

Abstract
There are now 24 antiepileptic drugs (AEDs) approved for use in epilepsy in the United States by the Food and Drug Administration. A literature search was conducted using PubMed, MEDLINE, and Google for all English-language articles that discuss newly approved AEDs and the use of AEDs in epilepsy in the United States from January 1, 2008, through December 31, 2011. Five new agents were identified that have come onto the market within the past 2 years. Moreover, 3 trends involving AEDs have become clinically important and must be considered by all who treat patients with epilepsy. These trends include issues of generic substitution of AEDs, pharmacogenomics predicting serious adverse events in certain ethnic populations, and the issue of the suicide risk involving the entire class of AEDs. This article discusses the most recent AEDs approved for use in the United States and the 3 important trends shaping the modern medical management of epilepsy.
Abbreviations and Acronyms: AED, antiepileptic drug; FDA, Food and Drug Administration; GABA, γ-aminobutyric acid; LGS, Lennox-Gastaut syndrome
Modern pharmacology has achieved a marked expansion in the number of therapies for conditions once thought to be unmanageable. In neurology, this growth has been particularly noted in options for the management of epilepsy. The number of antiepileptic drugs (AEDs) approved for use in the United States alone has more than doubled in the past 15 years (Table 1). Currently, 24 AEDs and 1 device are marketed in the United States, and additional agents are available worldwide. Given all of the drugs available for the management of epilepsy, it is important to consider the larger domain of epilepsy medications and to assess what new agents have been recently approved and what trends are occurring to best manage the considerable amount of information available regarding AEDs.
TABLE 1.
Antiepileptic Drugs and Devices Currently Approved by the Food and Drug Administration
| Before 1993 | 1993-2005 | 2009-2011 |
|---|---|---|
| Carbamazepine | Felbamate | Vigabatrin |
| Clonazepam | Gabapentin | Rufinamide |
| Diazepam | Lamotrigine | Lacosamide |
| Ethosuccimide | Levetiracetam | Clobazam |
| Lorazepam | Oxcarbazepine | Ezogabine |
| Phenobarbital | Pregabalin | |
| Phenytoin | Tiagabine | |
| Primidone | Topiramate | |
| Valproic acid | Vagus nerve stimulation | |
| Zonisamide |
To understand the current discipline of AEDs for epilepsy, a literature search was performed using PubMed, MEDLINE, and Google for all English-language articles published concerning either newly approved AEDs or use of AEDs for epilepsy from January 1, 2008, through December 31, 2011. Only studies that discussed newly approved AEDs in the United States or that discussed important clinical management issues of AEDs were included. The search found that during the past 2 years alone, 5 new AEDs have been introduced in the United States.
Given that seizures, the main symptom of epilepsy, are defined as abnormal paroxysmal electrical perturbations of cortical neural networks, most AEDs work via specific mechanisms that either diminish neuronal excitability (sodium and calcium channel modulation) or increase neuronal inhibition through interactions at various parts of γ-aminobutyric acid (GABA) receptors.1 Most AEDs are believed to exert their effect through a number of concurrent mechanisms and at both the excitatory and inhibitory synapses. A detailed discussion regarding AED mechanisms of action is beyond the scope of this article. However, because 2 of the AEDs have novel mechanisms of action, it is important to appreciate a somewhat simplistic overview of how AEDs work to appreciate the complexity of this therapy.
Figure 1 diagrams where AEDs are believed to exert their therapeutically relevant effects,1,2 with an emphasis on presynaptic effects. Currently available AEDs are thought to affect several molecules at the excitatory synapse. These molecules include voltage-gated Na+ channels, synaptic vesicle glycoprotein 2A, the α2δ subunit of the voltage-gated Ca2+ channel, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors, and N-methyl-d-aspartate receptors. The goal of targeting the excitatory receptors is to decrease depolarization-induced Ca+ influx and vesicular release of neurotransmitters. Lacosamide is thought to enhance slow inactivation of voltage-gated Na+ channels. This effect is different from that of other AEDs listed, which are thought to enhance fast inactivation, such as rufinamide (Figure 2). Levetiracetam is the only available drug that binds to synaptic vesicle glycoprotein 2A, which might have a role in neurotransmitter release (Tables 2 and 3). Gabapentin and pregabalin bind to the a2δ subunit of voltage-gated Ca2+ channels, which is thought to be associated with a decrease in neurotransmitter release. Ezogabine is the only drug known to reduce neuronal excitability by interacting with potassium channels. Excitatory neurotransmission at the postsynaptic membrane can be limited by topiramate (acting on α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid and kainate receptors) and felbamate (acting on N-methyl-d-aspartate receptors).
FIGURE 1.
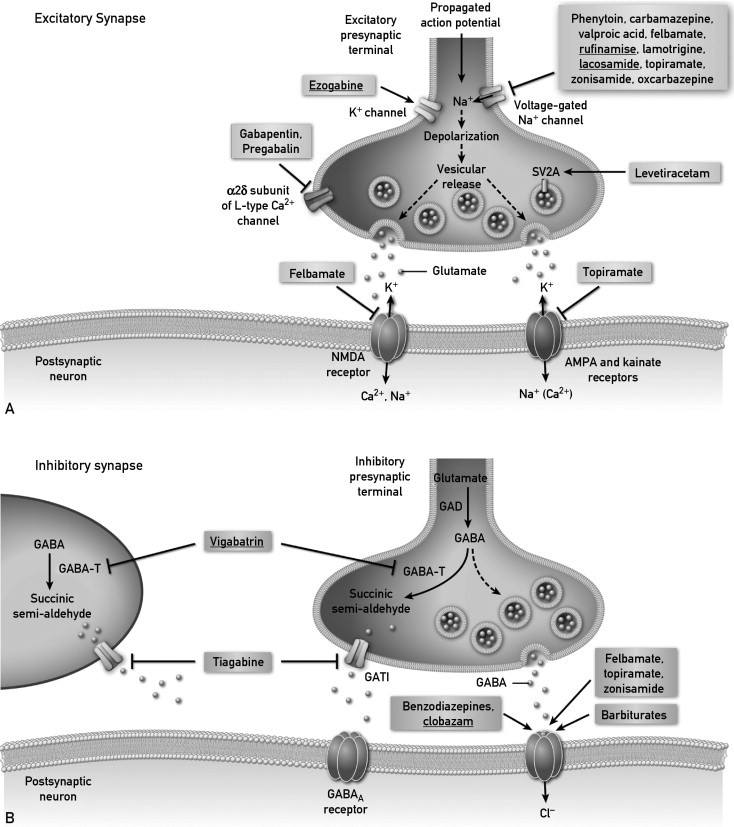
Figure and legend are modified and reprinted, with permission, from Bialer and White1 and Rho.2 Please see text for details. It is important to note that multiple mechanisms of action are ascribed to any given antiepileptic drug (AED). The diagram emphasizes the major mechanism of action for each AED. Part A shows drugs which may prevent seizures by acting upon excitatory synapses and Part B shows drugs which may affect inhibitory synapses. GABA = y-aminobutyric acid; GABA-T = GABA transaminase; GAD = glutamic acid decarboxylase.
