

Abstract
Osteosarcoma, Ewing sarcoma, and rhabdomyosarcoma are the most common malignant musculoskeletal tumors in children and adolescents. Today, most patients can be cured. Numerous factors have contributed to improved outcome for these patients over the past several decades. These include multidisciplinary care involving oncologists, radiation oncologists, surgeons, pathologists, and radiologists and enrollment of patients in clinical trials. Better understanding of molecular mechanisms of disease have resulted in studies using molecular targets in addition to standard chemotherapeutic agents, which hopefully will lead to better outcomes in the future. Moreover, new orthopedic techniques and devices as well as new technologies in radiation oncology hold promise for better local control of primary tumors and the potential for fewer late adverse effects. Despite this progress, patients must undergo lifelong follow-up for possible late effects of intense chemotherapy and radiation therapy. We review the diagnosis, prognosis, staging, multidisciplinary therapy, new directions in therapy, and long-term complications of treatment for these tumors. For this review, we searched MEDLINE using the terms rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, biology, and humans and limited the search to articles from 2000 to September 2011. Additional references found in these articles were utilized as appropriate, as well as references from the background information in current therapeutic studies of the Children's Oncology Group. The same database and time frame were searched for articles written by leading authorities in the field.
Abbreviations and Acronyms: ARMS, alveolar rhabdomyosarcoma; COG, Children's Oncology Group; EFS, event-free survival; ERMS, embryonal rhabdomyosarcoma; ES, Ewing sarcoma; FDG-PET, fluorodeoxyglucose positron emission tomography; IE, ifosfamide and etoposide; MAP, methotrexate, doxorubicin (Adriamycin), and cisplatin; OS, osteosarcoma; PNET, primitive neuroectodermal tumor; RMS, rhabdomyosarcoma; VDC, vincristine, doxorubicin, and cyclophosphamide
Osteosarcoma (OS), Ewing sarcoma/primitive neuroectodermal tumor (ES/PNET), and rhabdomyosarcoma (RMS) are the most common musculoskeletal tumors of childhood and adolescence. This article discusses advances in the multidisciplinary management of these tumors and reviews current understanding of some key biologic features with potential implications for future therapy.
Methods
We searched MEDLINE using the terms rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, biology, and humans and limited the search to articles from 2000 to September 2011. Additional references found in these articles were utilized as appropriate, as well as references from the background information in current therapeutic studies of the Children's Oncology Group (COG). The same database and time frame were searched for articles written by leading authorities in the field. Discussion in this review is restricted to articles pertinent to children and adolescents.
Epidemiology and Clinical Features
Osteosarcoma is the most common malignant bone tumor in children and adolescents, with an incidence of 4.4 per million.1 The peak age incidence is in the second decade of life, with a smaller peak in older adults. Ewing sarcoma/PNET is the second most common malignant bone tumor, with an incidence of 2.9 per million,2 and it also occurs in soft tissues. Bone tumors commonly manifest with pain and swelling in a bone or joint. Symptoms are often attributed to sports injuries in active adolescents. Night pain, systemic symptoms of fever and weight loss, or failure of symptoms to resolve after several weeks of conservative management should alert the clinician to a more serious underlying cause and prompt further investigation.
Rhabdomyosarcomas constitute more than half of soft tissue sarcomas in children, with an incidence of 4.5 cases per million children and adolescents per year.3 Because RMS can occur at any site, its presentation depends on site of origin. For example, orbital RMS may manifest as proptosis, bladder or prostate RMS may manifest with urinary retention, extremity RMS as a painless mass, and vaginal RMS with vaginal bleeding, especially in a prepubescent female.
Biology and Pathology
Osteosarcoma
High-grade OS is most likely derived from mesenchymal stem cells with at least partial osteoblastic lineage commitment, although the exact cell of origin is unclear.4 Patients with hereditary retinoblastoma, Rothmund-Thomson syndrome, Li-Fraumeni syndrome, and Werner syndrome are predisposed to development of OS, suggesting that alterations in the genes associated with these disorders (RB1, RECQL4, TP53, and WRN, respectively [for expansion of gene symbols, use search tool at www.genenames.org]) may play a role in the pathogenesis of OS.5 However, the vast majority of OSs arise in patients with no known germline abnormalities. At the cytogenetic level, OSs have highly complex karyotypes with many numerical and structural abnormalities; a consistent cytogenetic abnormality has not been identified.6 Three major subtypes of conventional OS are recognized: osteoblastic, chondroblastic, and fibroblastic, reflecting the predominant form of tumor matrix.7 Treatment and outcome of these subtypes are not different. Parosteal OS, central low-grade OS, and periosteal OS are morphologically and clinically distinct OS subtypes with an improved prognosis7and constitute less than 5% of cases of OS.8-14 The age at presentation for parosteal and periosteal OS is usually in the fourth and fifth decades of life (patients are usually in the 30- to 40-year age range). The microscopic diagnosis of OS rests on the identification of production of osteoid matrix by the neoplastic cells (Figure 1, top). There are no ancillary immunohistochemical or molecular genetic studies that are of value in the diagnosis of OS.
FIGURE 1.
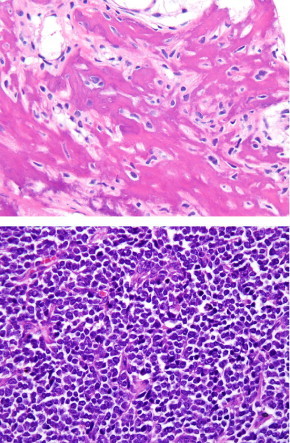
Top, Photomicrograph of osteosarcoma showing production of lacelike neoplastic osteoid by hyperchromatic spindle cells. Bottom, Photomicrograph of Ewing sarcoma/primitive neuroectodermal tumor consisting of a sheetlike proliferation of uniform, undifferentiated small round blue cells. Hematoxylin and eosin, magnification 200x.
Ewing Sarcoma/Primitive Neuroectodermal Tumor
Ewing sarcoma and PNET were originally described as distinct entities but are now recognized to represent ends along the morphologic spectrum of a single neoplasm.15 Histopathologically, ES consists in most cases of a vaguely lobular proliferation of uniform small blue round cells with clear to lightly eosinophilic cytoplasm, evenly dispersed chromatin, and indistinct nucleoli16 (Figure 1, bottom). Tumors falling closer to the PNET end of this spectrum may contain occasional pseudorosettes. By immunohistochemistry, ES/PNET strongly expresses CD99 (MIC2 gene product) in a membranous pattern and shows FLI1 expression in more than 80% of cases.16 Approximately 25% of ES/PNETs show aberrant expression of keratins, typically considered an epithelial marker. At the genetic level, approximately 95% of ES/PNETs contain the t(11;22)(q24;q12) translocation (EWSR1-FLI1), 4% contain the variant t(21;22)(q22;q12) (EWSR1-ERG), and the remainder show fusion of EWSR1 or very rarely FUS with a variety of other ETS family transcription factors.17 Fusion subtype is not known to be prognostically significant.18,19
Rhabdomyosarcoma
Although traditionally RMS has been considered to derive from primitive myoblasts, a large percentage of RMSs occur in locations normally lacking skeletal muscle (eg, bladder, prostate, vagina, bile ducts).15 Rhabdomyosarcomas are usually grouped into 3 subtypes: embryonal (ERMS) (including botryoid and spindle cell variants), alveolar (ARMS), and pleomorphic.20,21 Pleomorphic RMS is seen almost exclusively in adults and is not discussed here. The recently described sclerosing RMS most likely represents a variant of ERMS.22 Figure 2 shows typical light microscopic appearances of ERMS compared with ARMS. Both ERMS and ARMS are typically strongly desmin-positive, with ARMS usually showing strong myogenin expression and more limited MYOD1 expression and ERMS often showing the opposite pattern.23 Aberrant expression of epithelial and neuroendocrine markers is common in ARMS and may result in misdiagnosis.24 At the genetic level, ARMS is characterized in approximately 75% of cases by the t(2;13)(q35;q14) translocation (PAX3-FOX1A) and in approximately 10% of cases by t(1;13)(p36;q14) (PAX7-FOX1A).7 Although traditionally approximately 15% of ARMS have been considered to be fusion-negative, the noncanonical translocations t(2;2)(p23;q35) (PAX3-NCOA1) or t(2;8)(q35;q13) (PAX3-NCOA2) have recently been identified in subsets of these cases.25 In contrast to ARMS, ERMS possesses no distinct molecular signature, although molecular genetic analysis has shown frequent allelic loss on chromosome 11.26 Davicioni et al27 have recently identified a 34-metagene, based on expression patterns of 34 genes, that was highly predictive of outcome. It was not highly correlated with individual clinical risk factors such as patient age, tumor stage, tumor size, or histology. However, it was correlated with a risk classification used by the COG and the biologic subsets of alveolar histology tumors. Prospective clinical correlations are planned to verify these findings.
