

Abstract

Cyclic Py-Im polyamides containing two GABA turn units exhibit enhanced DNA binding affinity, but extensive studies of their biological properties have been hindered due to synthetic inaccessibility. A facile modular approach toward cyclic polyamides has been developed via microwave-assisted solid-phase synthesis of hairpin amino acid oligomer intermediates followed by macrocyclization. A focused library of cyclic polyamides 1–7 targeted to the androgen response element (ARE) and the estrogen response element (ERE) were synthesized in 12–17% overall yield. The Fmoc protection strategy also allows for selective modifications on the GABA turn units that have been shown to improve cellular uptake properties. The DNA binding affinities of a library of cyclic polyamides were measured by DNA thermal denaturation assays and compared to the corresponding hairpin polyamides. Fluorescein-labeled cyclic polyamides have been synthesized and imaged via confocal microscopy in A549 and T47D cell lines. The IC50 values of compounds 1–7 and 9–11 were determined, revealing remarkably varying levels of cytotoxicity.
Introduction
The selective modulation of eukaryotic gene expression by small molecules may have important implications in the field of chemical biology and human medicine. Pyrrole-imidazole polyamides are a class of synthetic ligands that can be programmed to bind the minor groove of specific DNA sequences.1 Antiparallel, side-by-side N-methylpyrrole (Py) and N-methylimidazole (Im) carboxamide (Im/Py) pairs distinguish G·C from C·G base pairs, N-methyl-3-hydroxypyrrole (Hp)/Py shows specificity for T·A over A·T, whereas Py/Py pairs are specific for both T·A and A·T.2−5 By linking two strands of these heterocyclic oligomers via a γ-amino butyric acid (GABA) turn unit, hairpin Py-Im polyamides can be programmed to bind a large library of DNA sequences with affinities comparable to natural DNA binding proteins.6−8 Hairpin polyamides have been shown to localize to the nuclei of living cells and regulate endogenous gene expression by disrupting protein/DNA interfaces.9−17 Cyclic polyamides containing a second GABA turn unit exhibit further enhanced DNA binding properties.18−21 We have recently demonstrated their gene regulatory effects on AR-activated gene expression in prostate cancer models.22
This discovery has opened a new area of research toward transcriptional regulation with small molecules, but the relative synthetic inaccessibility of cyclic polyamides has remained a bottleneck for examining libraries of structural variants that would modulate affinity, cell uptake, and biological activity. Initial solid-phase methods were low yielding and required substantial premodifications of the PAM resin.18−20 While the solution-phase synthesis of cyclic polyamides remains useful in large-scale target-oriented synthesis, it has limited practicality toward libraries for screening biological activities.22 The recent report by Morinaga et al. offers a modular approach to achieve cyclic polyamides by intramolecular coupling of a cysteine and a chloroacetyl residue, but the modification of the optimal three-carbon GABA turn into a sulfur-containing four-atom linker compromises its DNA binding affinity and may alter its biological properties.23
We report here a solid-phase polyamide synthesis of a key hairpin amino acid oligomer intermediate, which followed by intramolecular cyclization affords cyclic polyamides 1–8 in good yields. The polyamides were synthesized stepwise on 2-chlorotrityl resin. The modular approach led to rapid access of a focused library of cyclic polyamides 1–7 (Scheme 1) with various core sequences and turn unit modifications. The utilization of Fmoc chemistry allowed for differentially protected turn units, which were modified selectively to complement existing cellular imaging and cell uptake enhancement technologies.24 We examined the DNA binding properties and cytotoxicity profiles of compounds 1–11 and the cellular localization of cyclic polyamides 12–14 by fluorescence microscopy.
Scheme 1. Microwave-Assisted Synthesis of Cyclic Polyamides 1–7.

All PyBOP-mediated coupling conditions were performed under microwave-assisted conditions (see Table 1). Reagents and conditions: (i) 50% piperidine, DMF; (ii) FmocPyOH, PyBOP, DIEA, DMF; (iii) 50% piperidine, DMF; (iv) FmocPyOH, PyBOP, DIEA, DMF; (v) 50% piperidine, DMF; (vi) FmocImOH, PyBOP, DIEA, DMF; (vii) 50% piperidine, DMF; (viii) Z-β-Dab(Fmoc)-OH (for 1–6) or Boc-β-Dab(Fmoc)-OH (for 7), PyBOP, DIEA, DMF; (ix) 50% piperidine, DMF; (x) FmocPyOH, PyBOP, DIEA, DMF; (xi) 50% piperidine, DMF; (xii) FmocPyOH, PyBOP, DIEA, DMF; (xiii) 50% piperidine, DMF; (xiv) FmocPyOH (for 1–3) or FmocImOH (for 4–7), PyBOP, DIEA, DMF; (xv) 50% piperidine, DMF; (xvi) FmocImOH, PyBOP, DIEA, DMF; (xvii) 50% piperidine, DMF; (xviii) Z-β-Dab(Fmoc)-OH (for 1 and 4 and 7) or Fmoc-GABA-OH (for 2 and 5) or Boc-β-Dab(Fmoc)-OH (for 3 and 6), PyBOP, DIEA, DMF; (xix) 30% HFIP, DCM; (xx) 20% piperidine, DMF; (xxi) DPPA, DIEA, DMF; (xxii) 10% TFMSA, TFA (for 1, 2, 4, and 5) or TFA (for 3, 6, and 7).
Results and Discussion
Microwave-Assisted Solid-Phase Synthesis
Due to previously observed decomposition of the conjugated C-terminal free carboxylic acid in polyamide intermediates, 2-chlorotrityl chloride (2-Cl-Trt-Cl) resin was chosen for its mild synthesis and cleavage conditions. Polyamide synthesis on this resin has been previously reported by Aldrich-Wright and co-workers, but a resin-bound β-alanine linker was used in both instances, and a new loading procedure was therefore needed.25,26 2-Cl-Trt-Cl resin was first loaded with the Fmoc-protected Py monomer in N,N-dimethylformamide (DMF) and capped with methanol. Resin substitution levels were determined by the Fmoc test and confirmed by weighing the dry mass of the loaded resin. Fmoc deprotection was achieved using a 50% piperidine in DMF solution. In light of the recent improvements in both efficiency and yield, the couplings were performed under microwave-assisted conditions using the desired PyBOP-activated monomers.27 Initial syntheses performed at 60 °C led to premature cleavage of intermediates off the 2-Cl-Trt resin, and 50 °C couplings were therefore preferable. The challenging Im to Py coupling required an FmocPyImOH dimer, which was obtained via an optimized procedure by Weltzer and Wemmer.28 The deprotection and coupling conditions are detailed in Table 1. This two-step deprotection–coupling procedure was repeated until the desired polyamide sequence was achieved. To build the small library of polyamides in a modular fashion, the resin was split into different batches at corresponding steps for further derivatization. Upon completion, the N-terminal Fmoc-protected polyamide oligomer was cleaved from the resin with 30% hexafluoroisopropanol (HFIP) in dichloromethane (DCM), concentrated in vacuo, and the resulting residue was subjected to a 20% piperidine solution to remove the Fmoc group. Direct cleavage of the free amine polyamide oligomer was attempted, but found to be ineffective due to poor solubility of the zwitterion intermediate in the cleavage solution. After purification by high-performance liquid chromatography (HPLC), the desired polyamide intermediates 18–24 were obtained in 31–40% yields (Table 2).