Adapted from Nat Rev Drug Discov1 and Epilepsia,2 with permission.
FIGURE 2.
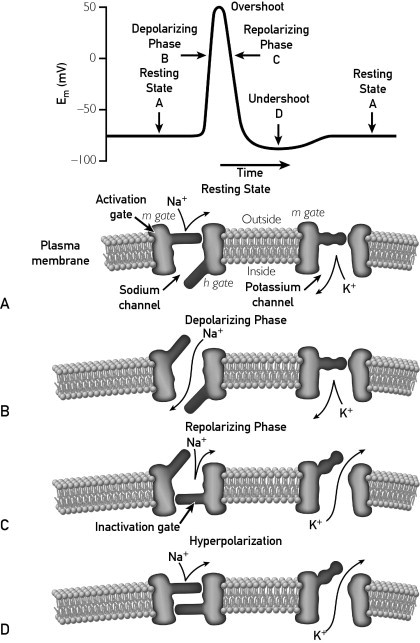
This illustration further conceptualizes the complex role of voltage gates sodium and potassium channels in neuronal excitability. There are 4 states of the voltage-gated channel opening and closing as it relates to the generation of an action potential: resting, depolarizing, repolarizing, and hyperpolarization. See A-D. The diagram conceptualizes what is occurring at the membrane level that corresponds to the action potential. One sees the sodium channel open correlating to the depolarizing phase. Repolarization occurs with closure of the inactivating gate and activating gate m. Potassium influx correlates with hyperpolarization stabilizing the membrane and bringing the system back to the resting state. Carbamazepine, felbamate lacosamide, lamotrigine oxcarbaxepine, phenytoin, rufinamide, topiramate, zonisamide, and valproic acid all modulate this channel to some degree. Lacosamide appears to modulate the complex slightly differently from the other sodium channels. Ezogabine appears to modulate the system at the potassium channel. Understanding the complexity of this channel helps to better demonstrate how varying antiepileptic drugs functioning at various parts of the channel may have complementary benefits. This has yet to be proven clinically.
Reprinted from Webanatomy.3
TABLE 2.
AEDs Approved in 2009-2011
| AED | FDA indication | Putative mechanism |
|---|---|---|
| Vigabatrin | Infantile spasms/add-on for partial epilepsy | Irreversible inhibition of GABA transaminase |
| Rufinamide | Atonic seizures | Sodium channel modulation |
| Lacosamide | Add-on for partial epilepsy | Sodium channel modulation |
| Clobazam | Lennox-Gastaut syndrome | GABAA binding |
| Ezogabine | Add-on for partial epilepsy | Potassium channel modulation |
AED = antiepileptic drug; FDA = Food and Drug Administration; GABA = γ-aminobutyric acid.
TABLE 3.
Summary of Characteristics of Recently Approved AEDs in the US
| Drug | Available doses | Age group | Initial dose | Titration | Maintenance dose | Drug interactions |
|---|---|---|---|---|---|---|
| Ezogabine | 50-, 200-, 300-, and 400-mg tablets | Adults | 100 mg 3 times daily (300 mg/d) | Increase by no more than 50 mg 3 times daily, no more than 150 mg/d weekly | 200-400 mg 3 times daily (600-1200 mg/d) | Carbamazepine and phenytoin may reduce ezogabine levels; ezogabine can inhibit clearance of digoxin |
| Vigabatrin | 500-mg tablets; 500-mg oral powder for solution | Adults and children | 500 mg twice daily; 50 mg/kg daily divided twice daily | 500 mg/wk; 25-50 mg/kg daily every 3 d | 1500 mg twice daily (maximum); 150 mg/d (maximum) | Vigabatrin may reduce phenytoin levels |
| Rufinamide | 40-mg/mL oral solution; 200- and 400-mg tablets | Adults and children | 200-400 mg twice daily; 10 mg/kg daily divided twice daily | 400-800 mg every other day; 10 mg/kg every other day | 1600 mg twice daily (maximum); 45 mg/kg daily divided twice daily (maximum, 3200 mg/d) | Rufinamide may decrease carbamazepine and lamotrigine levels; rufinamide may increase phenobarbital, phenytoin, and valproic acid levels |
| Lacosamide | 10-mg/mL intravenous solution; 10-mg/mL oral solution; 50-, 100-, 150-, and 200-mg tablets | Adults | 50 mg twice daily | 50 mg twice daily every wk | 200 mg twice daily (maximum) | None |
| Clobazam | 5-, 10-, and 20-mg tablets | Adults and children ≥2 y with weight >30 kg; adults and children ≥2 y with weight ≤30 kg | 5 mg twice daily; 2.5 mg twice daily | 10 mg twice daily at 7 d; 20 mg twice daily at 14 d; 5 mg twice daily at 7 d; 10 mg twice daily at 14 d | 20 mg twice daily; 10 mg twice daily | Hormonal contraceptives metabolized by CYP3A4 may have diminished efficacy when given with clobazam; fluconazole, fluvoxamine, ticlopidine, and omeprazole may increase serum levels of clobazam |
AED = antiepileptic drug.