FIGURE 2.
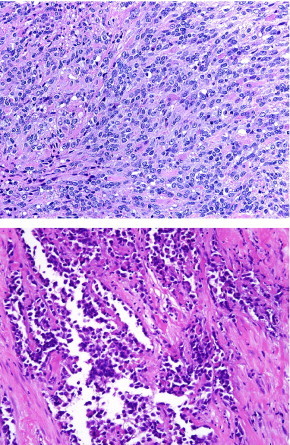
Top, Photomicrograph of embryonal rhabdomyosarcoma, a spindle cell sarcoma, showing a variable degree of rhabdomyoblastic differentiation in the form of “strap cells” and rounded rhabdomyoblasts. Bottom, Photomicrograph of alveolar rhabdomyosarcoma, a small blue round cell tumor with very limited rhabdomyoblastic differentiation, usually in the form of multinucleated giant cells and occasional cells with brightly eosinophilic cytoplasm. Hematoxylin and eosin, magnification 200x.
Diagnosis and Staging
Ideally, evaluation of the child and adolescent with a suspected bone or soft tissue tumor should be performed at the institution that ultimately will be providing multidisciplinary care for the patient. For bone tumors, the biopsy needle tract or incision should be placed such that it can be incorporated into the final surgical excision if surgery is the ultimate form of local tumor control.28 Planning of the biopsy should be coordinated between the radiologist and the surgeon for patients undergoing needle biopsy. Adequate tissue should be obtained not only for diagnosis but also for molecular and biologic studies. This includes obtaining tissue both for routine processing in formalin and for fresh tissue studies. Evaluation of the primary tumor consists of radiography and magnetic resonance imaging and/or computed tomography. Figures 3 and 4 show typical radiographic and magnetic resonance appearances of OS of the distal femur and ES/PNET of the proximal humerus, respectively. Rhabdomyosarcoma typically appears as a nonspecific soft tissue or visceral mass lesion.
FIGURE 3.
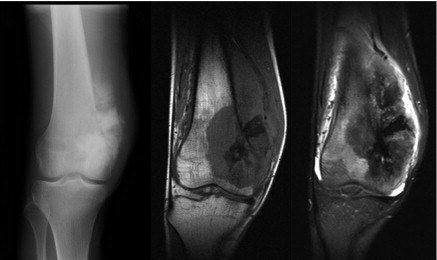
Left, Anteroposterior radiograph of osteosarcoma of the distal femur showing a destructive lesion in the metadiaphysis with neoplastic osteoid formation within the associated soft tissue mass. Middle and Right, Coronal T1- and T2-weighted magnetic resonance images demonstrate the anatomic extent of marrow involvement and soft tissue mass.
FIGURE 4.
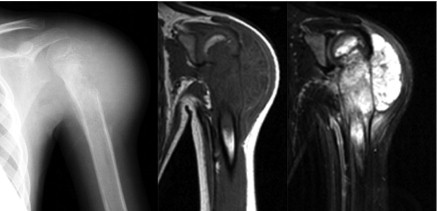
Left, Anteroposterior radiograph of Ewing sarcoma of the proximal humerus demonstrates a large permeative destructive lesion involving the metadiaphysis with extensive associated malignant periosteal new bone formation and large soft tissue mass laterally. Middle and Right, Coronal T1- and T2-weighted magnetic resonance images show the anatomic extent of the soft tissue mass and marrow infiltration.
Evaluation for metastatic disease consists of computed tomography of the chest, bone scan, and in the case of ES/PNET and RMS, bilateral bone marrow aspirates and biopsies. Lymph node sampling is also performed for selected primary sites in RMS. Fluorodeoxyglucose positron emission tomography (FDG-PET) is being used more routinely in the evaluation of metastatic disease and in evaluating response to therapy. A study by Völker et al29 found that adding FDG-PET to the staging process led to a change in therapy for 7 of 17 patients with ES. Fluorodeoxyglucose PET was nearly twice as sensitive for bone metastases compared with standard bone scintigraphy. In addition, the sensitivity of FDG-PET for lymph node involvement was 95% compared with 25% for conventional imaging modalities. Fluorodeoxyglucose PET has been correlated with histologic response to neoadjuvant chemotherapy in ES/PNET, with a standardized uptake value-2 less than 2.5 being predictive of progression-free survival.30 In extremity OS, FDG-PET was reported to be only partially correlated with histologic response to neoadjuvant chemotherapy; however, a standardized uptake value-2 less than 2.5 was associated with improved progression-free survival.31 The PET response to neoadjuvant chemotherapy and potential implications for outcome and subsequent therapy are being investigated prospectively in clinical trials of pediatric sarcoma.
Multidisciplinary Therapy and Prognosis
When evaluating a child or teenager newly diagnosed with one of these malignancies, it is important to involve a multidisciplinary team of oncologists, surgeons, and radiation oncologists from the outset so that the treatment planning can progress seamlessly and the timing of local control can be optimized so as not to delay chemotherapy. Because radiation treatments are based on the extent of tumor at the time of diagnosis, the radiation oncologist must see patients with ES/PNET and RMS promptly so that appropriate baseline imaging can be obtained and plans can be expedited for timing of simulation and radiation. It is beneficial to have simultaneous discussion and review of the case by members of all 3 disciplines to establish a unified approach to treatment. Most of the advances seen in the past several decades have been the result of randomized clinical trials performed by North American and European pediatric cooperative clinical trials groups such as the Children's Oncology Group (COG); the European Paediatric Soft Tissue Sarcoma Study Group; the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors Working Group; the European Intergroup Cooperative Ewing's Sarcoma Study; and the Cooperative Osteosarcoma Study Group. The multidisciplinary collaboration of pediatric oncologists, surgeons, and radiotherapists as well as the commitment of patients and physicians to clinical trials has significantly improved cure rates for these rare tumors.
Osteosarcoma
Despite several decades of clinical trials of chemotherapy in OS, the standard chemotherapeutic agents utilized in the treatment of OS remain methotrexate, doxorubicin (Adriamycin), and cisplatin (MAP) with or without ifosfamide. Surgical removal of all tumor (primary and metastatic) is required for cure. Most patients receive neoadjuvant chemotherapy followed by limb salvage surgery. With chemotherapy and surgery, the overall survival and event-free survival (EFS) for patients with nonmetastatic disease can be expected to be approximately 75% and 65%, respectively, at 5 years.32,33 Patients with lung or bone metastases fare less well, with only approximately 25% to 50% being alive at 5 years.34,35 The most recently published North American cooperative group trial evaluated the addition of ifosfamide and/or an immunoadjuvant, muramyl tripeptide phosphatidylethanolamine, to standard MAP therapy in a randomized trial.33 The outcome of the study showed that the addition of ifosfamide to standard MAP did not improve EFS or overall survival; however, the addition of muramyl tripeptide phosphatidylethanolamine to chemotherapy resulted in a statistically significant improvement in overall survival and a trend toward better EFS. In the largest study of prognostic factors in OS, tumor site and size, presence of metastases, surgical remission, and tumor necrosis after neoadjuvant chemotherapy emerged as the most important independent prognostic factors.36 Although the addition of ifosfamide and etoposide (IE) to the regimens of poor responders in one study seemed to improve their outcome to equal that of good responders, this study was not randomized.37 Intensification of chemotherapy with IE in patients who have suboptimal tumor necrosis after conventional therapy is being evaluated in an international randomized trial.38 Other areas of investigation for patients with metastatic disease at presentation have included the addition of trastuzumab to chemotherapy for patients with tumors expressing HER2/neu39 and the use of bisphosphonates.40,41
Wide surgical excision remains the mainstay of surgical treatment for OS. Advances in imaging, surgical techniques, and implants have dramatically reduced the need for amputation in the past 3 decades. Advances in survival have paralleled the increase in limb salvage.42 Surgical resection of OS in children poses unique challenges. Most tumors arise in the metaphyseal region and abut or involve the growth plate. Thus, surgical resection will lead to a limb length discrepancy in growing children. Modest limb length discrepancies can be managed with lengthening at the time of resection and shoe modifications; for children with greater anticipated discrepancies, extendible prostheses have been developed to allow in vivo lengthening as the child grows. Although initial systems required reoperation and had high rates of mechanical failure, several modern implant designs allow noninvasive expansion triggered by an external magnetic field43 (Figure 5).