Table 1. Standard Fmoc Deprotection and Microwave-Assisted Coupling Times for Solid-Phase Polyamide Synthesis.
| coupling
timesb (min) |
||||
|---|---|---|---|---|
| resin-bound nucleophile | deprotection timesa | Py | Im | GABA/β-Ala |
| Py | 3 × 10 min | 20 | 20 | 20 |
| Im | 3 × 10 min | IRc | 30 | 30 |
| GABA/β-Ala | 2 × 5 min | 20 | 20 | 20 |
All deprotections were performed in 50% piperidine in DMF.
All coupling reactions were conducted under microwave-assisted conditions at 50 °C with a 0.3 M solution of the activated monomers (3 equiv of monomer acid, 3 equiv of PyBOP, 8 equiv of DIEA, DMF).
FmocPyOH coupling onto resin with N-terminal Im was incomplete even at 60 °C for up to 1 h. Synthesis of polyamide sequences that require this linkage should use the FmocPyImOH dimer instead, demonstrated later in the synthesis of 8.
Table 2. Summary Table of MALDI-TOF Data and Synthetic Yields for Cyclic Polyamides 1–8 and Intermediates 18–25.

DPPA-Mediated Macrocyclization
The polyamide macrocyclization step was achieved by a DPPA-mediated ring-closing reaction between the N-terminal amino group and the C-terminal carboxylic acid. This method was first employed by Cho et al. in the synthesis of cyclic polyamides.18,29,30 In order to obtain a general workup procedure applicable to polyamides of various lipophilicities, diisopropylethylamine (DIEA) was used as the base in place of sodium bicarbonate (NaHCO3) in the original conditions.31 Deprotection of the turn units with trifluoromethanesulfonic acid (TFMSA) or trifluoroacetic acid (TFA), followed by HPLC purification, afforded polyamides 1–7 in 37–43% yields over two steps.
Selective Derivatization of Cyclic Polyamide Turn Units
By taking advantage of the Fmoc protection scheme, the two GABA β-amino groups in 3 were differentially protected. This is further highlighted in 6 and 7, which share the same asymmetric polyamide core targeted to the 5′-WGGWCW-3′ sequence, and allowed for the selective conjugation of a benzoic acid moiety on a single turn unit in polyamides 9–11 that has been recently developed to enhance the cellular localization properties of hairpin polyamides (Scheme 2).24 Monosubstituted benzyl carbamate (Cbz) polyamides 3, 6, and 7 were chosen as targets based on unpublished results indicating that Cbz-functionalized polyamides are biologically active (see Supporting Information). Cyclic polyamides 12–14 with a fluorescein dye were synthesized in a similar fashion and imaged in living cells via fluorescence microscopy (Scheme 3). Furthermore, both the solubility and the pharmacokinetic profiles of cyclic polyamides have been shown to be highly dependent on subtle structural modifications, and this method allows for the modular synthesis of these structural variants in an efficient manner.31−33
Scheme 2. Preparation of Cyclic Polyamides 9–11.

Reagents and conditions: (i) BzOH, PyBOP, DIEA, DMF; (ii) 10% TFMSA, TFA.
Scheme 3. Preparation of Cyclic Polyamides 12–14.

Reagents and conditions: (i) FITC, DIEA, DMF; (ii) 10% TFMSA, TFA.
Synthesis of Cyclic and Hairpin Polyamides with C-Terminal Imidazole Units
Previously established solid-phase polyamide synthesis methods have been generally limited to sequences beginning with a pyrrole monomer.34,35 Solid-phase synthesis of polyamides starting with imidazole units on the commonly used Kaiser oxime resin have been low yielding, mainly attributed to the sensitivity of the oxime–imidazole linkage that leads to premature cleavage of resin-bound intermediates. The addition of an aliphatic linker (e.g., Boc-β-Ala-PAM resin) circumvents this issue, but previous studies on hairpin polyamides with C-terminal β-alanine motifs have shown reduced cellular uptake properties and thus diminished gene regulatory effects of these products.9 Using the microwave-assisted conditions reported above, cyclic polyamide 8 targeted to the 5′-WCGWGW-3′ sequence found in E-Box binding sites has been synthesized in 13% yield overall.
Initial attempts starting with FmocImOH-loaded resin resulted in undesired cleavage during subsequent steps, and so syntheses began with loading of the FmocPyImOH dimer onto 2-chlorotrityl resin. After deprotection–coupling of the corresponding monomer units, followed by resin cleavage and Fmoc removal, polyamide intermediate 25 was isolated by HPLC purification in 34% yield. DPPA-mediated cyclization of 25, followed by Cbz deprotection afforded cyclic polyamide 8 in 39% yield over two steps (Scheme 4). Hairpin polyamide 17 was synthesized in a similar stepwise manner to afford the C-terminal acid intermediate, which was then coupled to a 3,3′-diamino-N-methyldipropylamine linker, followed by isophthalic acid conjugation, Boc deprotection, and isolated in 24% yield over 16 steps (see Scheme S1). This is a step forward which allows for the synthesis of both cyclic and non-β-alanine-linked hairpin polyamides with sequences beginning with an imidazole unit, further expanding the scope of targetable DNA sequences and inhibition of transcription-factor-mediated gene expression by Py-Im polyamides.
Scheme 4. Preparation of Cyclic Polyamide 8.

All PyBOP-mediated coupling conditions were performed under microwave-assisted conditions (see Table 1). Reagents and conditions: (i) 50% piperidine, DMF; (ii) FmocPyOH, PyBOP, DIEA, DMF; (iii) 50% piperidine, DMF; (iv) FmocPyOH, PyBOP, DIEA, DMF; (v) 50% piperidine, DMF; (vi) Z-β-Dab(Fmoc)-OH, PyBOP, DIEA, DMF; (vii) 50% piperidine, DMF; (viii) FmocImOH, PyBOP, DIEA, DMF; (ix) 50% piperidine, DMF; (x) Fmoc-β-Ala-OH, PyBOP, DIEA, DMF; (xi) 50% piperidine, DMF; (xii) FmocPyImOH, PyBOP, DIEA, DMF; (xiii) 50% piperidine, DMF; (xiv) Z-β-Dab(Fmoc)-OH, PyBOP, DIEA, DMF; (xv) 30% HFIP, DCM; (xvi) 20% piperidine, DMF; (xvii) DPPA, DIEA, DMF; (xviii) 10% TFMSA, TFA.
Thermal Stabilization of DNA Duplexes by Polyamides
Py-Im polyamide–DNA binding affinities and specificities have historically been measured by quantitative DNase I footprinting assays.36 As previously reported, however, cyclic polyamides have exceptionally high DNA binding affinities that exceed the detection limit of this experiment (i.e., Ka ≥ 2 × 1010 M–1).22,37,38 The DNA binding affinities of cyclic polyamides 1–14 have been rank ordered by magnitude of DNA thermal stabilization (ΔTm) and compared to the corresponding hairpin polyamides 14–16. Spectroscopic measurements were performed on 12-mer DNA duplexes with sequences 5′-CGATGTTCAAGC-3′, 5′-CGATGGTCAAGC-3′, and 5′-CGATCGTGAAGC-3′, each containing a match binding site for the corresponding polyamides.