In addition, AEDs affect inhibitory synapses.1 Vigabatrin irreversibly inhibits GABA transaminase, which decreases the metabolism of GABA in presynaptic terminals and glial cells. The benzodiazepines, including clobazam, barbiturates, topiramate, and felbamate, have been found to enhance inhibitory neurotransmission by allosterically modulating GABAA receptor–mediated Cl− currents. However, the action of each of these drugs is different and is dependent on the subunit conformation of the GABAA receptor complex.
Figure 2 conceptualizes the neuronal sodium channel, providing a better understanding of the complexity of the receptor and how various antiseizure drugs may potentially modulate activity at this location.1,3 Novel in the recently approved group of medications is the first AED to act by potassium channel modulation (ezogabine).4-8
Although most AEDs introduced in the past 2 decades were indicated for adjunctive therapy of partial epilepsy, 3 of the 5 newest AEDs have indications only for seizures associated with specific severe epilepsy syndromes (Table 1), particularly infantile spasms and Lennox-Gastaut syndrome (LGS). Infantile spasms are brief myoclonic or tonic seizures and a common manifestation of epileptic encephalopathies, such as West syndrome, consisting of a triad of spasms, hypsarrhythmic electroencephalography, and developmental failure or regression. Lennox-Gastaut syndrome represents a devastating pediatric condition characterized by multiple seizures types, slow spike-and-wave discharge on electroencephalography, and impaired intellectual function. Given the refractory nature of the seizures associated with these syndromes, expanding effective therapeutic options for these conditions is welcome news for the patients with these devastating afflictions.
Expanding therapeutic options challenges the physician to choose the optimal agent for an individual patient. Individual AEDs are not equal and differ by indication, mechanism of action, and adverse effect profile (Tables 2 and 3. In this review, we examine the 5 most recently US Food and Drug Administration (FDA)–approved AEDs: clobazam, ezogabine, lacosamide, rufinamide, and vigabatrin (Table 1). The discussion focuses on unique factors that distinguish these newer agents from older AEDs. We also examine 3 clinically meaningful trends in current AED therapy: the nascent application of pharmacogenomics to AED response, suicide warnings and risk to patients prescribed AEDs, and increasing problems associated with generic substitution of brand-name AEDs.
Vigabatrin
Vigabatrin has been available internationally for more than 2 decades. Release in the United States was delayed because of concerns about serious adverse effects. In 2010, the FDA approved vigabatrin for 2 forms of severe epilepsy: infantile spasms and as add-on therapy for refractory partial epilepsy in adults when other options have failed.9 Vigabatrin should be prescribed only to those individuals in whom the risks from uncontrolled seizures outweigh the risks associated with drug exposure. Infantile spasms, a catastrophic epilepsy syndrome beginning in the first 2 years of life, typically are associated with a poor prognosis for seizure outcome and development. Treatment options are limited: vigabatrin and corticotropin are recognized as the only first-line therapies for this condition.
Vigabatrin's exact mechanism of action is unknown; however, it is believed to act as an irreversible inhibitor of GABA transaminase, the enzyme responsible for the metabolism of the inhibitory neurotransmitter GABA.9-11 This action results in increased levels of GABA in the central nervous system. Unlike traditional AEDs in which serum concentration is an indirect measure of therapeutic efficacy, vigabatrin depends on an individual's physiologic resynthesis rate of GABA transaminase.9-11
Efficacy
The efficacy of vigabatrin monotherapy for infantile spasms has been established in 2 multicenter, controlled studies.9-11 In the first study, 221 infants younger than 2 years with new-onset infantile spasms were randomized to low-dose (18-36 mg/kg daily) vs high-dose (100-148 mg/kg daily) vigabatrin. The drug was titrated to target during 7 days, and seizure control was assessed during 21 days of therapy. The primary efficacy end point was the proportion of patients who were spasm free for 7 consecutive days. Seventeen patients in the high-dose group achieved a significant spasm freedom compared with 8 patients in the low-dose group.9-11
The second study was designed as a randomized, double-blind trial that enrolled a total of 40 patients from multiple centers. Patients were given 15 mg/kg daily of vigabatrin to a maximum titration of up to 150 mg/kg daily. The study had mixed results in that when a 2-hour spasm frequency was used as a primary outcome measure, no significant differences were noted. However, on a subsequent review using a 24-hour period to establish spasm frequency before and after treatment, a significant reduction was seen in spasm frequency in the vigabatrin group (68.9%) compared with placebo (17%). On the basis of these 2 studies, the drug was approved for the indication of infantile spasms.9-11
The efficacy of vigabatrin as adjunctive therapy in adults with complex partial seizures was established in 2 US multicenter, double-blind, placebo-controlled, parallel-group clinical studies.9-11 In the first study, a randomized, double-blind, placebo-controlled dose response study consisting of an 8-week baseline followed by an 18-week treatment period, patients were randomized to receive placebo or 1, 3, or 6 g of vigabatrin per day administered on a twice-daily schedule. A total of 357 adults, 18 to 60 years of age, with complex partial seizures with or without secondary generalization were enrolled. Patients were required to be taking an adequate and stable dose of an anticonvulsant and have a history of treatment failure on an adequate regimen of either carbamazepine or phenytoin. Enrolled patients were characteristically drug resistant, reporting a mean of 8 seizures per month for several years. Dose titration occurred during a 6-week period, starting at 1 g/d until either 3 or 6 g/d was reached, depending on which maximum dose group the patient was assigned. Both dose groups reduced seizures better than placebo; however, no difference in efficacy was found between the 3- and 6-g/d doses. Responder rates were nearly identical in both dose groups, with 51% of patients in the 3-g/d group and 53% of patients in the 6-g/d group experiencing a 50% or greater lessening in seizure frequency.
