

Abstract
The somatodendritic release of dopamine within the ventral tegmental area (VTA) and substantia nigra pars compacta activates inhibitory postsynaptic D2-receptors on dopaminergic neurons. The proposed mechanisms that regulate this form of transmission differ between electrochemical studies using rats and guinea pigs and electrophysiological studies using mice. This study examines the release and resulting dopamine D2-autoreceptor-mediated IPSCs (D2-IPSCs) in the VTA of mouse, rat, and guinea pig. Robust D2-IPSCs were observed in all recordings from neurons in slices taken from mouse, whereas D2-IPSCs in rat and guinea pig were observed less frequently and were significantly smaller in amplitude. In slices taken from guinea pig, dopamine release was more persistent under conditions of reduced extracellular calcium. The decline in the concentration of dopamine was also prolonged and not as sensitive to inhibition of reuptake by cocaine. This resulted in an increased duration of D2-IPSCs in the guinea pig. Therefore, unlike the mouse or the rat, the time course of dopamine in the extracellular space of the guinea pig determined the duration the D2-IPSC. Functionally, differences in D2-IPSCs resulted in inhibition of dopamine neuron firing only in slices from mouse. The results suggest that the mechanisms and functional consequences of somatodendritic dopamine transmission in the VTA vary among species. This highlights the complexity that underlies dopamine-dependent transmission in one brain area. Differences in somatodendritic transmission would be expected in vivo to affect the downstream activity of the mesocorticolimbic dopamine system and subsequent terminal release.
Introduction
Midbrain dopamine neurons play an essential role in goal-orientated motor and cognitive functions that include planning and initiating movements, and encoding the associations between rewards and salient environmental stimuli (Wise, 2004; Schultz, 2007). Disruptions in the mesocorticolimbic dopamine system may contribute to such psychiatric disorders as schizophrenia, attention-deficit and hyperactivity disorder, and drug addiction (Madras et al., 2005; Meisenzahl et al., 2007; Volkow et al., 2009).
Dopamine release in terminal regions is facilitated in vivo during bursts of activity driven by the appearance of unexpected rewards or by cues predicting rewards (Phillips et al., 2003; Tobler et al., 2005; Bromberg-Martin and Hikosaka, 2009). Dopamine neuron firing, in addition to driving axonal release, also drives the local release of dopamine from somatodendritic terminals in the midbrain (Geffen et al., 1976; Wilson et al., 1977; Cheramy et al., 1981; Groves and Linder, 1983; Rice et al., 1994; Nirenberg et al., 1996; Jaffe et al., 1998; Beckstead et al., 2004). Somatodendritic dopamine release in both the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc) activates D2-autoreceptors on postsynaptic dopamine neurons, leading to an inhibition via the activation of G-protein-coupled inwardly rectifying potassium (GIRK) channels (Lacey et al., 1987). Local release of dopamine in the VTA is therefore a form of feedback or lateral inhibition that can regulate dopamine neuron activity.
Somatodendritic release has often been examined using electrochemical or dialysis techniques to measure the bulk concentration of dopamine released into the extracellular space. Using these techniques in the rat and guinea pig, somatodendritic release has been found to differ from axonal release in several ways, including a prolonged duration lasting several seconds and a weak dependence upon extracellular calcium entry (Cragg and Greenfield, 1997; Rice et al., 1997; Hoffman and Gerhardt, 1999; Chen and Rice, 2001; Chen et al., 2011). In contrast, when measured using electrophysiology in the mouse, somatodendritic transmission in the VTA is short in duration and exhibits a steep dependence upon calcium entry (Beckstead et al., 2004; Ford et al., 2007, 2010). Differences in release could lead to differential activation of postsynaptic D2-receptors during somatodendritic transmission. Because D2-autoreceptors regulate dopaminergic excitability (Beckstead et al., 2004) and terminal release (Benoit-Marand et al., 2001; Phillips et al., 2002; Schmitz et al., 2002), alterations in the strength of somatodendritic transmission may have functional consequences for downstream dopamine neuron activity. Underlying this fact is recent work showing that D2-autoreceptor dysfunctions disrupt several dopamine-dependent behaviors, including locomotor activity, impulsivity, and motivation for rewards, in both mice and humans (Buckholtz et al., 2010; Bello et al., 2011).
To examine how differences in somatodendritic release regulate VTA neuron activity, the present study examined dopamine transmission in brain slices from the mouse, rat, and guinea pig. Using a combination of electrochemical and electrophysiological techniques, the results indicate that release across species varied dramatically in calcium dependence, strength, and time course. The functional consequences of these differences were apparent in the ability of phasic somatodendritic release to regulate dopamine neuron firing in the VTA.