Consistent with the findings of Chenoweth et al., the ΔTm value for bisamino cyclic polyamide 1 (ΔTm = 26.1 °C) was calculated to be significantly higher than that of hairpin polyamide 15 (ΔTm = 22.0 °C).22 So while 15 has an established binding affinity to the match androgen response element (ARE) half-site (5′-WGWWCW-3′) in the subnanomolar range, cyclic polyamide 1 provides even greater stabilization to such DNA duplexes. Mono-unsubstituted cyclic polyamide 2 provides less DNA stabilization compared to 1 (ΔTm = 20.4 °C), which is likely due to the loss of a positive charge and thus the loss of favorable electrostatic interactions with the negatively charged DNA backbone. Perhaps more surprising is the high ΔTm values retained by monoprotected cyclic polyamides 3 (ΔTm = 27.3 °C) and 9 (ΔTm = 28.0 °C), each containing a lone free amino group and net +1 charge. As shown in Figure 1, the benzoyl (Bz) group in 9 projects straight down the minor groove, avoiding unfavorable steric interactions with the groove wall, and may offer insight into the high degree of DNA stabilization by 3 and 9 comparable to the bisamino cycle 1. Fluorescein-conjugated 12 (ΔTm = 21.8 °C) has a DNA binding affinity lower than 3 and 9, perhaps due to increased steric clashes from the larger substitution group and unfavorable electrostatic interactions from the negatively charged fluorescein group, but still binds DNA at a similar level to 15 (Table 3).
Figure 1.
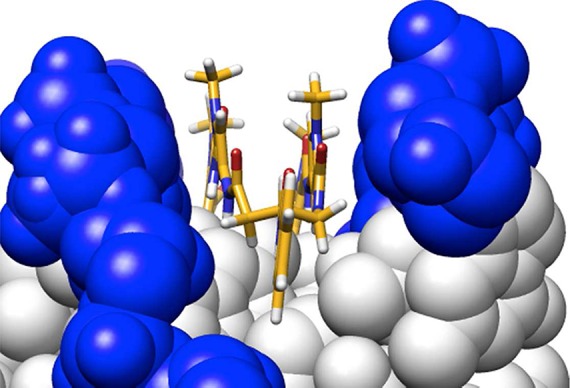
Molecular model of benzoyl-substituted cyclic polyamide turn along the DNA minor groove, based on published crystal structure by Chenoweth et al. (PDB ID: 3OMJ).
Table 3. Tm Values for the Polyamide Library.
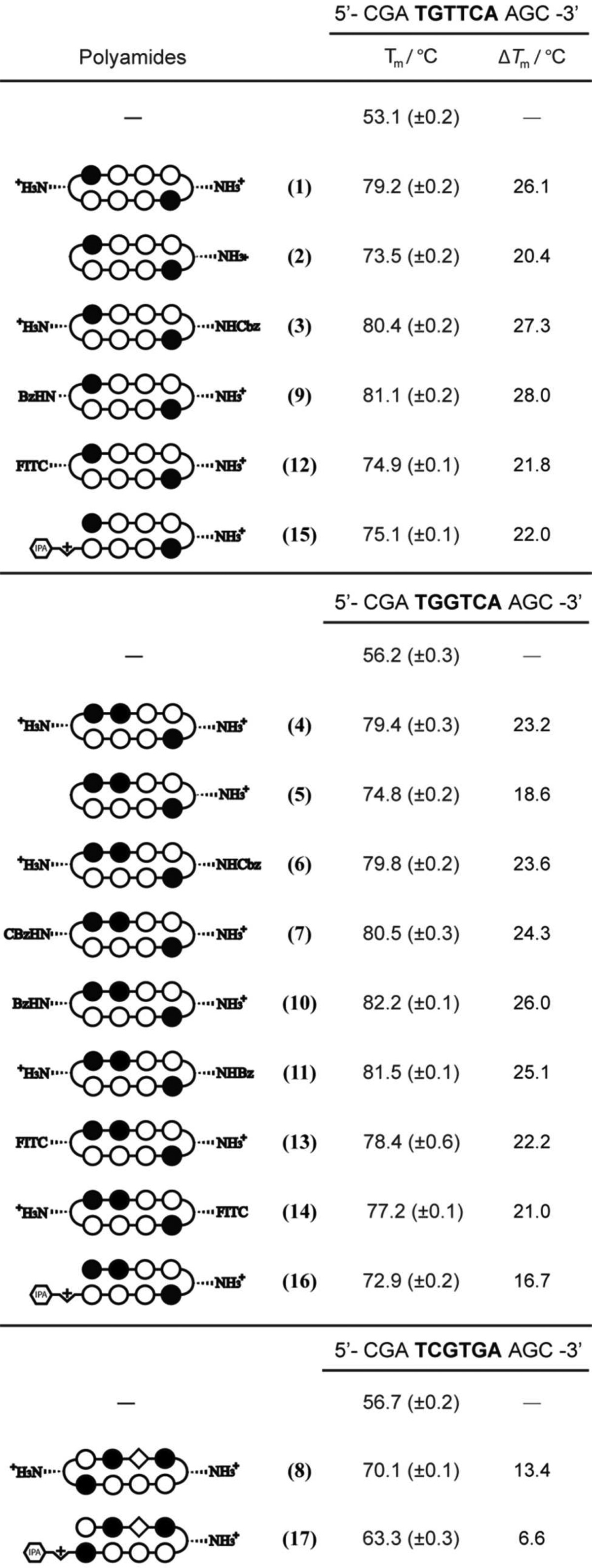
The magnitude of stabilization provided by cyclic polyamides 4–7, 10–11, and 13–14 targeted to estrogen response element (ERE) half-sites (5′-WGGWCW-3′) follows a similar pattern to the aforementioned ARE targeting series. Mono-unsubstituted cycle 5 (ΔTm = 18.6 °C) stabilizes the duplex at a comparable level to hairpin polyamide 16 (ΔTm = 16.7 °C), whereas bisamino compound 4 (ΔTm = 23.2 °C) has a higher ΔTm value. The mono-Cbz cycles 6 (ΔTm = 23.6 °C) and 7 (ΔTm = 24.3 °C), mono-Bz-substituted 10 (ΔTm = 26.0 °C) and 11 (ΔTm = 25.1 °C), and monofluorescein conjugates 13 (ΔTm = 22.2 °C) and 14 (ΔTm = 21.0 °C) each bind DNA similar to 4.
Cyclic polyamide 8 (ΔTm = 13.4 °C) binds the match 5′-WCGWGW-3′ sequence at an elevated level compared to hairpin 17 (ΔTm = 6.6 °C), which may prove important toward targeting oncogenic transcription factors such as c-Myc that act through binding canonical E-Box (5′-CACGTG-3′) sequences.
Sulforhodamine B Cytotoxicity Assay for Compounds 1–11
The cytotoxicity of compounds 1–11 was assessed in A549 human lung carcinoma and T47D human breast cancer cell lines (Tables 4 and 5). The cyclic polyamides targeted to the 5′-WGWWCW-3′ sequence generally exhibit a higher level of cytotoxicity than the 5′-WGGWCW-3′ series, which is consistent with the trends observed in the DNA thermal denaturation analysis and confocal microscopy studies. Detailed inspection of the IC50 values within the series of compounds and across the cell lines, however, offers some unanticipated insights into the different biological properties of these minor structural variants.
Table 4. SRB Cytotoxicity Data on Compounds 1–3, 9, and 15, in A549 and T47D Cells, 72 Hour Incubation.