The second study randomized 183 adults to add-on therapy with vigabatrin vs placebo during 16 weeks of treatment subsequent to 8 weeks of observation. Vigabatrin doses were started at 1 g/d and slowly titrated until a dose of 3 g/d was attained. A total of 39% of patients in the 3-g/d dose group had a 50% seizure reduction as opposed to only 21% of patients in the placebo group.9-11
Adverse Effects
Use of vigabatrin is limited to the indicated severe epilepsies because of serious potential adverse effects. There is an FDA black box warning for vigabatrin-induced, permanent, bilateral concentric visual field constriction in 30% or more of patients.9-13 Peripheral visual loss can range from mild to severe tunnel vision to within 10° of visual fixation. In some cases, vigabatrin may damage the central retina, decreasing visual acuity.9,12 Visual loss is irreversible, is unpredictable, and can occur any time during treatment. The risk typically increases with dose and cumulative exposure, but any exposure carries risk. The estimated risk of developing a visual field defect is 8% per year in adults.9,13 Because of this risk of permanent visual loss, vigabatrin is available only through a single national central pharmacy. A formal ophthalmologic assessment is required at baseline and every 3 months during therapy. Results of vision testing are tracked by the manufacturer per FDA mandate.9 Because of the risks to vision, a patient who does not show substantial benefit within 3 months of initiation of treatment should stop using the drug. Prescribing physicians must be prepared to counsel patients and manage these issues in patients with focal seizures.
Other adverse effects of vigabatrin include somnolence and fatigue, peripheral neuropathy, edema, and weight gain. The mean weight gain is 3.5 kg reported in 17% of patients.9
Summary
Given the limited indications, risk of visual loss, and controlled distribution, vigabatrin is most likely to be prescribed by a neurologist. It offers a new option for adults with disabling partial epilepsy who have not responded to other available AEDs. In children with infantile spasms, vigabatrin represents an alternative to corticotropin and expands the available treatment for this devastating problem.
Rufinamide
Rufinamide is a triazole derivative structurally unrelated to other currently marketed AEDs and was approved for treatment of atonic seizures in LGS in 2010.14 Lennox-Gastaut syndrome is a severe form of epilepsy characterized by childhood onset, cognitive impairment, and multiple seizure types of which atonic, or drop seizures, are often the most disabling. Seizures in LGS are characteristically drug resistant, and before rufinamide only 3 AEDs in the United States had a specific indication for LGS (felbamate, topiramate, and lamotrigine). The mechanism by which this drug exerts its antiepileptic effect is not fully known. However, in vitro studies suggest that the principal action is modulation of sodium channels, specifically, prolongation of time spent in the inactive state of the channel.1
Efficacy
The efficacy of rufinamide as adjunctive treatment for seizures associated with LGS was established in a single, multicenter, double-blind, placebo-controlled, randomized, parallel-group study with 138 individuals.15 Patients were between the ages of 4 and 30 years, were treated with 1 to 3 AEDs at baseline, and had at least 90 seizures in the month before study entry. Three end points that were addressed were the percent change in total seizure frequency for 28 days, the percent change in atonic seizure frequency in 28 days, and seizure severity based on apparent parent/guardian global evaluation. There was a 32.7% reduction in total seizure frequency in the rufinamide group compared with 11.7% in the placebo group, a 42.5% reduction in tonic seizures compared with 1.4% in the placebo group, and a 53.4% improvement in seizure severity rating from a caretaker. Because the trial end points were met, the drug was approved.
Adverse Effects
Common adverse effects are similar to those reported with other AEDs and include headache, dizziness, fatigue, and gastrointestinal distress. More unique to rufinamide is the potential for cardiac conduction disturbances with QT interval shortening.14,15 In the placebo-controlled trial, the observed degree of QT shortening was mild; nevertheless, the potential for increased risk of ventricular arrhythmia should be noted. Rufinamide should be avoided in patients with familial short QT syndrome and used with caution in combination with other drugs that shorten QT interval.14
Summary
Rufinamide is a limited-spectrum seizure drug, having a narrow therapeutic role in treating patients with LGS. However, for patients with atonic seizures and falls, rufinamide is an appropriate option.
Lacosamide
In 2009, the FDA approved lacosamide as adjunctive therapy for partial seizures in adults. Among the newest AEDs, lacosamide is the only one with both an oral and intravenous formulation.16 Lacosamide has rapidly increased in worldwide AED market share. It is believed that lacosamide stops seizures by enhancing slow inactivation of sodium channels. This is distinct from the action of other AEDs, such as phenytoin and carbamazepine, which act to block sodium channels in the fast inactivated state.1
Efficacy
The efficacy of lacosamide was established in three 12-week, randomized, double-blind, multicenter studies that enrolled adult patients.16-19 Patients had partial-onset seizures with or without secondary generalization that were not adequately controlled with 1 to 3 other AEDs. Patients averaged 4 or more seizures every 28 days and no seizure-free period exceeding more than 21 days. The median percentage reduction of seizure frequency was 35% to 39% in the 3 studies. A 50% reduction in seizure frequency was achieved in one-third of patients taking 200 mg of lacosamide and approximately 40% of those taking 400 mg. Doses higher than 400 mg led to more adverse effects than benefits; thus, 400 mg is the maximum recommended dose.
Intravenous lacosamide was also tested in a randomized, double-blind study with 60 patients.16 Efficacy of the intravenous formulation was consistent with the oral drug. Infusion rates of 10 to 50 minutes had similar adverse reactions. However, the recommended infusion rate is 300 mg for 30 to 60 minutes. One potential advantage of the intravenous formulation is that it is interchangeable milligram-to-milligram with its oral counterpart. No dosing adjustments are required.
Adverse Effects
Lacosamide's most common adverse effects included dizziness (25%) and ataxia (6%).16-19 Syncope was reported in a trial of lacosamide for diabetic neuropathy, an indication for which it was not approved. A small but measurable dose-dependent PR-interval prolongation has been observed in association with lacosamide; therefore, caution of its use is advised for patients with known cardiac conduction problems. A screening electrocardiogram is appropriate in patients with myocardial disease, patients with heart failure, or those taking other drugs known to affect PR interval.