FIGURE 5.
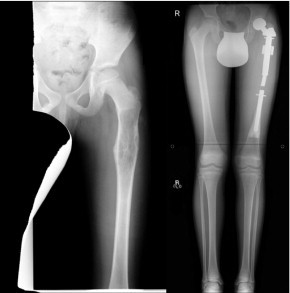
Expandable prosthesis to maintain limb length in a growing child. Left, Radiograph obtained on presentation of a 10-year-old child with a proximal femur osteosarcoma. Right, Radiograph at 4-year follow-up shows maintenance of equal leg lengths with the use of an expandable prosthesis.
Other innovative approaches to children with OS exist. When the joint surface can be spared, the use of intercalary allografts with or without vascularized fibula transfer yields an excellent outcome.44,45 New osseointegration methods may improve fixation in patients undergoing endoprosthetic reconstruction.46 In carefully selected patients whose tumors do not invade the physis, bone distraction can be used to create a buffer of newly formed bone in the distal metaphysis to provide a margin for tumor excision with intercalary joint-sparing reconstruction.47 However, poor outcomes have been reported in patients treated with bone distraction who had a poor response to neoadjuvant chemotherapy48; the uncertainties in predicting preoperatively the histologic response to chemotherapy have limited the adoption of this novel technique.
A minority of patients are unsuitable for limb salvage therapy because of the extent of the tumor, young age and an anticipated large leg length discrepancy, or the desire for high function more than cosmesis. Such patients are treated with rotationplasty (provided neurologic structures are free of tumor) or amputation. Although it is a dramatic-appearing procedure, rotationplasty offers excellent oncologic and functional results with normal psychosocial development.49,50 In patients requiring amputation, recent advances in microprocessor-controlled prosthetic joints improve the functional outcome compared with conventional prostheses.51
Although a minority of OSs arise in the axial skeleton, these pose particular treatment challenges. Advances in surgical techniques allow margin-free resection of tumors previously considered inoperable.52
Osteosarcoma has historically been considered to be a radioresistant tumor. However, advances in radiotherapy that allow safe administration of high radiation doses are showing promise for patients with unresectable or incompletely resected axial skeleton OS. In a recent report of 55 patients with unresectable or subtotally resected OS treated with proton radiotherapy, local control at 3 and 5 years was 82% and 72%, respectively, with a median tumor dose of 68.4 Gy.53 Heavier ions, such as carbon, have a radiobiologic advantage for radioresistant tumors and are being investigated in OS.54,55 Carbon ion doses of 70.4 Gy were associated with 92% local control at 3 years in patients with unresectable head and neck OS. Bone metastases from OS are often extremely painful. For patients without curative options, radiotherapy with external beam radiation or radioisotopes such as samarium and/or bisphosphonates can also be used to palliate symptoms of disease.56,57
In contrast to treatment of conventional OS with surgery and chemotherapy, treatment of low-grade central OS, parosteal OS, and periosteal OS consists of surgery only. These OS subtypes are associated with a much lower risk of distant metastases (except in the case of dedifferentiated parosteal OS), and chemotherapy is generally not used in these types of OS. It is important to recognize these subtypes so as not to subject the patient to unnecessary chemotherapy.8-14
Ewing Sarcoma/Primitive Neuroectodermal Tumor
The cure rate for patients with nonmetastatic ES/PNET has increased greatly with the advent of effective chemotherapy regimens and a series of randomized clinical trials beginning in the 1970s. Active agents for ES/PNET include vincristine, doxorubicin, cyclophosphamide, ifosfamide, etoposide, and actinomycin. Grier et al58 showed that the addition of IE to vincristine, doxorubicin, cyclophosphamide, and actinomycin improved the outcome of patients with nonmetastatic disease compared with those treated with vincristine, doxorubicin, cyclophosphamide, and dactinomycin alone. Since then, there have been 2 randomized North American studies evaluating the dose intensity of these agents in nonmetastatic ES/PNET. The first study randomized patients to receive the same total doses of vincristine, doxorubicin, and cyclophosphamide (VDC) alternating with IE given over 30 weeks vs 48 weeks by increasing dose per cycle and demonstrated no difference in outcome.59 The second study randomized patients with nonmetastatic disease to receive standard doses of VDC/IE either every 2 weeks or every 3 weeks and showed significantly improved EFS in those patients treated every 2 weeks compared with those treated every 3 weeks (79% 4-year EFS vs 70% EFS; P=.023).60 Thus, VDC alternating with IE on an every 2-week schedule has become the standard of care for patients with nonmetastatic disease in North America. The addition of topotecan and cyclophosphamide to VDC/IE is currently under evaluation by the COG in a randomized study.61
A somewhat different approach has evolved in Europe, where after a series of studies by the United Kingdom Children's Cancer Study Group and the German–Dutch–Swiss Cooperative Ewing's Sarcoma Studies, the current backbone for induction of therapy is vincristine, ifosfamide, doxorubicin, and etoposide followed by a randomized comparison of vincristine, actinomycin, and cyclophosphamide with vincristine, actinomycin, and ifosfamide for patients with good histologic response or small tumors treated with radiation. In patients who have poor histologic response, large tumors, and lung metastases, continuation chemotherapy with vincristine, actinomycin, and ifosfamide (plus lung radiotherapy for patients with lung metastases) is being compared with busulfan and melphalan megatherapy.62 The COG in North America is contributing patients with lung metastases to this study.
Patients with metastatic ES/PNET, especially with metastases outside the lung, have a dismal prognosis. The role of megatherapy (high-dose chemotherapy followed by stem cell rescue) in patients with high-risk and/or metastatic or relapsed ES/PNET remains quite controversial, with most studies reporting about a 20% to 30% 2- to 3-year EFS.63,64 Patients with metastatic ES/PNET and those who have experienced relapse remain the most challenging and discouraging to treat. Results from a recent COG study incorporating the addition of metronomic antiangiogenic therapy with vinblastine and celecoxib to the VDC/IE backbone are pending.
Local control in patients with ES/PNET may be via surgery, radiation, or both. No prospective randomized study has evaluated the merits of different local control modalities. Although nearly all prospective studies suggest improved local control and EFS outcomes for patients receiving surgery, patients receiving radiotherapy generally have adverse prognostic factors, such as large tumor size, that likely contribute to their poorer outcomes. Local control has improved significantly with the addition of IE chemotherapy. Local failure rates in the Intergroup INT-0091 study were remarkable for only 9% local recurrence after radiotherapy, 5% after surgery, and 2% after combined surgery and radiotherapy (P=NS).59 However, because of the risks of second malignancy and the long-term morbidity seen after radiation treatment for ES/PNET in young people,65 surgery is the treatment of choice in North America, with 65% of patients treated in the most recent COG trial receiving surgery alone for local therapy.66 Radiation may be used in addition to surgery when surgical margins are small and in patients who are opposed to amputation. In North America, this accounts for an additional 15% of patients, with the remaining 20% receiving radiotherapy alone for either unresectable tumors or tumors in which the morbidity associated with resection is unacceptable.
Approximately two-thirds of ES/PNET tumors occur in the appendicular skeleton, where the advances in limb salvage resection and reconstruction described previously (see “Osterosarcoma” in the “Multidisciplinary Therapy and Prognosis” section) may be employed. The selection of local control modality for axial ES is more difficult. The extent of these tumors, the complex local anatomy of the pelvis and spine, and the functional deficits associated with resection of involved neurologic structures increase both the morbidity associated with surgery and the uncertainty of achieving a wide margin of resection. Retrospective studies have suggested a benefit of surgery for pelvic ES/PNET but suffer from a potential selection bias in assigning treatment modalities.67 Analysis of patients treated through the COG INT-0091 study68 showed no significant difference in local control rates of pelvic ES/PNET between radiation or surgery, or both. In this study, treatment was not randomly assigned, and patients treated with both surgery and radiation had a risk of local recurrence of 10.5% compared with 25% for either surgery or radiation, a difference that did not reach statistical significance.