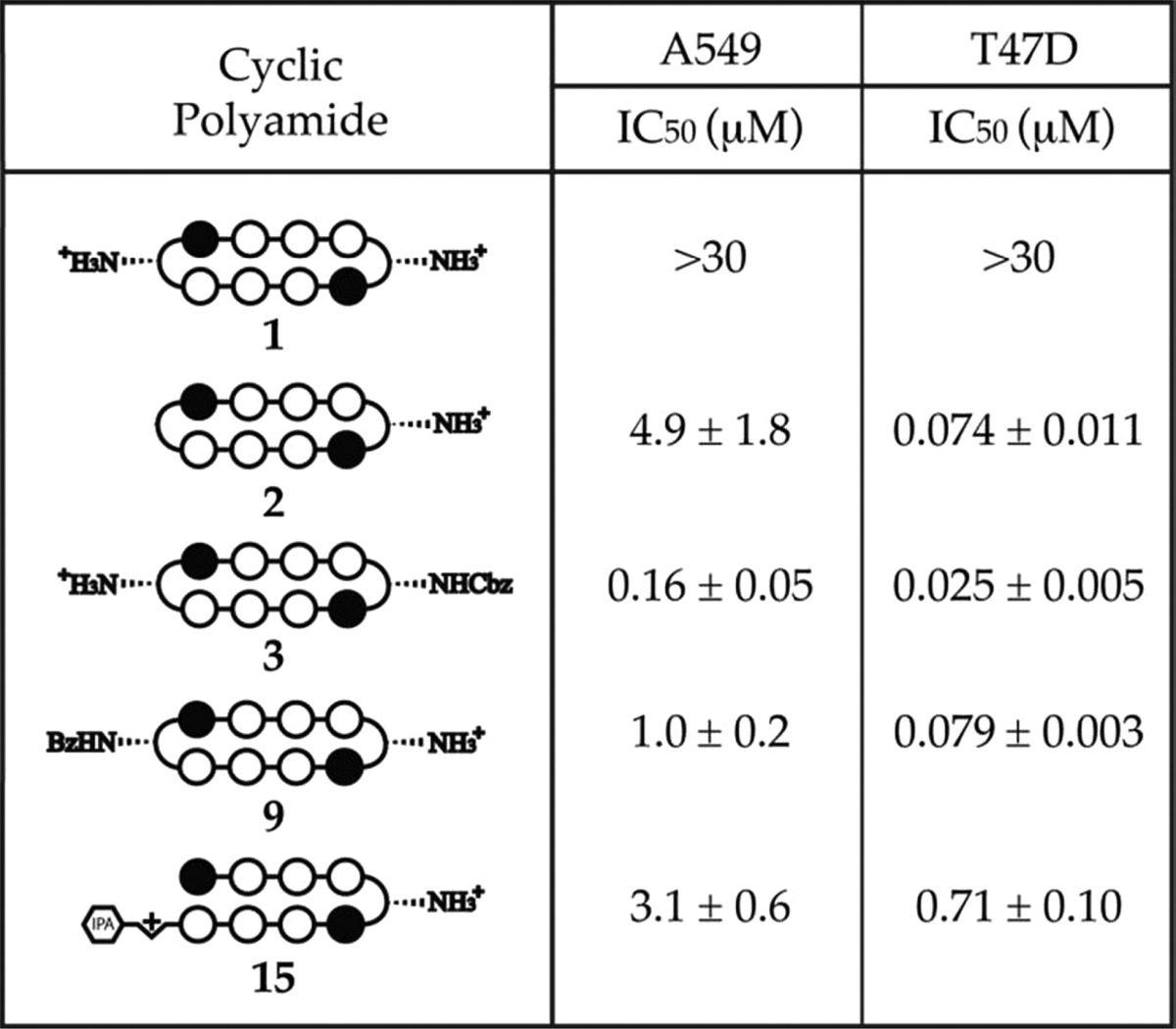
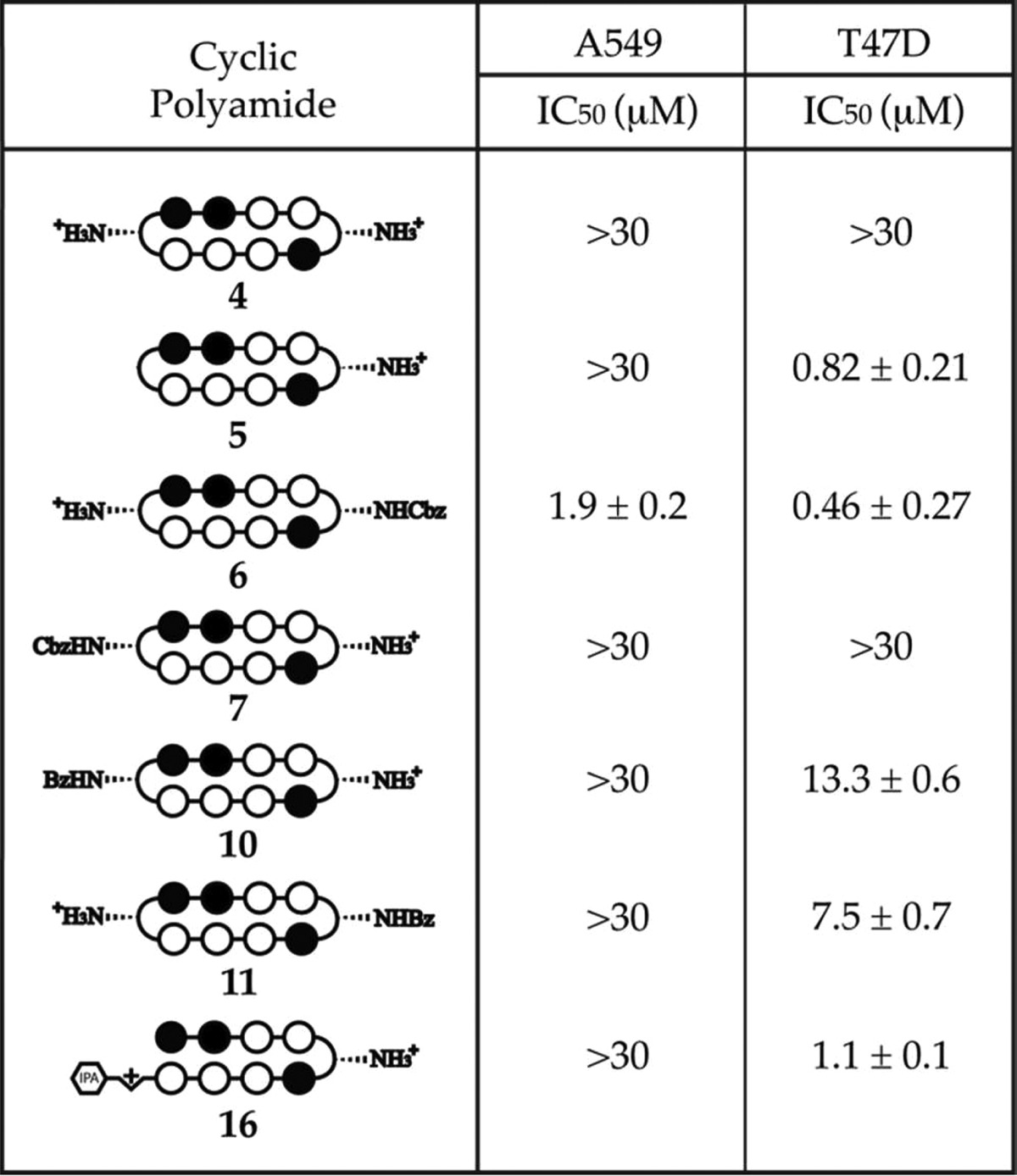
Table 5. SRB Cytotoxicity Data on Compounds 4–7, 10, 11, and 16, in A549 and T47D Cells, 72 Hour Incubation.