Summary
Lacosamide represents a novel medication for adults with uncontrolled partial seizures. Future studies will be required to determine whether it offers any therapeutic advantage over older AEDs. The intravenous formulation expands options for patients who require an AED and who are unable to receive oral medication. Any potential role for intravenous lacosamide for use in status epilepticus is still to be determined.
Clobazam
Clobazam was recently approved in the United States for adjunctive treatment of LGS in patients 2 years or older.20 Clobazam is a benzodiazepine, a family that includes drugs often used as abortive therapy for seizures, such as lorazepam (Ativan), diazepam (Valium), midazolam (Versed), and clonazepam (Klonopin). Sedation and development of tolerance typically limit long-term use of benzodiazepines in patients with epilepsy. Clobazam is unique because of relatively low tendency to produce sedation and possibly lower incidence of loss of therapeutic effect over time, rendering it appropriate for long-term maintenance therapy. Although new to the United States, worldwide clobazam is one of the most commonly used AEDs. The mechanism of action for clobazam, like other benzodiazepines, is potentiation of GABAergic neurotransmission via binding to the GABAA receptor.1
Efficacy
The FDA indication for clobazam is add-on therapy for LGS in persons 2 years or older.20 Effectiveness was established in 2 multicenter controlled studies performed specifically to bring the drug to the US market.20,21 The first study was a randomized, double-blind, placebo-controlled study of patients aged 2 to 54 years with LGS.20,21 Dosing was determined first based on weight (>30 kg or <30 kg), then with a low, medium, and high dose for each weight-based category. The primary outcome measure was percent reduction of total seizures, drop seizures, tonic seizures, or myoclonic seizures during a 4-week baseline to a 12-week period of observation. Total seizure reduction was 41% in the low-dose group, 50% in the medium-dose group, and 68% in the high-dose group compared with 12% with placebo. No significant tolerance issues were noted.
A second randomized, double-blind study compared high- and low-dose clobazam in patients 2 to 25 years old with LGS.20 Dosing was again determined first by body weight: low dose for weight less than 30 kg was 5 mg, and high dose was 10 mg for those over 30 kg. High dose was 20 or 40 mg in the 2 weight groups, respectively. Seizure reduction was significantly greater in the high-dose (93%) compared with the low-dose group (29%). On the basis of these analyses, the drug was approved.
Adverse Effects
The most commonly reported adverse effects include tiredness and sedation.20,21 In general, these effects tend to be dose related. Because clobazam is a benzodiazepine, patients need to avoid other depressant drugs or alcohol and abrupt discontinuation of use. Discontinuing use of clobazam must be done slowly; otherwise, withdrawal symptoms can occur. The most common adverse effects that led to discontinuation of clobazam therapy in studies included lethargy, somnolence, ataxia, aggression, fatigue, and insomnia.
Summary
Clobazam has an advantage of extensive clinical experience in the global market before introduction in the United States. Worldwide, clobazam is a frequently used AED for patients with difficult-to-manage epilepsy. Popularity in the United States may be tempered by high expense. As with other benzodiazepines, sedation and tolerance are important issues that must be balanced against benefit in seizure control.
Ezogabine
Ezogabine was approved for use as adjunctive treatment of partial epilepsy in November 2011 and is expected to be in US pharmacies sometime in 2012.6,8,22 Its mechanism of action appears to be enhancement of potassium currents mediated by a particular family of ion channels known as KCNQ.6,8,22 By activating these specific channels on neurons, ezogabine is thought to reduce brain excitability. This drug is the first AED to control seizures by modulation of potassium channels. It may also potentiate GABAA receptors.6,8,22
Efficacy
Ezogabine has been evaluated for efficacy as adjunctive therapy in partial-onset seizures in 3 multicenter, randomized, double-blind, placebo-controlled trials in 1239 adult patients.7,22-24 The primary end point was percent change in seizure frequency from baseline. Enrolled patients experienced a minimum of 4 partial seizures per month despite use of up to 3 AEDs or vagus nerve stimulation. More than 75% of patients were taking 2 or 3 AEDs, and the mean duration of epilepsy was 22 years. Patients were randomized to daily maintenance doses of 600, 900, or 1200 mg/d, administered in 3 equally divided doses. Compared with placebo, ezogabine reduced seizure frequency at 600 mg/d by 27%, 900 mg/d by 25%, and 1200 mg/d by 24%.7,22-24
Adverse Effects
The most concerning adverse effects of ezogabine are urinary retention, neuropsychiatric symptoms, dizziness and somnolence, and QT-interval lengthening.4,22-24 Urinary retention was reported in approximately 2% of patients treated with ezogabine in epilepsy trials.22 Half of the patients with retention required catheterization. After discontinuation of ezogabine therapy, 1 of 14 patients who needed catheterization during treatment required ongoing, intermittent self-catheterization. As a result, patients at high risk for urinary symptoms, particularly urinary obstruction, need to be carefully assessed. This need is particularly true for patients with benign prostatic hypertrophy or those individuals taking other drugs that can affect urination. Moreover, vulnerable patients who are unable to communicate need to be closely watched for urinary problems.22
Neuropsychiatric symptoms, such as psychotic states associated with confusion and hallucinations, are frequently noted in patients taking ezogabine.22-24 Most psychiatric symptoms resolved rapidly after discontinuation of drug use. Rapid titration at greater than the recommended doses appears to increase the risk of these adverse effects. Ezogabine has a potential for abuse and dependence and is classified by the FDA as a controlled substance.22
In healthy volunteers, the use of 1200 mg/d of ezogabine led to a mean QT prolongation of 7.7 milliseconds. Electrocardiographic monitoring of QT intervals is appropriate, and caution should be used in patients with known preexisting cardiac conduction abnormalities or using medicines known to increase QT intervals.