Materials and Methods
Slice preparation and solutions.
All procedures were in accordance with guidelines of the Institutional Animal Care and Use Committee at Case Western Reserve University. Following anesthesia, horizontal midbrain slices containing the VTA (220 μm) or coronal forebrain slices containing the dorsal striatum (240 μm) were made from 3–5-week-old male and female C57BL6 mice (Jackson Laboratories), Sprague Dawley rats (Charles River), or Hartley guinea pigs (Charles River). Brain slices were cut in ice-cold sucrose cutting solution containing the following (in mm): 75 NaCl, 2.5 KCl, 6 MgCl2, 0.1 CaCl2, 1.2 NaH2PO4, 25 NaHC03, 2.5 d-glucose, and 50 sucrose; bubbled with 95% O2 and 5% CO2. For 45 min before recording, slices were incubated postcutting at 35°C in oxygenated 95% O2 and 5% CO2 ACSF solution containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11.1 mm d-glucose. During incubation, 10 μm MK-801 was included to reduce excitotoxicity and increase slice viability. Following incubation, slices were placed in a recording chamber and constantly perfused with warm ACSF (34 ± 2°C) containing 100 μm picrotoxin, 10 μm DNQX, and 200 nm CGP 55845 [(2S-3-([(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl)phosphinic acid] at 2 ml/min. For experiments testing the calcium dependence of dopamine release, the reduced Ca2+ was substituted for an equal molar amount of Mg2+ to maintain a constant divalent cation concentration (3.7 mm). Dopamine neurons were visualized with a BXWI51 microscope (Olympus) with infrared custom-built gradient contrast optics. The VTA was defined as the area within and medial to the fibers of the medial lemniscus (Ford et al., 2006, 2007). Because dopamine neurons projecting to the prefrontal cortex are insensitive to the inhibitory actions of dopamine due to a low expression of D2-receptors and GIRK channels (Chiodo et al., 1984, Lammel et al., 2008), we avoided recording from dopamine neurons in the most medial portions of the VTA.
Electrophysiology.
Whole-cell current-clamp or voltage-clamp recordings were made from VTA dopamine neurons using an Axopatch 200B amplifier (Molecular Devices). Patch pipettes (1.5–2 MΩ) were pulled from borosilicate glass (World Precision Instruments). The intracellular pipette solution contained 115 mm K-methylsulphate, 20 mm NaCl, 1.5 mm MgCl2, 10 mm HEPES(K), 10 mm BAPTA-tetrapotassium, 1 mg/ml ATP, 0.1 mg/ml GTP, and 1.5 mg/ml phosphocreatine, pH 7.4, 275 mOsm. Data were acquired using an ITC-18 interface (Instrutech) and Axograph X (Axograph Scientific) at 10 kHz and filtered to 2 KHz for voltage-clamp recordings. For voltage-clamp experiments, neurons were held at −60 mV. Series resistance was not compensated and cells were discarded if the access resistance exceeded 15 MΩ. Action potentials were recorded in current-clamp mode at a membrane potential set with constant current injection to ensure tonic pacemaker firing. VTA dopamine neurons were identified by the presence of a D2-receptor-sensitive dopamine conductance, pacemaker firing (0.5–5 Hz), an h-current, an input resistance of <300 MΩ, and an input capacitance of >25 pF (Ford et al., 2006; Chieng et al., 2011). To pharmacologically isolate evoked D2-receptor-mediated GIRK currents, 100 μm picrotoxin, 10 μm DNQX, and 200 nm CGP 55845 were added to the ACSF solution. The high concentration (10 mm) of BAPTA in the pipette solution and MK-801 in the incubation solution were used to block mGluR signaling and NMDA receptors. The D2-receptor antagonist sulpiride (200 nm) was used to confirm that dopamine currents were mediated through the D2 receptor.
Dopamine release was elicited with an ACSF-filled extracellular glass monopolar stimulating electrode placed ∼50–100 μm from the neuron recorded. A train of stimuli (5 pulses, 0.6 ms, 40 Hz, 40–60 μA) was used to evoke release. All drugs except dopamine were applied by bath perfusion. Dopamine was applied by iontophoresis. Dopamine (1 m) was ejected as a cation (160 nA) for 1 s using an Ion-100 Iontophoresis Generator (Dagan) from thin-walled iontophoretic electrodes placed ∼10 μm from the cell. A retention current of 3–20 nA was applied to prevent the leakage of dopamine.