Bisamino cycle 1, which has previously been shown to be biologically active, did not display any significant level of cytotoxicity in either cell line (IC50 > 30 μM). Mono-unsubstituted compound 2 (IC50 = 4.9 μM) and mono-Bz 9 (IC50 = 1.0 μM) were comparably cytotoxic to hairpin polyamide 15 (IC50 = 3.1 μM) in A549 cells but an order of magnitude more cytotoxic (IC50 = 74 and 79 nM, respectively) than 15 (IC50 = 710 nM) in T47D cells. The mono-Cbz cycle 3 was consistently the most cytotoxic compound in both A549 (IC50 = 160 nM) and T47D (IC50 = 25 nM) cell lines.
For the 5′-WGGWCW-3′ targeting polyamides, the only compound that exhibited an IC50 value lower than 30 μM in A549 cells was the mono-Cbz compound 6 (IC50 = 1.9 μM). In T47D cells, consistent with the 5′-WGWWCW-3′ series, bisamino cycle 4 (IC50 > 30 μM) was found to be not significantly cytotoxic and 6 (IC50 = 460 nM) was the most cytotoxic compound. Mono-unsubstituted cycle 5 (IC50 = 0.82 μM) again shares a comparable level of cytotoxicity with the reference hairpin 16 (IC50 = 1.1 μM), whereas mono-Bz compounds 10 (IC50 = 13.3 μM) and 11 (IC50 = 7.5 μM) are both an order of magnitude less cytotoxic.
Perhaps most interestingly, mono-Cbz compound 7 did not exhibit observable levels of cytotoxicity (IC50 > 30 μM) in either A549 or T47D cells. Considering that 7 is a regioisomer of 6, where the Cbz group is simply swapped onto the other turn, and that 7 only differs from 3 by a single −CH to −N substitution, it is rather surprising that 7 is more than 15- to 65-fold less cytotoxic than 6 and at least 180- to 1200-fold less cytotoxic than 3 in the two examined cell lines. Given the comparable DNA stabilization properties between 6 and 7, and their common core sequence, we would not have predicted this vast discrepancy in cytotoxicity.
This study has demonstrated the large and somewhat unpredictable effects in biological activity induced by small structural variations of cyclic polyamides. On the basis of our preliminary work, the aggregation and pharmacokinetic properties of polyamides also vary greatly depending on structural modifications.31−33 All this combines to highlight the importance of a fast and reliable method to generate focused libraries of cyclic polyamides for future research.
Confocal Microscopy of Cyclic Polyamide–Fluorescein Conjugates 12–14
To directly examine the cellular localization of cyclic polyamides, fluorescein conjugates 12–14 were synthesized and visualized in living cells via confocal microscopy (Figures 2 and 3). The selective conjugation of a single fluorescein molecule not only helped retain a free amino group for solubility purposes but also allowed for the qualitative comparison of the two fluorescein conjugates 13 and 14 that share the same asymmetric polyamide core.
Figure 2.

Confocal microscopy of cyclic polyamide–fluorescein conjugates 12 (top), 13 (middle), and 14 (bottom) in A549 cells. In order to confirm nuclear localization, the fluorescence panel (left) was compared with Hoechst 33258 staining (middle) and overlay (right).
Figure 3.

Confocal microscopy of cyclic polyamide–fluorescein conjugates 12 (top), 13 (middle), and 14 (bottom) in T47D cells. In order to confirm nuclear localization, the fluorescence panel (left) was compared with Hoechst 33258 staining (middle) and overlay (right).
In each of the cases examined, cyclic polyamides 12–14 appear to permeate through the cellular membrane and localize in the cell nucleus, which was confirmed by the colocalization with Hoechst 33258 DNA stain. For ease of qualitatively assessing compound uptake, all A549 images were taken at a 660 fluorescence gain level, and all T47D images were taken at 600 fluorescence gain. Comparing Figures 2 and 3, the fluorescence levels of compounds 12–14 in T47D cells are all significantly higher than in A549, which is only further amplified by this difference in gain levels.
Compound 12 matched to the 5′-WGWWCW-3′ sequence exhibits the highest level of nuclear localization in both cell lines. Among the two 5′-WGGWCW-3′ targeting cycles, polyamide 14 qualitatively appears to have a relatively higher fluorescence signal in the cell nuclei in both A549 and T47D cells. This may help explain the cytotoxicity data reported above, where compounds 6 and 11 with Cbz and Bz substitutions on the same side as the fluorescein in 14 consistently display larger biological effects than 7 and 10 that are more structurally similar to cyclic polyamide 13.
Conclusion
We have described a modular solid-phase synthesis method, which, when combined with an established DPPA-mediated macrocyclization step, afforded cyclic polyamides in a high-yielding and time-efficient manner. Using this method, we have overcome previous limitations and synthesized both cyclic and hairpin polyamides that start with an imidazole unit. The binding affinities of all synthesized cycles have been assessed by DNA thermal denaturation assays and compare favorably to hairpin polyamides that bind their match DNA sequences at subnanomolar concentrations. Furthermore, the protection strategy of our method allows for selective modification of the GABA turn units, which we have used to rapidly generate a focused library of compounds. The cytotoxicity and uptake analysis of the cyclic polyamides revealed unexpected properties that further highlight the need for an efficient method to synthesize structural variants of cyclic polyamides for future studies.
Experimental Section
General Experimental Methods
2-Chlorotrityl chloride (2-Cl-Trt-Cl) resin was purchased from Bachem. FmocPyOH and FmocImOH monomers were purchased from Wako. PyBOP was purchased from NovaBioChem. Boc-β-Dab(Fmoc)-OH was purchased from Peptides International. All DNA oligomers were purchased HPLC purified from Integrated DNA Technologies. Cell culture medium was purchased from Gibco. Fetal bovine serum was purchased from Omega Scientific. Microwave-assisted coupling reactions were conducted on a Biotage Initiatior Eight synthesizer. Polyamide concentrations were measured in 20% MeCN in 0.1% (v/v) aqueous TFA using an approximated extinction coefficient of 69 200 M–1 cm–1 at λmax near 310 nm, unless otherwise specified.32,37
Monomer Loading onto 2-Cl-Trt Resin
Prior to manual microwave-assisted synthesis, 2-Cl-Trt-Cl resin (1.0 g, 1.59 mmol/g) was first loaded by mixing with 576 mg (1.59 mmol, 1 equiv) of FmocPyOH monomer, followed by addition of 6 mL of dimethylformamide (DMF) and 1.38 mL of diisopropylethylamine (DIEA) (7.59 mmol, 5 equiv). The suspended mixture was stirred for 18 h, then capped by addition of 1 mL of methanol (MeOH) and stirred for 1 h. The orange-colored, loaded resin was then collected on a fritted peptide synthesis vessel, washed with DMF (2×), MeOH (2×), DMF (2×), MeOH (2×), and diethylether (Et2O). [Owing to the sensitivity of the 2-Cl-Trt resin toward hydrolysis/methanolysis, this final Et2O wash was found to be essential and all loaded resin was sealed and stored at −20 °C.] The loading efficiency was quantitated via the Fmoc test and confirmed by measuring the mass of the dried resin. Typical monomer loading was calculated to be 0.4–0.8 mmol/g.