Ezogabine has important drug interactions. Carbamazepine and phenytoin decrease ezogabine serum concentrations by 31% to 34%, respectively.22 Therefore, one needs to consider an increase in the dose of ezogabine when adding either carbamazepine or phenytoin. Ezogabine can also inhibit glycoprotein binding protein–mediated transport of digoxin in a concentration-dependent manner and may inhibit renal clearance of digoxin, leading to increased serum digoxin levels. When prescribing ezogabine with digoxin, it is important to monitor digoxin concentrations. Ethanol use can increase serum ezogabine levels and can lead to increased adverse effects.
Summary
Ezobagine may be helpful for patients with partial epilepsy when other medications have failed. This drug works by a novel mechanism of action. Serious adverse effects, including urinary retention and potential for QT-interval prolongation, mandate careful monitoring. It is too early to tell when ezogabine should be used compared with other choices for seizures.
Trends
Pharmacogenomics
One of the more exciting trends in the world of AEDs is use of pharmacogenetic markers. Pharmacogenomics is the prediction of drug response or adverse effects based on genetic markers. It is considered the fundamental underpinning of what is known as individualized medicine. The discipline as it applies to epilepsy is still in early development. The hope is that pharmacogenomics can help predict drug-resistant epilepsy early in the disease and help refine AED choice based on predicted seizure response and the likelihood of idiosyncratic adverse effects.
Stevens-Johnson syndrome is a serious dermatologic adverse effect that can more likely occur with certain AEDs. Pharmacogenomics has already affected this issue. Certain HLA-B haplotypes predict serious rash when the patient is exposed to carbamazepine. There is a strong association in the Han Chinese and other Southeast Asian populations with Stevens-Johnson syndrome and the HLA-B*1502 marker.25,26 HLA-A*3101 has subsequently been identified as a risk factor for carbamazepine-induced hypersensitivity reactions in Europeans.27 Other biomarkers need to be identified to help predict response to therapy. Haplotype testing is currently available.
Suicide Behavior and Ideation
In 2008, the FDA issued a safety alert regarding the association of AEDs and suicidal behavior and ideation.27-29 The warning was based on a large pooled analysis composed of 199 randomized controlled trials involving both adjunctive and monotherapy drug trial designs, enrolling 43,892 patients in trials of 11 different AEDs in which 4 suicides had occurred in patients taking AEDs and none in those taking placebo. The adjusted relative risk for suicide or suicidal ideation was reported as 1.8 (95% confidence interval, 1.2-2.7). There was a doubling of the incidence rate of suicide or suicidal ideation, with 0.43% rate in patients taking AEDs compared with a 0.24% rate in those taking a placebo, representing an increase of approximately 1 case of suicidal ideation for every 530 patients treated.27-29 The increased risk of suicidal thoughts or behavior with the use of AEDs was observed as early as 1 week after starting treatment and persisted for the assessed treatment duration. The median treatment duration in the trials analyzed was only 12 weeks, and risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
This issue remains far from settled. The meta-analysis has been criticized for failure to include older AEDs and inadequate sample size. Most AED studies of epilepsy would have excluded patients with known active psychiatric disorders, and some argue that this may have minimized the appearance of risk. Subsequent population-based studies have suggested that suicide risk may be unique to a subset of patients with preexisting comorbid depression who were taking an AED.30,31 This finding was particularly underscored in the study by Arana et al,30 who evaluated nearly 5 million patients in a United Kingdom database and found that nondepressed patients with epilepsy who were taking an AED did not have an increased risk of suicide. Nonetheless, the need for physicians to understand the effect on mood of epilepsy and other conditions for which AEDs are used should be reinforced. Practitioners should screen for mood disorders because anxiety and depression are frequent comorbidities of epilepsy that may need to be treated as aggressively as the seizures.
Generic Substitution of AEDs
Generic drug substitution is a significant public health issue. Generic entry into the US pharmaceutical market has saved billions of dollars in health care costs. The FDA has supported bioequivalence of approved brand-name and generic AEDs, suggesting that generic drugs can be safely interchanged with brand-name or other generic products. However, this may not be the case in epilepsy. Neurologists and epilepsy advocacy groups have made a significant effort to show switching brand-name to generic AEDS may result in breakthrough seizures.32 A comprehensive cost-savings analysis may need to include the cost of missed work, seizure-related injury, and seizure-associated increased use of medical resources, rather than the price of drug alone.
In November 2006 the American Academy of Neurology objected to the substitution of generic AEDs without a physician's approval.33 This objection was then followed by a large Epilepsy Foundation survey of more than 1000 individuals with epilepsy.34 This survey found that seizures worsened for 59% of individuals who had switched from brand-name to generic AEDs. The survey findings were further underscored when Berg et al35 published a retrospective analysis of breakthrough seizures in individuals who had switched from brand-name to generic AEDs. In a study of 150 physicians who completed a case review form regarding a patient who experienced a sudden loss of seizure control due to a generic AED, 69 physicians reported having had patients who had breakthrough seizures. Moreover, 50 patients with well-controlled conditions who changed to a generic drug without any other confounding variables reported a seizure recurrence.35 The sample size was too small to identify a particular generic agent that accounted for the problem. This study is concerning given that a seizure has an important effect on quality of life. In fact, 30 individuals lost driving privileges and 9 were unable to attend school or work. These findings raised the possibility that the current FDA provisions establishing bioequivalence may not be easily translatable to generic AEDs. This study ultimately led an advisory committee for pharmaceutical science to recommend to the FDA that the FDA needs to reassess bioequivalence topics relevant to generic drug approval in April 2010.36 In 2011, Krauss et al37 studied differences in total drug exposure and maximum concentration ratios of generic and reference formulation during fasting and fed bioequivalence studies. They found that in simulating switches between 595 pairs of generic AED formulations, estimated areas under the curve varied by more than 15% for 17% of pairs, and an estimated maximum concentration differed by more than 15% for 39%. The AEDs with low bioavailability and solubility, such as oxcarbazepine, had the greatest variability. The investigators concluded that most generic AED products provide total drug delivery or area under the curve similar to reference products; however, differences in peak concentrations among formulations are more common. Switches between generic AED products may cause greater changes in plasma drug concentrations than generic substitutions of reference products.37 Because the study by Krauss et al did not settle the issue, the FDA began awarding grants to pharmacy schools to research the effect of switching of federally approved generic and brand-name AEDs. The final answer on this important topic is yet to be determined. The best advice to physicians who prescribe AEDs to patients with epilepsy is to always question patients about generic switches should seizures occur without any clear explanation.