Despite limitations of retrospective evaluations and lack of statistical significance, multiple studies suggest a benefit of surgery for axial lesions.67-70 The best local control outcomes appear to be associated with combined surgery and radiotherapy.68 This finding has led to a divergence in the treatment paradigm in the European studies such that 62% of patients receive surgery and radiation for local therapy and only 19% and 18%, respectively, receive surgery or radiotherapy alone.71 Local control outcomes are similar between North American and European studies, however, even with significantly fewer North American patients receiving radiotherapy. Because of the desire to avoid radiotherapy in young patients, surgery remains the first-line treatment for local control of axial skeletal ES/PNET in North America. If a wide surgical margin is anatomically possible (1 cm of uninvolved bone, anatomic fascial boundary, or 2 cm of free tissue), radiation is not used. If postoperative analysis demonstrates a small margin or poor response to chemotherapy, postoperative radiotherapy is employed. The current COG study is investigating the importance of chemotherapy response and margin width to further evaluate the need for adjuvant radiotherapy after good response, even with close margins. Radiotherapy alone is reserved for patients presenting with disease still unresectable after induction chemotherapy or those who are unwilling to accept the morbidity and functional loss associated with surgical resection.
The poor overall survival in patients with metastatic disease has not changed significantly despite decades of research and intense therapy. Because of the poor survival in this group, radiotherapy is often the treatment of choice for local control because long-term risks for second malignancy and morbidity are less of a concern. In addition, short-term or acute toxicity is often less with radiotherapy than with surgery. The exception is patients with lung-only metastases amenable to surgical resection. Current protocols recommend local therapy for all known sites of metastatic disease if possible. Recent evidence suggests that the approach of aggressive local therapy may be associated with improved EFS in this group of patients.72,73
Rhabdomyosarcoma
Treatment for RMS incorporating risk-adapted treatment approaches; multidisciplinary care utilizing chemotherapy, surgery, and radiotherapy; improved supportive care; and participation in clinical trials has resulted in approximately 70% of patients being cured. Prognostic factors that are taken into consideration for treatment stratification by the COG include tumor site (favorable vs unfavorable), size, and histology; completeness of resection before chemotherapy; and lymph node and metastatic status. In North America, staging for RMS utilizes a modified TNM approach that takes into account primary site of tumor (favorable vs unfavorable), lymph node status, size of tumor (≤5 cm or >5 cm), and clinical group before beginning chemotherapy (group I, complete resection; group II, microscopic residual disease; group III, gross residual disease; and group IV, metastases). European groups also take age into consideration when allocating patients to risk groups for treatment.74,75
The chemotherapy backbone utilized in North America is vincristine, actinomycin, and cyclophosphamide, whereas the European studies have substituted ifosfamide for cyclophosphamide in their studies.75-77 Approximately 90% of patients with favorable histology, completely resected (or only microscopic residual disease) tumors in favorable sites (eg, the female genital tract, paratesticular area, nonparameningeal head and neck region), or nonresected orbital tumors (all low-risk disease) can be cured with minimal therapy consisting of vincristine and actinomycin, with or without lower doses of cyclophosphamide, and radiation in the case of residual disease.78,79
Treatment for intermediate-risk patients (patients with nonmetastatic disease with unfavorable [alveolar] histology of any stage or patients with unresectable tumor in unfavorable primary sites) is more intense, and North American studies are exploring the role of topoisomerase inhibitors. However, the most recent randomized North American study of intermediate-risk RMS did not show improved outcome for patients treated with a regimen of vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide.80 The current randomized North American intermediate-risk RMS trial is evaluating the role of irinotecan as well as early radiotherapy for this group of patients.81 The overall 4-year failure-free survival for intermediate-risk RMS is approximately 70%.80
For patients with very high-risk disease, in particular those with disease metastatic to 3 or more sites, bone or marrow involvement, alveolar histology, and unfavorable sites, the outcome unfortunately remains dismal, with less than 20% of patients surviving.82 Megatherapy has not resulted in improved outcome in these patients.83-85 The COG is exploring dose-intense multiagent chemotherapy with vincristine, doxorubicin, cyclophosphamide, ifosfamide, temozolomide, and cixutumumab for these patients, whereas the European group is adding doxorubicin to vincristine, dactinomycin, and ifosfamide and then evaluating the addition of vinorelbine and cyclophosphamide as maintenance therapy in a randomized fashion.
The approach to local tumor control in RMS is to avoid mutilating or disfiguring surgical procedures. Radiotherapy is generally used for patients whose tumor cannot be completely resected. There have been philosophical differences in the use of radiotherapy between some of the European and North American groups. Because some patients whose tumors are initially incompletely resected but who have good response to chemotherapy (and in some situations second-look surgical procedures) can in fact be cured without radiotherapy, some European study groups have attempted to omit radiotherapy for certain subsets of patients. This has resulted in higher local recurrence rates but in some cases the same overall survival rates with aggressive salvage therapy, and thus an increased “total burden of therapy.”86 The dilemma is whether to accept a higher local failure rate by omitting radiotherapy for some patients, who when or if they experience recurrence will need more intense retreatment for salvage and thus more therapy overall, or whether to administer radiotherapy to all patients with group III tumors to decrease local recurrence rates. Until a more detailed molecular and genetic understanding of tumors is available, it is difficult to determine in which patients one can safely omit radiotherapy and avoid local recurrence and which patients are at a higher risk for local recurrence that then results in significant additional therapy with its attendant short-term and long-term morbidity.87
Late Effects
Despite the advances in cure rates for children and adolescents with these tumors, the long-term consequences of aggressive chemotherapy, radiotherapy, and surgery can be substantial. Survivors are much more likely than controls to have at least one, and often multiple, chronic medical conditions. Examples are cardiac toxicity from anthracyclines, hearing loss or tinnitus from cisplatin, infertility from alkylator therapy or radiation to the pelvis, endocrine complications usually from radiation to reproductive organs or the hypothalamic-pituitary axis, bladder dysfunction from chemotherapy and radiation, and second malignant neoplasms due to radiation and/or chemotherapy.88-92 Postpubertal boys should be offered semen cryopreservation before initiation of therapy. Cryopreservation of oocytes or embryos (the latter requires use of donor sperm) is possible for postpubertal females but may delay treatment initiation. Experimental cryopreservation of testicular or ovarian tissue has been reported in the literature but is not a current standard of care.
All children who receive radiotherapy as a component of treatment are at risk for secondary radiation-induced malignancy. The average latency period is 7 years, but the risk does not appear to decline over time. Results from the Childhood Cancer Survivor Study indicate a risk of 8% over 30 years for tumors excluding nonmelanoma skin cancers and an additional 9% risk of skin cancer; ES patients appear to be particularly at risk.90,93,94 Radiation to soft tissues and bones within the treatment field can lead to skeletal deformity and growth retardation. Radiation to the thorax can result in pulmonary fibrosis and cardiac dysfunction, and radiation to the pelvis can result in infertility.90,95 Long-term follow-up of these patients is mandatory. At the end of therapy, patients should be provided with a comprehensive treatment summary detailing what therapies were received to assist clinicians in tailoring follow-up appropriately. Patients can be followed up in survivorship clinics or by primary care physicians who can all have access to the treatment summary as well as the COG long-term follow-up guidelines. The COG has developed treatment-based long-term follow-up guidelines that are available online for help in caring for survivors of childhood cancer.96
New Directions
Recently, the importance of insulin-like growth factor receptor in promoting tissue growth and the contribution of the insulin-like growth factor system to cellular proliferation and immortality in a wide variety of cancers have been recognized. Overexpression of IGF1 and IGF2 have been reported in ES, OS, and RMS, and high expression of IGF1R has also been reported in these and other cancers. In vitro data have demonstrated the key roles of IGF1R in pediatric sarcomas in particular.97-99 Blockade of insulin-like growth factor receptor pathways as therapeutic targets are being explored in current therapeutic studies.100-102 Targeting angiogenesis using inhibitors of vascular endothelial growth factor such as bevacizumab and inhibition of the mTOR (mammalian target of rapamycin) pathway with agents such as temsirolimus are also being explored on the basis of both preclinical and early clinical data.103-107 Small-molecule tyrosine kinase inhibitors, some of which are approved for treatment of renal cell cancer, are also being investigated in pediatric phase 1 studies for refractory solid tumors.
Advances in orthopedic techniques hold promise for patients who require amputations. Continued advancements in transcutaneous osseointegration allow the potential for firm prosthetic fixation even in patients with short residual limbs; initial results of this technique are promising, and infectious complications are surprisingly rare.108,109 Because of the need to minimize the risk of infection during chemotherapy for OS and ES, the formation of the transcutaneous attachment is probably best delayed until completion of chemotherapy in patients treated in this manner.