Microwave-Assisted Solid-Phase Synthesis (18–24)
All solid-phase polyamide coupling reactions were performed manually on a Biotage Initiator Eight microwave synthesizer on a 200–500 mg scale of loaded resin. Prior to each monomer coupling reaction, the N-terminal Fmoc group was first removed in a piperidine solution. The Fmoc deprotections were performed in a fritted peptide synthesis vessel at room temperature, and the specific conditions for each N-terminal monomer are as follows:
N-Fmoc-Pyrrole/Imidazole:
(a) swell resin in DCM; (b) wash with DMF; (c) add 50% piperidine in DMF; (d) shake suspension for 10 min; (e) wash with DMF; (f) repeat steps a–e twice.
N-Fmoc-GABA/β-Alanine:
(a) swell resin in DCM; (b) wash with DMF; (c) add 50% piperidine in DMF; (d) shake suspension for 5 min; (e) wash with DMF; (f) repeat steps a–e once.
Following Fmoc removal, the resin was deswelled in MeOH, washed with Et2O, dried in vacuo, and transferred to a microwave synthesis vessel as a dry powder. The corresponding monomer acid (3 equiv) was activated with PyBOP (3 equiv) and DIEA (6 equiv) in DMF (0.3 M concentration of monomer), and added to the resin. The coupling reactions were then setup in the microwave reactor at 50 °C for the time durations described in Table 1. After the listed microwave-assisted coupling times, the reaction mixture was filtered into a peptide synthesis vessel, and the collected resin was washed with DMF (3×), MeOH (3×), Et2O, and dried in vacuo. To ensure completion of each deprotection and coupling step, analytical HPLC spectra were taken by cleaving a small resin sample in 30% hexafluoroisopropanol (HFIP) in DCM.
The polyamide core was synthesized on 2-Cl-Trt resin in an iterative manner by repeating the deprotection–coupling procedures described above using the corresponding monomeric units. Upon completion of the sequence, 100–200 mg of the resin was suspended in 1 mL of 30% HFIP in DCM and stirred for 1 h to yield the crude N-terminal Fmoc-protected polyamide intermediate. The reaction mixture was then run through a cotton filter to remove the resin, and the filtrate was concentrated in vacuo. The residual oil was resuspended in 5 mL of a 1:1 MeOH/DCM mixture and reconcentrated in vacuo to give an off-white/beige solid. To remove the N-terminal Fmoc group, the solid was redissolved in 800 μL of DMF, followed by addition of 200 μL of piperidine, and the solution was stirred for 30 min. Upon confirmation of complete deprotection by analytical HPLC, the solution was added to 4 mL of 30% MeCN in 0.1% aqueous TFA. The precipitated 9-methylenefluorene side product was then removed by centrifugation and washed twice with 2 mL of 30% MeCN in 0.1% aqueous TFA. The combined aqueous solution was purified by reverse-phase HPLC and lyophilized to dryness to yield precyclic polyamide intermediates 18–24. All dried samples of 18–24 were stored at −20 °C prior to DPPA-mediated macrocyclization.
Synthetic yields and MALDI-TOF characterization data for 18–24 are summarized below:
(18): 12.6 μmol recovered (38.0 μmol theoretical, 33% yield). MALDI-TOF [M + H]+ calcd for C70H77N22O15+ = 1465.4, observed = 1465.9.
(19): 15.2 μmol recovered (38.0 μmol theoretical, 40% yield). MALDI-TOF [M + H]+ calcd for C62H70N21O13+ = 1316.5, observed = 1316.9.
(20): 12.8 μmol recovered (38 μmol theoretical, 34% yield). MALDI-TOF [M + Na]+ calcd for C67H79N22NaO15+ = 1453.6, observed = 1453.9.
(21): 15.1 μmol recovered (49 μmol theoretical, 31% yield). MALDI-TOF [M + H]+ calcd for C69H76N23O15+ = 1466.5, observed = 1466.9.
(22): 17.2 μmol recovered (51 μmol theoretical, 34% yield). MALDI-TOF [M + H]+ calcd for C61H69N22O13+ = 1317.5, observed = 1317.2.
(23): 16.2 μmol recovered (51 μmol theoretical, 32% yield). MALDI-TOF [M + Na]+ calcd for C66H77N23NaO15+ = 1454.5, observed = 1455.0.
(24): 8.2 μmol recovered (25 μmol theoretical, 33% yield). MALDI-TOF [M + Na]+ calcd for C66H78N23NaO15+ = 1454.5, observed = 1454.9.
DPPA-Mediated Macrocyclization (1–7)
The macrocyclization reactions were run on a 2–16 μmol scale. Intermediates 18–24 were first dissolved in DMF (0.25 mM) in a round-bottom flask equipped with a magnetic stir bar, followed by addition of DIEA (200 equiv), and purged with argon for 15 min. Diphenylphosporyl azide (DPPA) (50 equiv) was then added to the reaction mixture in a dropwise manner, while rapidly stirring. Upon full addition of the DPPA, the solution was allowed to react and stirred at room temperature for 16–20 h. After confirmation of reaction completion by analytical HPLC, the reaction mixture was concentrated in vacuo and the resulting oil residue was dissolved in 3 mL of MeCN and transferred to a 15 mL Falcon tube. The MeCN was then removed with air flow, and 3 mL of 0.1% aqueous TFA was added to the remaining oil layer to yield an off-white suspension, which was isolated via centrifugation and lyophilized to dryness.
For reactions starting with 18, 19, 21, and 22, the lyophilized residue was submitted to 1 mL of 10% trifluoromethanesulfonic acid (TFMSA) in TFA, stirred for 5 min, frozen in LN2, and thawed by layering 1 mL of DMF. For reactions starting with 20, 23, and 24, the lyophilized residue was submitted to 1 mL of neat TFA, stirred for 15 min, frozen in LN2, and thawed by layering 1 mL of DMF. All of the thawed solutions were then diluted with 6 mL of 0.1% aqueous TFA, purified by reverse-phase HPLC, and lyophilized to dryness to yield cyclic polyamides 1–7.
Synthetic yields and MALDI-TOF characterization data for 1–7 are summarized below:
(1): 4.5 μmol recovered (12.9 μmol theoretical, 35% yield). MALDI-TOF [M + H]+ calcd for C54H63N22O10+ = 1179.5, observed = 1179.9.
(2): 0.84 μmol recovered (2.0 μmol theoretical, 42% yield). MALDI-TOF [M + H]+ calcd for C54H62N21O10+ = 1164.5, observed = 1164.6.
(3): 3.2 μmol recovered (6.7 μmol theoretical, 47% yield). MALDI-TOF [M + H]+ calcd for C62H69N22O12+ = 1313.6, observed = 1314.0.
(4): 3.1 μmol recovered (8.0 μmol theoretical, 39% yield). MALDI-TOF [M + H]+ calcd for C53H62N23O10+ = 1180.5, observed = 1180.9.
(5): 0.96 μmol recovered (2.0 μmol theoretical, 48% yield). MALDI-TOF [M + H]+ calcd for C53H61N22O10+ = 1165.5, observed = 1165.5.
(6): 3.7 μmol recovered (10.0 μmol theoretical, 37% yield). MALDI-TOF [M + H]+ calcd for C61H68N23O12+ = 1314.5, observed = 1314.5.
(7): 2.2 μmol recovered (5.7 μmol theoretical, 38% yield). MALDI-TOF [M + H]+ calcd for C61H68N23O12+ = 1314.5, observed = 1314.8.