Electrochemistry.
Fast-scan cyclic voltammetry (FSCV) recordings were made with glass-encased carbon fiber electrodes as previously described (Ford et al., 2009) using custom-built hardware (Electronics and Materials Engineering Shop, University of Washington, Seattle, WA) and software (Tarheel CV, Labview). Carbon fibers (34-700; 7 μm diameter; Goodfellow) were cut to a final length of 50–100 μm. Once cut, the tip was placed in isopropanol with activated carbon for 10 min before use. The exposed tip of the electrode was placed in the VTA ∼50 μm below the surface of the slice. A triangular waveform from −0.4 V to +1.3 V versus Ag/AgCl at changing voltage of 400 V/s at 10 Hz was used for all recordings. Between scans, the electrode was maintained at −0.4 V (vs Ag/AgCl). Background-subtracted cyclic voltammogram currents were obtained by subtracting the average of 10 cyclic voltammograms obtained before stimulation. The time course of dopamine release was obtained by plotting the current at the peak of oxidation (0.6–0.7 V) against time. The chemical identity of the FSCV recordings was examined by comparing cyclic voltammograms produced from the evoked release of dopamine to voltammograms produced by the exogenous iontophoretic application of dopamine hydrochloride (1 m) or serotonin hydrochloride (0.5 m). The peak reduction values of evoked voltammograms (−0.2 to −0.3 V) were similar across VTA of mice, rats, and guinea pigs (p > 0.7, one-way ANOVA) and were similar to the peak reduction value produced by exogenous dopamine (p > 0.5, one-way ANOVA) but not serotonin (p < 0.0001, one-way ANOVA), indicating that serotonin release did not contribute significantly to FSCV signals in the VTA. Currents were calibrated against dopamine standards ranging from 0.1 to 10 μm.
Because serotonergic terminals are also present in the SNc and the release of serotonin contributes to the FSCV signal detected in the SNc (Cragg et al., 1997; John et al., 2006; Ford et al., 2010), only the VTA was examined in the present study. This prevented the release of serotonin from axon terminals from interfering with FSCV measurements of dopamine release.
Chemicals.
Picrotoxin, DNQX, and MK-801 were from Ascent Scientific. CGP 55845 and S-(-)-sulpiride were from Tocris Bioscience. K-methylsulphate was from Acros Organics. BAPTA was from Invitrogen. Cocaine hydrochloride was from the National Institute of Drug Abuse. All other chemicals were from Sigma-Aldrich.
Statistics and analysis.
All data are shown as mean ± SE (SEM). Statistical significance (p < 0.05) was assessed by a one-way ANOVA or a Student's unpaired t test unless as noted (InStat 3.0, Graphpad). If ANOVA showed statistical significance (p < 0.05), all pairwise post hoc analysis stated was performed using a Tukey's post hoc test. Decay kinetics of D2-autoreceptor-mediated IPSCs (D2-IPSCs) and extracellular dopamine transients were fit with a single or double exponential using a Simplex algorithm optimized by the sum of squared errors in Axograph X (Axograph Scientific).
Results
The kinetics of dopamine release and transmission in the VTA
Carbon fiber electrodes were used with FSCV to compare the kinetics and the amount of dopamine released in VTA brain slices from the mouse, rat, and guinea pig. A train of five stimuli (40 Hz) evoked a rapid increase in the concentration of dopamine in the extracellular space ([DA]o). Nearly twice as much dopamine could be detected within the VTA of the mouse (145 ± 15 nm, n = 11) and guinea pig (153 ± 18 nm, n = 11) as in the VTA of the rat (86 ± 8 nm, n = 10, p < 0.05 vs mouse; p < 0.01 vs rat) (Fig. 1A). Despite the range in [DA]o that was evoked, the rise time and time to peak of the dopamine transients were similar among rats, mice, and guinea pigs (time-to-peak mouse: 536 ± 74 ms, n = 11; rat: 570 ± 73, n = 10; guinea pig: 545 ± 67 ms, n = 11; p > 0.9, one-way ANOVA) (Fig. 1A). Following release, however, the [DA]o remained elevated for longer in the guinea pig VTA than in the mouse or rat VTA. The [DA]o had a τdecay of 2.3 ± 0.2 s (n = 11) in the guinea pig VTA, which was slower than in the VTA of the rat (1.3 ± 0.2 s, n = 10) or mouse (1.4 ± 0.2 s, n = 11; p < 0.001) (Fig. 1B). Thus, despite a similarity in the rate of release, the amount and duration over which dopamine was present in the extracellular space varied between species with the concentration of dopamine being elevated for the longest in the VTA of the guinea pig.