Advanced radiotherapy techniques are also expected to improve the outcomes for unresectable tumors while minimizing morbidity and, potentially, late effects of therapy for most patients. Particle radiotherapy, the most common being proton radiotherapy, is currently being utilized for treatment of pediatric sarcomas.110,111 In contrast to photons, protons and other charged particles have a discrete stopping distance that minimizes exposure of normal tissues to unnecessary radiotherapy112 as well as allowing dose escalation and improved local control in tumors adjacent to critical structures such as the spine and skull base.113 Second malignancy risk reduction is one of the expected long-term improvements in late effects because the radiation exposure is generally reduced by 60% to 70% in comparison to photon plans.114 Heavier, larger ions, such as carbon, have the additional benefit of increased radiobiologic effectiveness; that is, they can overcome radiation resistance, in addition to their precise stopping edge. Heavier particles are being explored in unresectable OSs with promising results.55,115 Access to particle therapy is currently very limited. In centers that do not have accesss to particle therapy, advances in patient immobilization and image guidance are allowing dose escalation with photon techniques, albeit with an increased low-dose radiation exposure compared with that associated with particle therapy. Stereotactic body radiotherapy is the use of highly conformal radiation treatment delivered with daily image guidance to allow for reduction in margins of error.116 Robotic tables that adjust patient position to within 1 mm or 1 degree of rotation and periodic imaging even during treatment are used to minimize risk of setup error. Treatments can be given in the traditional fractionation pattern (ie, over 5-6 weeks) or in shorter courses (hypofractionation). Hypofractionated treatment is another technique to overcome radioresistance and is being investigated with sarcomas in the axial skeleton with promising results.117,118
Conclusion
Recent advances in the multidisciplinary management of musculoskeletal tumors in children and adolescents have enabled most children and adolescents with nonmetastatic disease to be cured; however, late effects are not inconsequential. It is hoped that during the next decade we will discover which patients can have therapy reduced and still have the same good outcome, and new approaches with targeted agents and new surgical and radiotherapeutic techniques will be developed for patients with metastatic disease.
Article Highlights.
-
■
The most common malignant musculoskeletal tumors of childhood and adolescence are osteosarcoma, Ewing sarcoma, and rhabdomyosarcoma.
-
■
With multimodality therapy, including chemotherapy and/or surgery and radiotherapy, the majority of patients with nonmetastatic disease can be expected to survive. Patients with metastatic disease have a much worse prognosis.
-
■
Long-term sequelae of therapy, including second malignancy from radiation and certain chemotherapeutic agents, infertility due to radiotherapy and alkylating agents, cardiac toxicity from anthracyclines, and other late effects, are common, making long-term monitoring of these patients mandatory.
-
■
Understanding of molecular mechanisms and pathways of disease has resulted in the development of studies exploring targeted therapies in hopes of improving the outcome. Examples include inhibition of the insulin-like growth factor pathway and inhibition of angiogenesis.
-
■
New techniques of radiation therapy such as proton therapy have the potential to decrease late effects of radiation.
-
■
Close collaboration among members of the multidisciplinary team including oncologists, radiation oncologists, surgeons, pathologists, and radiologists is imperative in managing these complex patients to ensure the best comprehensive approach.
Acknowledgments
The authors would like to thank Karen Fasbender for help in manuscript preparation and submission.
Supplemental Online Material
Author Interview Video
References
- 1.Mirabello L., Troisi R.J., Savage S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009;115(7):1531–1543. doi: 10.1002/cncr.24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esiashvili N., Goodman M., Marcus R.B., Jr Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance, Epidemiology, and End Results data. J Pediatr Hematol Oncol. 2008;30(6):425–430. doi: 10.1097/MPH.0b013e31816e22f3. [DOI] [PubMed] [Google Scholar]
- 3.Ognjanovic S., Linabery A.M., Charbonneau B., Ross J.A. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer. 2009;115(18):4218–4226. doi: 10.1002/cncr.24465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohseny A.B., Szuhai K., Romeo S. Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. J Pathol. 2009;219(3):294–305. doi: 10.1002/path.2603. [DOI] [PubMed] [Google Scholar]
- 5.Gorlick R. Current concepts on the molecular biology of osteosarcoma. Cancer Treat Res. 2009;152:467–478. doi: 10.1007/978-1-4419-0284-9_27. [DOI] [PubMed] [Google Scholar]
- 6.Guillou L., Aurias A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 2010;456(2):201–217. doi: 10.1007/s00428-009-0853-4. [DOI] [PubMed] [Google Scholar]
- 7.Folpe A.L., Inwards C.Y. Saunders/Elsevier; Philadelphia, PA: 2010. Bone and Soft Tissue Pathology. [Google Scholar]
- 8.Cesari M., Alberghini M., Vanel D. Periosteal osteosarcoma: a single-institution experience. Cancer. 2011;117(8):1731–1735. doi: 10.1002/cncr.25718. [DOI] [PubMed] [Google Scholar]
- 9.Kurt A.-M., Unni K.K., McLeod R.A., Pritchard D.J. Low-grade intraosseous osteosarcoma. Cancer. 1990;65(6):1418–1428. doi: 10.1002/1097-0142(19900315)65:6<1418::aid-cncr2820650629>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 10.Han I., Oh J.H., Na Y.G., Moon K.C., Kim K.S. Clinical outcome of parosteal osteosarcoma. J Surg Oncol. 2008;97(2):146–149. doi: 10.1002/jso.20902. [DOI] [PubMed] [Google Scholar]
- 11.Okada K., Frassica F.J., Sim F.H., Beabout J.W., Bond J.R., Unni K.K. Parosteal osteosarcoma: a clinicopathological study. J Bone Joint Surg Am. 1994;76(3):366–378. doi: 10.2106/00004623-199403000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Raymond A.K. Surface osteosarcoma. Clin Orthop Relat Res. 1991;270:140–148. [PubMed] [Google Scholar]
- 13.Sheth D.S., Yasko A.W., Raymond A.K. Conventional and dedifferentiated parosteal osteosarcoma: diagnosis, treatment, and outcome. Cancer. 1996;78(10):2136–2145. [PubMed] [Google Scholar]
- 14.Rose P.S., Dickey I.D., Wenger D.E., Unni K.K., Sim F.H. Periosteal osteosarcoma: long-term outcome and risk of late recurrence. Clin Orthop Relat Res. 2006;453:314–317. doi: 10.1097/01.blo.0000229341.18974.95. [DOI] [PubMed] [Google Scholar]
- 15.Weiss S.W., Goldblum J.R. 5th ed. Mosby Elsevier; Philadelphia, PA: 2008. Enzinger and Weiss' Soft Tissue Tumors. [Google Scholar]
- 16.Folpe A.L., Goldblum J.R., Rubin B.P. Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol. 2005;29(8):1025–1033. [PubMed] [Google Scholar]
- 17.Khoury J.D. Ewing sarcoma family of tumors: a model for the new era of integrated laboratory diagnostics. Expert Rev Mol Diagn. 2008;8(1):97–105. doi: 10.1586/14737159.8.1.97. [DOI] [PubMed] [Google Scholar]
- 18.Le Deley M.C., Delattre O., Schaefer K.L. Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol. 2010;28(12):1982–1988. doi: 10.1200/JCO.2009.23.3585. [DOI] [PubMed] [Google Scholar]
- 19.van Doorninck J.A., Ji L., Schaub B. Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol. 2010;28(12):1989–1994. doi: 10.1200/JCO.2009.24.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parham D.M. Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod Pathol. 2001;14(5):506–514. doi: 10.1038/modpathol.3880339. [DOI] [PubMed] [Google Scholar]
- 21.Parham D.M. Correspondence re: Parham DM: Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod Pathol. 2001;14(10):1068. doi: 10.1038/modpathol.3880339. [Mod Pathol. 2001;14:506-514] [DOI] [PubMed] [Google Scholar]
- 22.Croes R., Debiec-Rychter M., Cokelaere K., De Vos R., Hagemeijer A., Sciot R. Adult sclerosing rhabdomyosarcoma: cytogenetic link with embryonal rhabdomyosarcoma. Virchows Arch. 2005;446(1):64–67. doi: 10.1007/s00428-004-1131-0. [DOI] [PubMed] [Google Scholar]
- 23.Dias P., Chen B., Dilday B. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol. 2000;156(2):399–408. doi: 10.1016/S0002-9440(10)64743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bahrami A., Gown A.M., Baird G.S., Hicks M.J., Folpe A.L. Aberrant expression of epithelial and neuroendocrine markers in alveolar rhabdomyosarcoma: a potentially serious diagnostic pitfall. Mod Pathol. 2008;21(7):795–806. doi: 10.1038/modpathol.2008.86. [DOI] [PubMed] [Google Scholar]
- 25.Sumegi J., Streblow R., Frayer R.W. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer. 2010;49(3):224–236. doi: 10.1002/gcc.20731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallego Melcón S., Sánchez de Toledo Codina J. Molecular biology of rhabdomyosarcoma. Clin Transl Oncol. 2007;9(7):415–419. doi: 10.1007/s12094-007-0079-3. [DOI] [PubMed] [Google Scholar]