Selective Conjugation of Benzoic Acid Derivatives (9–11)
A solution of benzoic acid (3.0 mg, 0.025 mmol, 25 equiv) and PyBOP (13 mg, 0.025 mmol, 25 equiv) in DMF (0.5 mL) and DIEA (44 μL, 0.25 mmol, 250 equiv) was stirred at room temperature for 10 min. The activated solution was then added to 3 (1.0 μmol) and stirred for 3 h. After confirmation of complete reaction by analytical HPLC, 12 mL of cold Et2O was added to the reaction mixture and cooled at −20 °C for 16 h. The precipitate was then isolated by centrifugation and allowed to air-dry. The resulting residue was submitted to 1 mL of 10% trifluoromethanesulfonic acid (TFMSA) in TFA, stirred for 5 min, frozen in LN2, and thawed by layering 1 mL of DMF. The thawed solution was then diluted with 6 mL of 0.1% aqueous TFA, purified by reverse-phase HPLC, and lyophilized to dryness to yield cyclic polyamide 9 (684 nmol, 68% yield). Using the same procedure described above, starting with 6 (1.60 μmol) and 7 (750 nmol), yielded monobenzoyl-substituted cyclic polyamides 10 (1.05 μmol, 67% yield) and 11 (367 nmol, 49% yield), respectively.
(9): MALDI-TOF [M + H]+ calcd for C61H67N22O11+ = 1283.5, observed = 1284.1.
(10): MALDI-TOF [M + H]+ calcd for C60H66N23O11+ = 1284.5, observed = 1284.5.
(11): MALDI-TOF [M + H]+ calcd for C60H66N23O11+ = 1284.5, observed = 1284.9.
Cyclic Polyamide–Fluorescein Conjugates (12–14)
A solution of fluorescein isothiocyanate (FITC) (2.7 mg, 7.0 μmol, 25 equiv) in DMF (0.2 mL) and DIEA (12 μL, 0.07 mmol, 250 equiv) was added to 3 (0.28 μmol) and stirred for 2 h. After confirmation of complete reaction by analytical HPLC, 12 mL of cold Et2O was added to the reaction mixture and cooled at −20 °C for 16 h. The precipitate was then isolated by centrifugation and allowed to air-dry. The resulting residue was submitted to 1 mL of 10% trifluoromethanesulfonic acid (TFMSA) in TFA, stirred for 5 min, frozen in LN2, and thawed by layering 1 mL of DMF. The thawed solution was then diluted with 6 mL of 0.1% aqueous TFA, purified by reverse-phase HPLC, and lyophilized to dryness to yield cyclic polyamide 12 (65 nmol, 23% yield). Using the same procedure described above, starting with 6 (0.40 μmol) and 7 (0.40 μmol), yielded cyclic polyamide–fluorescein-conjugate 13 (345 nmol, 86% yield) and 14 (118 nmol, 29% yield), respectively.
(12): ESI-MS [M + H]+ calcd for C75H74N23O15S+ = 1568.6, observed = 1568.3.
(13): ESI-MS [M + H]+ calcd for C74H73N24O15S+ = 1569.5, observed = 1569.2.
(14): ESI-MS [M + H]+ calcd for C74H73N24O15S+ = 1569.5, observed = 1569.3.
Cyclic and Hairpin Polyamides Targeted to 5′-WCGWGW-3′ Sequence (8 and 17)
2-Cl-Trt-Cl resin (200 mg, 1.59 mmol/g) was first loaded with FmocPyImOH dimer (96 mg, 0.20 mmol), which was obtained from published procedures.28 Experimental details were analogous to the monomer loading protocol reported above. The obtained Fmoc-Py-Im-(2-Cl-Trt) resin (265 mg, 0.59 mmol/g) was subjected to the previously described microwave-assisted solid-phase synthesis conditions to build the corresponding polyamide sequence. A quarter of the resin (0.15 μmol theoretical) was then cleaved and purified by reverse-phase HPLC to yield precyclic polyamide intermediate 25 as an off-white powder (13.3 μmol, 34% yield). The isolated 25 (2.0 μmol) was subjected to DPPA-mediated macrocyclization conditions analogous to that for compounds 1–7, with the Cbz groups removed with 10% TFMSA in TFA, and purified by reverse-phase HPLC to afford cyclic polyamide 8 (773 nmol, 39% yield).
(25): MALDI-TOF [M – CO2 + H]+ calcd for C65H675N22O13+ = 1371.6, observed = 1371.7.
(8): MALDI-TOF [M + H]+ calcd for C61H67N22O11+ = 1129.5, observed = 1130.0.
Detailed experimental procedures and characterization data for 17 are provided in the Supporting Information.
Thermal Denaturation Analysis
Melting temperature analysis was performed on a Varian Cary 100 spectrophotometer equipped with a thermocontrolled cell holder possessing a cell path length of 1 cm. A degassed aqueous solution of 10 mM sodium cacodylate, 10 mM KCl, 10 mM MgCl2, and 5 mM CaCl2 at pH 7.0 was used as analysis buffer. DNA duplexes and polyamides were mixed in 1:1 stoichiometry to a final concentration of 2 μM for each experiment. Prior to analysis, samples were heated to 95 °C and cooled to a starting temperature of 25 °C with a heating rate of 5 °C/min for each ramp. Denaturation profiles were recorded at λ = 260 nm from 25 to 95 °C with a heating rate of 0.5 °C/min. The reported melting temperatures were defined as the maximum of the first derivative of the denaturation profile.
Cell Culture
Cell lines were cultured at 37 °C under 5% CO2 using standard cell culture and sterile techniques. Cell medium was supplemented with 10% fetal bovine serum. Ham’s F-12K (Kaighn’s) medium was used for A549 cells, and RPMI 1640 was used for T47D cells.
Confocal Microscopy
For each experiment, cells were plated in 200 μL of the proper medium onto glass-bottom cell culture plates at a density of 1 × 105 (A549) or 1.5 × 105 cells/mL (T47D). Cells were grown for 24 h, and media were replaced with fresh media containing polyamide to give a final DMSO concentration of 0.1%. Next, cells were incubated for 16 h, followed by removal of media, washing, and addition of fresh media. Hoechst 33258 was added 2 h prior to imaging. Imaging was performed at the Caltech Beckman Imaging Center using a Zeiss LSM 510 Meta NLO two-photon inverted laser scanning microscope equipped with a 40× oil-immersion objective lens. Polyamide–fluorescein conjugates 12–14 were imaged in multi-track mode using 488 nm laser excitation at 15% output with a pinhole of 375 μm and a standard fluorescein filter set. Hoechst was imaged using 800 nm two-photon excitation with an HFT KP680 dichroic and a 390 to 465 nm band-pass filter with a fully open pinhole. All images were analyzed using Zeiss LSM Zen software.
