-2) [DOI] [PubMed] [Google Scholar]
- 60.Kærn M, Elston TC, Blake WJ, Collins JJ. 2005. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464 10.1038/nrg1615 (doi:10.1038/nrg1615) [DOI] [PubMed] [Google Scholar]
- 61.Paulsson J. 2004. Summing up the noise in gene networks. Nature 427, 415–418 10.1038/nature02257 (doi:10.1038/nature02257) [DOI] [PubMed] [Google Scholar]
- 62.Dekel E, Alon U. 2005. Optimality and evolutionary tuning of the expression level of a protein. Nature 436, 588–592 10.1038/nature03842 (doi:10.1038/nature03842) [DOI] [PubMed] [Google Scholar]
- 63.Fraser HB, Hirsh AE, Giaever G, Kumm J, Eisen MB. 2004. Noise minimization in eukaryotic gene expression. PLoS Biol. 2, e137. 10.1371/journal.pbio.0020137 (doi:10.1371/journal.pbio.0020137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eldar A, Elowitz MB. 2010. Functional roles for noise in genetic circuits. Nature 467, 167–173 10.1038/nature09326 (doi:10.1038/nature09326) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.So L-h, Ghosh A, Zong C, Sepúlveda LA, Segev R, Golding I. 2011. General properties of transcriptional time series in Escherichia coli. Nat. Genet. 43, 554–560 10.1038/ng.821 (doi:10.1038/ng.821) [DOI] [PMC free article] [PubMed] [Google Scholar]