- 27.Davicioni E., Anderson J.R., Buckley J.D., Meyer W.H., Triche T.J. Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: a report from the Children's Oncology Group. J Clin Oncol. 2010;28(7):1240–1246. doi: 10.1200/JCO.2008.21.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mankin H.J., Mankin C.J., Simon M.A. The hazards of biopsy, revisited: for the members of the Musculoskeletal Tumor Society. J Bone Joint Surg Am. 1996;78(5):656–663. doi: 10.2106/00004623-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Völker T., Denecke T., Steffen I. Positron emission tomography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol. 2007;25(34):5435–5441. doi: 10.1200/JCO.2007.12.2473. [DOI] [PubMed] [Google Scholar]
- 30.Hawkins D.S., Schuetze S.M., Butrynski J.E. [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. J Clin Oncol. 2005;23(34):8828–8834. doi: 10.1200/JCO.2005.01.7079. [DOI] [PubMed] [Google Scholar]
- 31.Hawkins D.S., Conrad E.U., III, Butrynski J.E., Schuetze S.M., Eary J.F. [F-18]-fluorodeoxy-D-glucose-positron emission tomography response is associated with outcome for extremity osteosarcoma in children and young adults. Cancer. 2009;115(15):3519–3525. doi: 10.1002/cncr.24421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bielack S., Jürgens H., Jundt G. Osteosarcoma: the COSS experience. Cancer Treat Res. 2009;152:289–308. doi: 10.1007/978-1-4419-0284-9_15. [DOI] [PubMed] [Google Scholar]
- 33.Meyers P.A., Schwartz C.L., Krailo M.D., Children's Oncology Group Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival—a report from the Children's Oncology Group. J Clin Oncol. 2008;26(4):633–638. doi: 10.1200/JCO.2008.14.0095. [DOI] [PubMed] [Google Scholar]
- 34.Bacci G., Fabbri N., Balladelli A., Forni C., Palmerini E., Picci P. Treatment and prognosis for synchronous multifocal osteosarcoma in 42 patients. J Bone Joint Surg Br. 2006;88(8):1071–1075. doi: 10.1302/0301-620X.88B8.17809. [DOI] [PubMed] [Google Scholar]
- 35.Kager L., Zoubek A., Pötschger U., Cooperative German-Austrian-Swiss Osteosarcoma Study Group Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol. 2003;21(10):2011–2018. doi: 10.1200/JCO.2003.08.132. [DOI] [PubMed] [Google Scholar]
- 36.Bielack S.S., Kempf-Bielack B., Delling G. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol. 2002;20(3):776–790. doi: 10.1200/JCO.2002.20.3.776. [DOI] [PubMed] [Google Scholar]
- 37.Bacci G., Ferrari S., Bertoni F. Long-term outcome for patients with nonmetastatic osteosarcoma of the extremity treated at the Istituto Ortopedico Rizzoli according to the Istituto Ortopedico Rizzoli/Osteosarcoma-2 Protocol: an updated report. J Clin Oncol. 2000;18(24):4016–4027. doi: 10.1200/JCO.2000.18.24.4016. [DOI] [PubMed] [Google Scholar]
- 38.Euramos I Trial. http://www.ctu.mrc.ac.uk/euramos/euramos_i_trial.asp The European and American Osteosarcoma Study Group Web site. Accessed April 4, 2012.
- 39.National Cancer Institute Phase II study of chemotherapy with or without trastuzumab (Herceptin®) in patients with metastatic osteosarcoma. http://www.cancer.gov/clinicaltrials/search/view?cdrid=68882&version=HealthProfessional&protocolsearchid=9547220 National Cancer Institute Web site. Accessed April 9, 2012.
- 40.Meyers P.A., Healey J.H., Chou A.J. Addition of pamidronate to chemotherapy for the treatment of osteosarcoma. Cancer. 2011;117(8):1736–1744. doi: 10.1002/cncr.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.National Cancer Institute Pilot study of zoledronic acid in combination with standard chemotherapy in patients with newly diagnosed metastatic osteosarcoma: National Cancer Institute Web site. http://www.cancer.gov/clinicaltrials/search/view?cdrid=612613&version=HealthProfessional&protocolsearchid=9547220 Accessed April 9, 2012.
- 42.Ayerza M.A., Farfalli G.L., Aponte-Tinao L., Muscolo D.L. Does increased rate of limb-sparing surgery affect survival in osteosarcoma? Clin Orthop Relat Res. 2010;468(11):2854–2859. doi: 10.1007/s11999-010-1423-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neel M.D., Wilkins R.M., Rao B.N., Kelly C.M. Early multicenter experience with a noninvasive expandable prosthesis. Clin Orthop Relat Res. 2003;415:72–81. doi: 10.1097/01.blo.0000093899.12372.25. [DOI] [PubMed] [Google Scholar]
- 44.Musculo D.L., Ayerza M.A., Aponte-Tinao L., Farfalli G.L. Allograft reconstruction after sarcoma excision in children younger than 10 years old. Clin Orthop Relat Res. 2008;466(8):1856–1862. doi: 10.1007/s11999-008-0303-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abed Y.Y., Beltrami G., Campanacci D.A., Innocenti M., Scoccianti G., Capanna R. Biological reconstruction after resection of bone tumours around the knee: long-term follow-up. J Bone Joint Surg Br. 2009;91(10):1366–1372. doi: 10.1302/0301-620X.91B10.22212. [DOI] [PubMed] [Google Scholar]
- 46.Farfalli G.L., Boland P.J., Morris C.D., Athanasian E.A., Healey J.H. Early equivalence of uncemented press-fit and Compress femoral fixation. Clin Orthop Relat Res. 2009;467(11):2792–2799. doi: 10.1007/s11999-009-0912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cañadell J., Forriol F., Cara J.A. Removal of metaphyseal bone tumours with preservation of the epiphysis: physeal distraction before excision. J Bone Joint Surg Br. 1994;76(1):127–132. [PubMed] [Google Scholar]
- 48.Teplyakov V., Solovyev J., Aliev M. Transosseous osteosynthesis by Ilizarov in the treatment of patients with primary malignant tumours of long bones. Proceedings of the 17th Annual Meeting of the European Musculoskeletal Oncology Society; June 9-11, 2004. Oslo, Norway. [Google Scholar]
- 49.Rödl R.W., Pohlmann U., Gosheger G., Lindner N.J., Winkelmann W. Rotationplasty—quality of life after 10 years in 22 patients. Acta Orthop Scand. 2002;73(1):85–88. doi: 10.1080/000164702317281468. [DOI] [PubMed] [Google Scholar]
- 50.Fuchs B., Kotajarvi B.R., Kaufman K.R., Sim F.H. Functional outcome of patients with rotationplasty about the knee. Clin Orthop Relat Res. 2003;415:52–58. doi: 10.1097/01.blo.0000093896.12372.c1. [DOI] [PubMed] [Google Scholar]
- 51.Highsmith M.J., Kahle J.T., Bongiorni D.R., Sutton B.S., Groer S., Kaufman K.R. Safety, energy efficiency, and cost efficacy of the C-Leg for transfemoral amputees: a review of the literature. Prosthet Orthot Int. 2010;34(4):362–377. doi: 10.3109/03093646.2010.520054. [published correction appears in Prosthet Orthot Int. 2011;35(1):113] [DOI] [PubMed] [Google Scholar]
- 52.Fuchs B., Yaszemski M.J., Sim F.H. Combined posterior pelvis and lumbar spine resection for sarcoma. Clin Orthop Relat Res. 2002;397:12–18. doi: 10.1097/00003086-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 53.Ciernik I.F., Niemierko A., Harmon D.C. Proton-based radiotherapy for unresectable or incompletely resected osteosarcoma. Cancer. 2011;117(19):4522–4530. doi: 10.1002/cncr.26037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blattmann C., Oertel S., Schulz-Ertner D. Non-randomized therapy trial to determine the safety and efficacy of heavy ion radiotherapy in patients with non-resectable osteosarcoma. BMC Cancer. 2010;10:96. doi: 10.1186/1471-2407-10-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jingu K., Tsujii H., Mizoe J.E., Organizing Committee for the Working Group for Head-and-Neck Cancer Carbon ion radiation therapy improves the prognosis of unresectable adult bone and soft-tissue sarcoma of the head and neck. Int J Radiat Oncol Biol Phys. 2012;82(5):2125–2131. doi: 10.1016/j.ijrobp.2010.08.043. [DOI] [PubMed] [Google Scholar]
- 56.Hillegonds D.J., Franklin S., Shelton D.K., Vijayakumar S., Vijayakumar V. The management of painful bone metastases with an emphasis on radionuclide therapy. J Natl Med Assoc. 2007;99(7):785–794. [PMC free article] [PubMed] [Google Scholar]
- 57.Schwarz R., Bruland O., Cassoni A., Schomberg P., Bielack S. The role of radiotherapy in oseosarcoma. Cancer Treat Res. 2009;152:147–164. doi: 10.1007/978-1-4419-0284-9_7. [DOI] [PubMed] [Google Scholar]
- 58.Grier H.E., Krailo M.D., Tarbell N.J. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 59.Granowetter L., Womer R., Devidas M. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol. 2009;27(15):2536–2541. doi: 10.1200/JCO.2008.19.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Womer R.B., West D.C., Krailo M.D., Dickman P.S., Pawel B., Children's Oncology Group AEWS0031 Committee Randomized comparison of every-two-week v. every-three-week chemotherapy in Ewing sarcoma family tumors (ESFT) J Clin Oncol. 2008;26(15, suppl):10504. [abstract] [Google Scholar]
- 61.National Cancer Institute Phase III randomized study of adding vincristine sulfate, topotecan hydrochloride, and cyclophosphamide to standard chemotherapy in patients with non-metastatic extracranial Ewing sarcoma. http://www.cancer.gov/clinicaltrials/search/view?cdrid=687639&version=HealthProfessional&protocolsearchid=9547232 National Cancer Institute Web site.