Conclusion
The management of epilepsy has been transformed, with more than 24 agents available for use in the United States and even more options elsewhere in the world. The issue that remains is how to best tailor AED choice to the individual patient with epilepsy. Although there are a considerable number of choices of AEDs for the management of both the first seizure and chronic epilepsy, there is a paucity of comparative effectiveness data to offer evidence-based recommendations of which drug should be best used in a given clinical scenario. Therefore, much work is needed to answer vital common clinical questions regarding AEDs, such as (1) Which drug should be used (first-generation or a new agent)? (2) Are there certain agents that should be tried before surgery? (3) Can a certain combination of medications yield better seizure control than a single agent? (4) Are the trial designs used to approve new AEDs clinically meaningful?
Pharmacogenomics has begun to identify specific populations in whom certain AEDs may cause serious adverse effects. There has been a greater focus on the psychiatric comorbidities of epilepsy and the role that AEDs may play in serious mood disorders. As financial constraints force improved efficiencies from our therapies, comparative benefit studies among AEDs are now needed. Studies reviewing the generic substitution of brand-name AEDs have suggested that not all generic AEDs are equivalent to brand-name drugs and that the consequences of those differences are not small and may include breakthrough seizures in patients whose conditions had been previously controlled. More work is needed to understand how to best counsel our patients regarding these important topics.
Article Highlights.
-
■
Five new antiepileptic drugs (AEDs) have been recently approved in 2009-2011 in the United States for the treatment of epilepsy.
-
■
Two of the new agents, ezogabine and vigabatrin, are the first agents that have novel mechanisms of action—potassium channel modulation for ezogabine and irreversible inhibition of γ-aminobutyric acid transaminase for vigabatrin.
-
■
Three new trends regarding AEDs have been identified: use of pharmacogenomics to predict a serious adverse effect, the limitations of generic substitution of brand-name AEDs, and the Food and Drug Administration warning issued for AEDs regarding suicidal ideation.
Footnotes
Potential Competing Interests: Dr Sirven has received research support from Eisai, Epilepsy Therapy Project, MAP, National Institutes of Health, Neuropace, UCB, Upsher Smith, and Vertex.
Supplemental Online Material
Author Interview Video
References
- 1.Bialer M., White H.S. Key factors in the discovery and development of new antiepileptic drugs. Nat Rev Drug Discov. 2010;9(1):68–82. doi: 10.1038/nrd2997. [DOI] [PubMed] [Google Scholar]
- 2.Rho J.M., Sankar R. The pharmacologic basis of antiepileptic drug action. Epilepsia. 1999;40(11):1471–1483. doi: 10.1111/j.1528-1157.1999.tb02029.x. [DOI] [PubMed] [Google Scholar]
- 3.Webanatomy: Excitability and Membrane Electropotential. 2012. http://webanatomy.net/237/channels/channels.htm Accessed July 27, 2012.
- 4.Brickel N, HJ, DeRossett S. Pharmacological effects of retigabine on bladder function: results from phase 2/3 studies. In: Proceedings from the American Epilepsy Society; December 2010; San Antonio, TX. Abstract 1.272.
- 5.Brandt C., Heile A., Potschka H., Stoehr T., Löscher W. Effects of the novel antiepileptic drug lacosamide on the development of amygdala kindling in rats. Epilepsia. 2006;47(11):1803–1809. doi: 10.1111/j.1528-1167.2006.00818.x. [DOI] [PubMed] [Google Scholar]
- 6.Gunthorpe M.J., Large C.H., Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K(+) channel opener for the treatment of epilepsy. Epilepsia. 2012;53(3):412–424. doi: 10.1111/j.1528-1167.2011.03365.x. [DOI] [PubMed] [Google Scholar]
- 7.Large C.H., Sokal D.M., Nehlig A. The spectrum of anticonvulsant efficacy of retigabine (ezogabine) in animal models: implications for clinical use. Epilepsia. 2012;53(3):425–436. doi: 10.1111/j.1528-1167.2011.03364.x. [DOI] [PubMed] [Google Scholar]
- 8.Wuttke T.V., Seebohm G., Bail S., Maljevic S., Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the K 7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67(4):1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- 9.Food and Drug Administration . Lundbeck; Deerfield, IL: 2009. Sabril [product label] [Google Scholar]
- 10.Willmore L.J., Abelson M.B., Ben-Menachem E., Pellock J.M., Shields W.D. Vigabatrin: 2008 update. Epilepsia. 2009;50(2):163–173. doi: 10.1111/j.1528-1167.2008.01988.x. [DOI] [PubMed] [Google Scholar]
- 11.Wohlrab G., Leiba H., Kastle R. Vigabatrin therapy in infantile spasms: solving one problem and inducing another? Epilepsia. 2009;50(8):2006–2008. doi: 10.1111/j.1528-1167.2009.02167.x. [DOI] [PubMed] [Google Scholar]
- 12.Conway M., Cubbidge R.P., Hosking S.L. Visual field severity indices demonstrate dose-dependent visual loss from vigabatrin therapy. Epilepsia. 2008;49(1):108–116. doi: 10.1111/j.1528-1167.2007.01249.x. [DOI] [PubMed] [Google Scholar]