Figure 1.
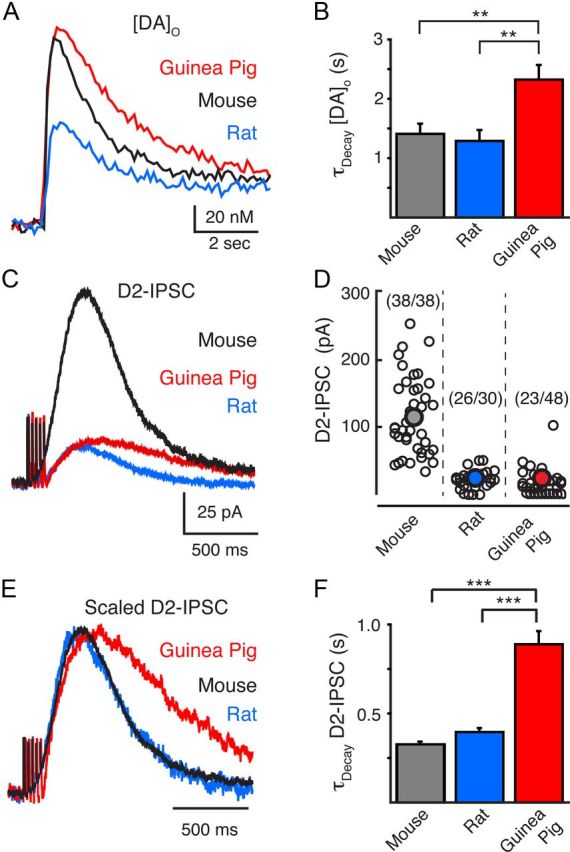
Interspecies characterization of dopamine release and D2-IPSCs in the VTA. A, FSCV measurements of the average evoked [DA]o by a train of stimuli (5 pulses, 0.5 ms, 40 Hz) in the mouse (black, n = 11), rat (blue, n = 10), and guinea pig (red, n = 11). Stimulation evoked less dopamine release in the rat than in the guinea pig or mouse. B, Summary data illustrating that the decay time constants for the [DA]o was longer in the guinea pig VTA than in the rat or mouse VTA. C, Representative traces of D2-IPSCs evoked by a train of stimuli (5 pulses, 0.5 ms, 40 Hz) in the mouse (black), rat (blue), and guinea pig (red). Synaptic currents were larger in amplitude in the mouse VTA than in the guinea pig or rat VTA. D, Summary distribution of D2-IPSC amplitude of all neurons recorded in the VTA of the mouse, rat, and guinea pig. Numbers in parenthesis refer to the proportion of neurons recorded in the VTA of each species that expressed a measurable D2-IPSC. Large filled circle illustrates the average synaptic currents of responding neurons. E, Normalized D2-IPSCs illustrated in D. D2-IPSCs have been scaled to the peak of their amplitude. D2-IPSCs from the mouse and rat exhibited similar kinetics while D2-IPSCs in the guinea pig had a slower rise time, time to peak, and time of decay. F, Summary data illustrating the slower time constant of decay of D2-IPSCs from the guinea pig (n = 23) than from the mouse (n = 37) or rat (n = 26). **p < 0.01; ***p < 0.001.
The extended extracellular presence of dopamine has often been implicated to indicate that dopamine is not constrained within the synapse following release. This has led to the hypothesis that spillover of dopamine or volume transmission may play a major role in shaping the time course of somatodendritic signaling. However, this may not be the case in the VTA of the mouse because the time course of the dopamine transient underlying somatodendritic transmission may be brief (Ford et al., 2009). As the amount and duration of dopamine released into the extracellular space varied between species, we next examined what effect this difference in [DA]o had on synaptic transmission. Whole-cell recordings were made from VTA dopamine neurons in brain slices from mice, rats, and guinea pigs. Dopamine D2-IPSCs were evoked by stimulating somatodendritic dopamine release (train of 5 pulses at 40 Hz) from adjacent presynaptic dopamine neurons with an extracellular stimulating electrode placed in the VTA. Only dopamine neurons in the central and lateral portions of the VTA were recorded because these neurons do not project to the prefrontal cortex and are known to exhibit autoreceptor-mediated inhibitory responses (Ford et al., 2006; Lammel et al., 2008). Robust D2-IPSCs were observed in all experiments from mouse VTA slices (n = 38). D2-IPSCs in slices from rat and guinea pig, in contrast, were smaller and observed less frequently (Fig. 1D). The average amplitude of D2-IPSCs in mouse dopamine cells was 115 ± 9 pA (n = 38) (Fig. 1D). The amplitude of the synaptic currents in rat VTA neurons was less than a fifth of that seen in the mouse (24 ± 2 pA, n = 26, p < 0.001). Surprisingly, the high [DA]o that could be detected in the guinea pig VTA did not reliably produce D2-IPSCs. IPSCs were observed in only 23 of 48 neurons (Fig. 1D) despite the exogenous application of dopamine evoking a D2-receptor-mediated outward current in all cells examined. Of the D2-IPSCs recorded, the amplitude was also small relative to those in the mouse (24 ± 3 pA, n = 23, p < 0.001) (Fig. 1C,D). Thus between species, the [DA]o detected by FSCV did not correlate with the amplitude of D2-IPSCs.