- 62.National Cancer Institute Phase III randomized study of standard induction chemotherapy comprising vincristine, dactinomycin, ifosfamide, and etoposide followed by consolidation chemotherapy comprising vincristine, dactinomycin, and ifosfamide versus high-dose busulfan and melphalan followed by autologous peripheral blood stem cell support with or without radiotherapy and/or surgery in patients with tumor of the Ewing's family. http://www.cancer.gov/clinicaltrials/search/view?cdrid=68608&version=HealthProfessional&protocolsearchid=9547232 National Cancer Institute Web site. Accessed April 9, 2012.
- 63.Rosenthal J., Pawlowska A.B. High-dose chemotherapy and stem cell rescue for high-risk Ewing's family of tumors. Expert Rev Anticancer Ther. 2011;11(2):251–262. doi: 10.1586/era.10.215. [DOI] [PubMed] [Google Scholar]
- 64.Balamuth N.J., Womer R.B. Ewing's sarcoma. Lancet Oncol. 2010;11(2):184–192. doi: 10.1016/S1470-2045(09)70286-4. [DOI] [PubMed] [Google Scholar]
- 65.Fuchs B., Valenzuela R.G., Inwards C., Sim F.H., Rock M.G. Complications in long-term survivors of Ewing sarcoma. Cancer. 2003;98(12):2687–2692. doi: 10.1002/cncr.11891. [DOI] [PubMed] [Google Scholar]
- 66.Granowetter L., Womer R., Devidas M. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol. 2009;27(15):2536–2541. doi: 10.1200/JCO.2008.19.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Donati D., Yin J., Di Bella C. Local and distant control in non-metastatic pelvic Ewing's sarcoma patients. J Surg Oncol. 2007;96(1):19–25. doi: 10.1002/jso.20752. [DOI] [PubMed] [Google Scholar]
- 68.Yock T.I., Krailo M., Fryer C.J., Children's Oncology Group Local control in pelvic Ewing sarcoma: analysis from INT-0091—a report from the Children's Oncology Group. J Clin Oncol. 2006;24(24):3838–3843. doi: 10.1200/JCO.2006.05.9188. [DOI] [PubMed] [Google Scholar]
- 69.Yang R.S., Eckardt J.J., Eilber F.R. Surgical indications for Ewing's sarcoma of the pelvis. Cancer. 1995;76(8):1388–1397. doi: 10.1002/1097-0142(19951015)76:8<1388::aid-cncr2820760814>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 70.Frassica F.J., Frassica D.A., Pritchard D.J., Schomberg P.J., Wold L.E., Sim F.H. Ewing sarcoma of the pelvis: clinicopathologic features and treatment. J Bone Joint Surg Am. 1993;75(10):1457–1465. doi: 10.2106/00004623-199310000-00006. [DOI] [PubMed] [Google Scholar]
- 71.Dunst J., Schuck A. Role of radiotherapy in Ewing tumors. Pediatr Blood Cancer. 2004;42(5):465–470. doi: 10.1002/pbc.10446. [DOI] [PubMed] [Google Scholar]
- 72.Liu A.K., Stinauer M., Albano E., Greffe B., Tello T., Maloney K. Local control of metastatic sites with radiation therapy in metastatic Ewing sarcoma and rhabdomyosarcoma. Pediatr Blood Cancer. 2011;57(1):169–171. doi: 10.1002/pbc.23063. [DOI] [PubMed] [Google Scholar]
- 73.Haeusler J., Ranft A., Boelling T. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES) Cancer. 2010;116(2):443–450. doi: 10.1002/cncr.24740. [DOI] [PubMed] [Google Scholar]
- 74.Ferrari A., Casanova M. Current chemotherapeutic strategies for rhabdomyosarcoma. Expert Rev Anticancer Ther. 2005;5(2):283–294. doi: 10.1586/14737140.5.2.283. [DOI] [PubMed] [Google Scholar]
- 75.Sultan I., Ferrari A. Selecting multimodal therapy for rhabdomyosarcoma. Expert Rev Anticancer Ther. 2010;10(8):1285–1301. doi: 10.1586/era.10.96. [DOI] [PubMed] [Google Scholar]
- 76.Crist W.M., Anderson J.R., Meza J.L., Intergroup Rhabdomyosarcoma Study Group (IRSG) Intergroup Rhabdomyosarcoma Study-IV: results for patients with nonmetastatic disease. J Clin Oncol. 2001;19(12):3091–3102. doi: 10.1200/JCO.2001.19.12.3091. [DOI] [PubMed] [Google Scholar]
- 77.Stevens M.C., Rey A., Bouvet N. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology—SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol. 2005;23(12):2618–2628. doi: 10.1200/JCO.2005.08.130. [DOI] [PubMed] [Google Scholar]
- 78.Raney R.B., Walterhouse D.O., Meza J.L. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J Clin Oncol. 2011;29(10):1312–1318. doi: 10.1200/JCO.2010.30.4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Walterhouse D., Pappo A.S., Meza J.L. Shorter duration therapy that includes vincristine (V), dactinomycin (A), and lower doses of cyclophosphamide (C) with or without radiation therapy for patients with newly diagnosed low-risk embryonal rhabdomyosarcoma (ERMS): a report from the Children's Oncology Group (COG) J Clin Oncol. 2011;29(15, suppl):9516. [abstract] [Google Scholar]
- 80.Arndt C.A., Stoner J.A., Hawkins D.S. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children's Oncology Group Study D9803. J Clin Oncol. 2009;27(31):5182–5188. doi: 10.1200/JCO.2009.22.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.National Cancer Institute Phase III randomized study of vincristine, dactinomycin, and cyclophosphamide (VAC) versus VAC alternating with vincristine and irinotecan hydrochloride in combination with radiotherapy in patients with newly diagnosed, intermediate-risk rhabdomyosarcoma. http://www.cancer.gov/clinicaltrials/search/view?cdrid=487560&protocolsearchid=9398356&version=healthprofessional National Cancer Institute Web site. Accessed April 9, 2012.