Sulforhodamine B Cytotoxicity
For cytotoxicity assays, cell lines were plated in 96-well cell culture plates in 100 μL of media at a density of 1 × 104 (A549) or 5 × 104 cells/mL (T47D). IC50 values were determined using the sulforhodamine B (SRB) colorimetric assay as previously described.39 Cells were grown for 24 h, before polyamides in 100 μL of media were added in serial dilution, in quadruplicate for each concentration. After incubation for 72 h, cell medium was replaced with 100 μL of fresh media, and cells were allowed to recover for an additional 24 h. Cells were then fixed by adding 100 μL of 10% trichloroacetic acid directly to each well and stored at 4 °C for 1 h, before being washed, dried, stained with 100 μL of 0.057% SRB solution per well for 30 min, and washed and dried again as described. After solubilizing the bound dye with 200 μL of 10 mM Tris (pH 10.5) per well, absorbance at 490 nm was measured on a PerkinElmer Victor microplate reader. The data are charted as a percentage of untreated controls, corrected for background absorbance. IC50 is defined as the concentration that inhibits 50% of control cell growth. These values were determined by nonlinear least-squares regression fit to Y = A + (B – A)/(1 + 10∧((log EC50 – X) × H)), where A = max, B = min, and H = Hill slope. All calculations were performed using Prism 4 (GraphPad) software. Stated IC50 values represent the mean and standard deviation of three independent biological replicates.
Acknowledgments
This work was supported by the National Institutes of Health (GM27681). B.C.L. is grateful to the California Tobacco-Related Disease Research Program (18DT-0015) for a dissertation award. D.C.M. is thankful to the National Institutes of Health for a predoctoral research training grant (5T32GM007616). The authors would like to thank Dr. Amanda E. Hargrove for helpful discussions, and Dr. Jordan L. Meier for assistance with molecular modeling.
Supporting Information Available
Experimental procedures and synthetic scheme for 17, HPLC spectra for 1–8 and 17–26, detailed confocal microscopy data for 12–14, and cytotoxicity data for compound S1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Dedication
Dedicated to Robert E. Ireland.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dervan P. B. Bioorg. Med. Chem. 2001, 9, 2215–2235. [DOI] [PubMed] [Google Scholar]
- Trauger J. W.; Baird E. E.; Dervan P. B. Nature 1996, 382, 559–561. [DOI] [PubMed] [Google Scholar]
- White S.; Szewczyk J. W.; Turner J. M.; Baird E. E.; Dervan P. B. Nature 1998, 391, 468–470. [DOI] [PubMed] [Google Scholar]
- Kielkopf C. L.; Baird E. E.; Dervan P. B.; Rees D. C. Nat. Struct. Biol. 1998, 5, 104–109. [DOI] [PubMed] [Google Scholar]
- Kielkopf C. L.; White S.; Szewczyk J. W.; Turner J. M.; Baird E. E.; Dervan P. B.; Rees D. C. Science 1998, 282, 111–115. [DOI] [PubMed] [Google Scholar]
- Mrksich M.; Parks M. E.; Dervan P. B. J. Am. Chem. Soc. 1994, 116, 7983–7988. [Google Scholar]
- Herman D. M.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1998, 120, 1382–1391. [Google Scholar]
- Hsu C. F.; Phillips J. W.; Trauger J. W.; Farkas M. E.; Belitsky J. M.; Heckel A.; Olenyuk B. Z.; Puckett J. W.; Wang C. C. C.; Dervan P. B. Tetrahedron 2007, 63, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best T. B.; Edelson B. S.; Nickols N. G.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12063–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dervan P. B.; Edelson B. S. Curr. Opin. Struct. Biol. 2003, 13, 284–299. [DOI] [PubMed] [Google Scholar]
- Hsu C. F.; Dervan P. B. Bioorg. Med. Chem. Lett. 2008, 18, 5851–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olenyuk B. Z.; Zhang G. J.; Klco J. M.; Nickols N. G.; Kaelin W. G. Jr.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 16768–16773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y.; Sugiyama H.; Ayame H.; Iwai A.; Fujii Y.; Huang L. E.; Kizaka-Kondoh S.; Hiraoka M.; Kihara K. Acta Oncol. 2006, 45, 317–324. [DOI] [PubMed] [Google Scholar]
- Nickols N. G.; Jacobs C. S.; Farkas M. E.; Dervan P. B. ACS Chem. Biol. 2007, 2, 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols N. G.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10418–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H.; Fukuda N.; Ueno T.; Tahira Y.; Ayame H.; Zhang W.; Bando T.; Sugiyama H.; Saito S.; Matsumoto K.; Mugishima H.; Serie H. J. Am. Soc. Nephrol. 2006, 17, 422–432. [DOI] [PubMed] [Google Scholar]
- Raskatov J. A.; Meier J. L.; Puckett J. W.; Yang F.; Ramakrishnan P.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J.; Parks M. E.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 10389–10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman D. M.; Turner J. M.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1999, 121, 1121–1129. [Google Scholar]
- Melander C.; Herman D. M.; Dervan P. B. Chem.—Eur. J. 2000, 6, 4487–4497. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Dwyer T. J.; Tsui V.; Case D. A.; Cho J.; Dervan P. B.; Wemmer D. E. J. Am. Chem. Soc. 2004, 126, 7958–7966. [DOI] [PubMed] [Google Scholar]
- Chenoweth D. M.; Harki D. A.; Phillips J. W.; Dose C.; Dervan P. B. J. Am. Chem. Soc. 2009, 131, 7182–7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaga H.; Bando T.; Takagaki T.; Yamamoto M.; Hashiya K.; Sugiyama H. J. Am. Chem. Soc. 2011, 133, 18924–18930. [DOI] [PubMed] [Google Scholar]
- Meier J. L.; Montgomery D. C.; Dervan P. B. Nucleic Acids Res. 2012, 40, 2345–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taleb R. I.; Jaramillo D.; Wheate N. J.; Aldrich-Wright J. R. Chem.—Eur. J. 2007, 13, 3177–3186. [DOI] [PubMed] [Google Scholar]
- van Holst M.; Le Pevelen D.; Aldrich-Wright J. R. Eur. J. Inorg. Chem. 2008, 29, 4608–4615. [Google Scholar]
- Puckett J. W.; Green J. T.; Dervan P. B. Org. Lett. 2012, 14, 2774–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weltzer M.; Wemmer D. E. Org. Lett. 2010, 12, 3488–3490. [DOI] [PubMed] [Google Scholar]
- Brady S. F.; Varga S. L.; Freidinger R. M.; Schwenk D. A.; Mendlowski M.; Holly F. W.; Veber D. F. J. Org. Chem. 1979, 44, 3101–3105. [Google Scholar]
- Boger D. L.; Yohannes D. J. Org. Chem. 1988, 53, 487–499. [Google Scholar]
- Raskatov J. A.; Hargrove A. E.; So A. Y.; Dervan P. B. J. Am. Chem. Soc. 2012, 134, 7995–7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove A. E.; Raskatov J. A.; Meier J. L.; Montgomery D. C.; Dervan P. B. J. Med. Chem. 2012, 55, 5425–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synold T. W.; Xi B.; Wu J.; Yen Y.; Li B. C.; Yang F.; Phillips J. W.; Nickols N. G.; Dervan P. B. Cancer Chemother. Pharmacol. 2012, 70, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1996, 118, 6141–6146. [Google Scholar]
- Wurtz N. R.; Turner J. M.; Baird E. E.; Dervan P. B. Org. Lett. 2001, 3, 1201–1203. [DOI] [PubMed] [Google Scholar]
- Trauger J. W.; Dervan P. B. Methods Enzymol. 2001, 340, 450–466. [DOI] [PubMed] [Google Scholar]
- Dose C.; Farkas M. E.; Chenoweth D. M.; Dervan P. B. J. Am. Chem. Soc. 2009, 130, 6859–6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas M. E.; Li B. C.; Dose C.; Dervan P. B. Bioorg. Med. Chem. Lett. 2009, 19, 3919–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichai V.; Kirtikara K. Nat. Protoc. 2006, 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.