- 13.Maguire M.J., Hemming K., Wild J.M., Hutton J.L., Marson A.G. Prevalence of visual field loss following exposure to vigabatrin therapy: a systematic review. Epilepsia. 2010;51(12):2423–2431. doi: 10.1111/j.1528-1167.2010.02772.x. [DOI] [PubMed] [Google Scholar]
- 14.Food and Drug Administration . Eisai Inc; Baltimore, MD: 2011. Banzel [product label] [Google Scholar]
- 15.Glauser T., Kluger G., Sachdeo R., Krauss G., Perdomo C., Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70(21):1950–1958. doi: 10.1212/01.wnl.0000303813.95800.0d. [DOI] [PubMed] [Google Scholar]
- 16.Food and Drug Administration . UCB Inc; Brussels, Belgium: 2009. Vimpat [product label] [Google Scholar]
- 17.Ben-Menachem E., Biton V., Jatuzis D., Abou-Khalil B., Doty P., Rudd G.D. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48(7):1308–1317. doi: 10.1111/j.1528-1167.2007.01188.x. [DOI] [PubMed] [Google Scholar]
- 18.Chung S., Sperling M.R., Biton V. Lacosamide as adjunctive therapy for partial-onset seizures: a randomized controlled trial. Epilepsia. 2010;51(6):958–967. doi: 10.1111/j.1528-1167.2009.02496.x. [DOI] [PubMed] [Google Scholar]
- 19.Halasz P., Kalviainen R., Mazurkiewicz-Beldzinska M. Adjunctive lacosamide for partial-onset seizures: efficacy and safety results from a randomized controlled trial. Epilepsia. 2009;50(3):443–453. doi: 10.1111/j.1528-1167.2008.01951.x. [DOI] [PubMed] [Google Scholar]
- 20.Food and Drug Administration . Lundbeck; Deerfield, IL: 2011. Onfi [product label] [Google Scholar]
- 21.Ng Y.T., Conry J.A., Drummond R., Stolle J., Weinberg M.A., OV-1012 Study Investigators Randomized, phase III study results of clobazam in Lennox-Gastaut syndrome. Neurology. 2011;77(15):1473–1481. doi: 10.1212/WNL.0b013e318232de76. [DOI] [PubMed] [Google Scholar]
- 22.Food and Drug Administration . GlaxoSmithKline; Columbia, MD: 2011. Potiga [product label] [Google Scholar]
- 23.French J.A., Abou-Khalil B., Leroy R.F. Randomized placebo-controlled trial ezogabine (retigabine) in partial epilepsy. Neurology. 2011;76(18):1555–1563. doi: 10.1212/WNL.0b013e3182194bd3. [DOI] [PubMed] [Google Scholar]
- 24.Porter R.J., Partiot A., Sachdeo R., Nohria V., Alves W.M., 205 Study Group Randomized, multicentre dose ranging trial of retigabine for partial onset seizures. Neurology. 2007;68(15):1197–2004. doi: 10.1212/01.wnl.0000259034.45049.00. [DOI] [PubMed] [Google Scholar]
- 25.Chen P., Lin J.J., Lu C.S. Carbamazepine-induced toxic effects and HLA B*1502 screening in Taiwan. N Engl J Med. 2011;364(12):1126–1133. doi: 10.1056/NEJMoa1009717. [DOI] [PubMed] [Google Scholar]
- 26.Chung W.H., Hung S.I., Hong H.S. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486. doi: 10.1038/428486a. [DOI] [PubMed] [Google Scholar]
- 27.McCormack M., Alfirevic A., Bourgeois S. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364(12):1134–1143. doi: 10.1056/NEJMoa1013297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Research CfDEa Antiepileptic Drugs and Suicidality. 2008. www.fda.gov/ohrms/dockets/ac/08/slides/2008-4372s1-03-FDA Accessed August 9, 2012.
- 29.Treatment CfDEa: Antiepileptic Drugs and Suicidality. 2008. http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008-4372b1-01-FDA.pdf Accessed August 9, 2012.
- 30.Arana A., Wentworth C.E., Ayuso-Mateos J.L., Arellano F.M. Suicide-related events in patients treated with antiepileptic drugs. N Engl J Med. 2010;363(6):542–551. doi: 10.1056/NEJMoa0909801. [DOI] [PubMed] [Google Scholar]
- 31.Andersohn F., Schade R., Willich S.N., Garbe E. Use of antiepileptic drugs in epilepsy and the risk of self-harm or suicidal behavior. Neurology. 2010;75(4):335–340. doi: 10.1212/WNL.0b013e3181ea157e. [DOI] [PubMed] [Google Scholar]
- 32.Andermann F., Duh M.S., Gosselin A., Paradis P.E. Compulsory generic switching of antiepileptic drugs: high switchback rates to branded compounds compared with other drug classes. Epilepsia. 2007;48(3):464–469. doi: 10.1111/j.1528-1167.2007.01007.x. [DOI] [PubMed] [Google Scholar]
- 33.Liow K., Barkley G.L., Pollard J.R. Position statement on the coverage of anticonvulsant drugs for the treatment of epilepsy. Neurology. 2007;68(16):1249–1250. doi: 10.1212/01.wnl.0000259400.30539.cc. [DOI] [PubMed] [Google Scholar]
- 34.Berg M.J., Gross R.A., Haskins L.S. Generic substitution in the treatment of epilepsy: patient and physician perceptions. Epilepsy Behav. 2008;13(4):693–699. doi: 10.1016/j.yebeh.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Berg M.J., Gross R.A., Tomaszewski K.J., Zingaro W.M., Haskins L.S. Generic substitution in the treatment of epilepsy: case evidence of breakthrough seizures. Neurology. 2008;71(7):525–530. doi: 10.1212/01.wnl.0000319958.37502.8e. [DOI] [PubMed] [Google Scholar]
- 36.Committee PSaCPA . Food and Drug Administration; Washington, DC: 2010. Meeting Materials for Advisory Committee for Pharmaceutical Science and Clinical Pharmacology. [Google Scholar]
- 37.Krauss G.L., Caffo B., Chang Y.T., Hendrix C.W., Chuang K. Assessing bioequivalence of generic antiepilepsy drugs. Ann Neurol. 2011;70(2):221–228. doi: 10.1002/ana.22452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Author Interview Video