- 82.Oberlin O., Rey A., Lyden E. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol. 2008;26(14):2384–2389. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carli M., Colombatti R., Oberlin O. European intergroup studies (MMT4-89 and MMT4-91) on childhood metastatic rhabdomyosarcoma: final results and analysis of prognostic factors. J Clin Oncol. 2004;22(23):4787–4794. doi: 10.1200/JCO.2004.04.083. [published correction appears in J Clin Oncol. 2005;23(1):248] [DOI] [PubMed] [Google Scholar]
- 84.Bisogno G., Ferrari A., Prete A. Sequential high-dose chemotherapy for children with metastatic rhabdomyosarcoma. Eur J Cancer. 2009;45(17):3035–3041. doi: 10.1016/j.ejca.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 85.Weigel B.J., Breitfeld P.P., Hawkins D., Crist W.M., Baker K.S. Role of high-dose chemotherapy with hematopoietic stem cell rescue in the treatment of metastatic or recurrent rhabdomyosarcoma. J Pediatr Hematol Oncol. 2001;23(5):272–276. doi: 10.1097/00043426-200106000-00007. [DOI] [PubMed] [Google Scholar]
- 86.Stevens M.C. Treatment for childhood rhabdomyosarcoma: the cost of cure. Lancet Oncol. 2005;6(2):77–84. doi: 10.1016/S1470-2045(05)01733-X. [DOI] [PubMed] [Google Scholar]
- 87.Donaldson S.S., Anderson J.R. Rhabdomyosarcoma: many similarities, a few philosophical differences. J Clin Oncol. 2005;23(12):2586–2587. doi: 10.1200/JCO.2005.11.909. [editorial] [DOI] [PubMed] [Google Scholar]
- 88.Ritchey M., Ferrer F., Shearer P., Spunt S.L. Late effects on the urinary bladder in patients treated for cancer in childhood: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2009;52(4):439–446. doi: 10.1002/pbc.21826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nagarajan R., Kamruzzaman A., Ness K.K. Twenty years of follow-up of survivors of childhood osteosarcoma: a report from the Childhood Cancer Survivor Study. Cancer. 2011;117(3):625–634. doi: 10.1002/cncr.25446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ginsberg J.P., Goodman P., Leisenring W. Long-term survivors of childhood Ewing sarcoma: report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2010;102(16):1272–1283. doi: 10.1093/jnci/djq278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aksnes L.H., Bauer H.C., Dahl A.A. Health status at long-term follow-up in patients treated for extremity localized Ewing Sarcoma or osteosarcoma: a Scandinavian sarcoma group study. Pediatr Blood Cancer. 2009;53(1):84–89. doi: 10.1002/pbc.22027. [DOI] [PubMed] [Google Scholar]
- 92.Punyko J.A., Mertens A.C., Gurney J.G. Long-term medical effects of childhood and adolescent rhabdomyosarcoma: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer. 2005;44(7):643–653. doi: 10.1002/pbc.20310. [DOI] [PubMed] [Google Scholar]
- 93.Friedman D.L., Whitton J., Leisenring W. Subsequent neoplasms in 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2010;102(14):1083–1095. doi: 10.1093/jnci/djq238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Navid F., Billups C., Liu T., Krasin M.J., Rodriguez-Galindo C. Second cancers in patients with the Ewing sarcoma family of tumours. Eur J Cancer. 2008;44(7):983–991. doi: 10.1016/j.ejca.2008.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Armstrong G.T., Stovall M., Robison L.L. Long-term effects of radiation exposure among adult survivors of childhood cancer: results from the Childhood Cancer Survivor Study. Radiat Res. 2010;174(6):840–850. doi: 10.1667/RR1903.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Children's Oncology Group Long-term follow-up guidelines for survivors of childhood, adolescent, and young adult cancers. http://www.survivorshipguidelines.org/ Children's Oncology Group Web site. Accessed April 9, 2012.
- 97.Scotlandi K., Manara M.C., Nicoletti G. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res. 2005;65(9):3868–3876. doi: 10.1158/0008-5472.CAN-04-3192. [DOI] [PubMed] [Google Scholar]
- 98.Maloney E.K., McLaughlin J.L., Dagdigian N.E. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res. 2003;63(16):5073–5083. [PubMed] [Google Scholar]
- 99.Kolb E.A., Gorlick R., Houghton P.J. Initial testing (stage 1) of a monoclonal antibody (SCH 717454) against the IGF-1 receptor by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2008;50(6):1190–1197. doi: 10.1002/pbc.21450. [DOI] [PubMed] [Google Scholar]
- 100.Juergens H., Daw N.C., Geoerger B. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol. 2011;29(34):4534–4540. doi: 10.1200/JCO.2010.33.0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maki R.G. Small is beautiful: insulin-like growth factors and their role in growth, development, and cancer. J Clin Oncol. 2010;28(33):4985–4995. doi: 10.1200/JCO.2009.27.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pappo A.S., Patel S.R., Crowley J. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. 2011;29(34):4541–4547. doi: 10.1200/JCO.2010.34.0000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chawla S.P., Staddon A.P., Baker L.H. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol. 2012;30(1):78–84. doi: 10.1200/JCO.2011.35.6329. [DOI] [PubMed] [Google Scholar]
- 104.Ferrara N., Hillan K.J., Gerber H.P., Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 105.Gerber H.P., Kowalski J., Sherman D., Eberhard D.A., Ferrara N. Complete inhibition of rhabdomyosarcoma xenograft growth and neovascularization requires blockade of both tumor and host vascular endothelial growth factor. Cancer Res. 2000;60(22):6253–6258. [PubMed] [Google Scholar]
- 106.Houghton P.J., Morton C.L., Kolb E.A. Initial testing (stage 1) of the mTOR inhibitor rapamycin by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2008;50(4):799–805. doi: 10.1002/pbc.21296. [DOI] [PubMed] [Google Scholar]
- 107.Kurmasheva R.T., Dudkin L., Billups C., Debelenko L.V., Morton C.L., Houghton P.J. The insulin-like growth factor-1 receptor-targeting antibody, CP-751,871, suppresses tumor-derived VEGF and synergizes with rapamycin in models of childhood sarcoma. Cancer Res. 2009;69(19):7662–7671. doi: 10.1158/0008-5472.CAN-09-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tillander J., Hagberg K., Hagberg L., Brånemark R. Osseointegrated titanium implants for limb prostheses attachments: infectious complications. Clin Orthop Relat Res. 2010;468(10):2781–2788. doi: 10.1007/s11999-010-1370-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hagberg K., Brånemark R. One hundred patients treated with osseointegrated transfemoral amputation prostheses—rehabilitation perspective. J Rehabil Res Dev. 2009;46(3):331–344. [PubMed] [Google Scholar]
- 110.Childs S.K., Kozak K.R., Friedmann A.M. Proton radiotherapy for parameningeal rhabdomyosarcoma: clinical outcomes and late effects. Int J Radiat Oncol Biol Phys. 2012;82(2):635–642. doi: 10.1016/j.ijrobp.2010.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rombi B., DeLaney T.F., MacDonald S.M. Proton radiotherapy for pediatric Ewing's sarcoma: initial clinical outcomes. Int J Radiat Oncol Biol Phys. 2012;82(3):1142–1148. doi: 10.1016/j.ijrobp.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 112.Merchant T.E. Proton beam therapy in pediatric oncology. Cancer J. 2009;15(4):298–305. doi: 10.1097/PPO.0b013e3181b6d4b7. [DOI] [PubMed] [Google Scholar]
- 113.Patel S., DeLaney T.F. Advanced-technology radiation therapy for bone sarcomas. Cancer Control. 2008;15(1):21–37. doi: 10.1177/107327480801500104. [DOI] [PubMed] [Google Scholar]
- 114.Miralbell R., Lomax A., Cella L., Schneider U. Potential reduction of the incidence of radiation-induced second cancers by using proton beams in the treatment of pediatric tumors. Int J Radiat Oncol Biol Phys. 2002;54(3):824–829. doi: 10.1016/s0360-3016(02)02982-6. [DOI] [PubMed] [Google Scholar]
- 115.DeLaney T.F., Liebsch N.J., Pedlow F.X. Phase II study of high-dose photon/proton radiotherapy in the management of spine sarcomas. Int J Radiat Oncol Biol Phys. 2009;74(3):732–739. doi: 10.1016/j.ijrobp.2008.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Timmerman R.D., Kavanagh B.D., Cho L.C., Papiez L., Xing L. Stereotactic body radiation therapy in multiple organ sites. J Clin Oncol. 2007;25(8):947–952. doi: 10.1200/JCO.2006.09.7469. [DOI] [PubMed] [Google Scholar]
- 117.Levine A.M., Coleman C., Horasek S. Stereotactic radiosurgery for the treatment of primary sarcomas and sarcoma metastases of the spine. Neurosurgery. 2009;64(2, suppl):A54–A59. doi: 10.1227/01.NEU.0000339131.28485.4A. [DOI] [PubMed] [Google Scholar]
- 118.Bilsky M.H., Yenice K., Lovelock M., Yamada J. Stereotactic intensity-modulation radiation therapy for vertebral body and paraspinal tumors. Neurosurg Focus. 2001;11(6):e7. doi: 10.3171/foc.2001.11.6.8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Author Interview Video