Despite the variation in amplitude, the time course of D2-IPSCs was remarkably similar between the rat and mouse (Fig. 1E). In agreement with previous results, D2-IPSCs from the mouse VTA peaked in 362 ± 7 ms (n = 38) (Fig. 1E) (Ford et al., 2009). D2-IPSCs from the rat had similar rise times to those in mice (time to peak: 349 ± 9 ms, n = 26, p > 0.2) (Fig. 1E). The time constant of decay was also similar between D2-IPSCs from rat and mouse VTA cells (mouse: τdecay = 327 ± 14 ms, n = 37; rat: τdecay = 396 ± 21 ms, n = 26; p > 0.05 one-way ANOVA) (Fig. 1F). These results indicate that, while large differences occur in the amount of dopamine released and in the amplitude of D2-IPSCs between rats and mice, the mechanisms controlling the time course of transmission are similar; in both species, the duration of D2-receptor activation appears to be tightly regulated. The reduction in both the [DA]o and amplitude of D2-IPSCs in the rat compared with that in the mouse may result from a difference in the number of release sites and/or points of synaptic contact in the rat.
D2-IPSCs in guinea pig VTA dopamine neurons were slower. The time to peak was 547 ± 22 ms (n = 23; p < 0.001) (Fig. 1E). Likewise the τdecay of D2-IPSCs was slower in dopamine cells from the guinea pig VTA than in dopamine cells from rat or mouse (889 ± 73 ms; n = 22; p < 0.0001). Thus, when scaled to their peak amplitude, D2-IPSCs from guinea pig VTA neurons did not overlap with those in rat and mouse dopamine cells (Fig. 1E). These results suggest dopamine is more loosely regulated within guinea pig VTA somatodendritic synapses than within rat and mouse VTA somatodendritic synapses. These observations are consistent with the idea that the extent of transmitter spillover or volume transmission varies widely at different somatodendritic synapses among species. The difference in time course in D2-IPSCs may result from postsynaptic differences between guinea pigs and rats and mice. This possibility is unlikely, however, because the dopamine transient measured electrochemically was also prolonged in the guinea pig VTA relative to that in the rat VTA and the mouse VTA (Fig. 1A,B).
Calcium dependence of dopamine release in the VTA and striatum
Somatodendritic dopamine release in the guinea pig persists under conditions where extracellular calcium is reduced (Rice et al., 1997; Chen and Rice, 2001; Chen et al., 2011). Because dopamine transmission in the mouse is inhibited in low extracellular calcium (Ford et al., 2010), we next asked whether lowering calcium would similarly affect somatodendritic release in the guinea pig, rat, and mouse. Dopamine release in both the mouse and rat VTA was strongly inhibited in 0.5 mm extracellular calcium. Lowering the extracellular calcium concentration ([Ca2+]o) from control values (2.5 mm) to 0.5 mm led to an inhibition of 91 ± 4% (n = 7) and 94 ± 2% (n = 11) of [DA]o and D2-IPSCs respectively in the mouse VTA (Fig. 2A,D). The inhibition was similar to that observed in the mouse SNc (percentage inhibition D2-IPSC: 91 ± 2%, n = 7; p > 0.2; data not shown). Lowering [Ca2+]o to 0.5 mm led to a similar reduction of 90 ± 3% (n = 10) and 94 ± 2% (n = 11) of [DA]o and D2-IPSCs in the rat VTA (Fig. 2B,D). Thus, dopamine release in the rat and mouse midbrain exhibits a strong dependence on extracellular calcium.
Figure 2.
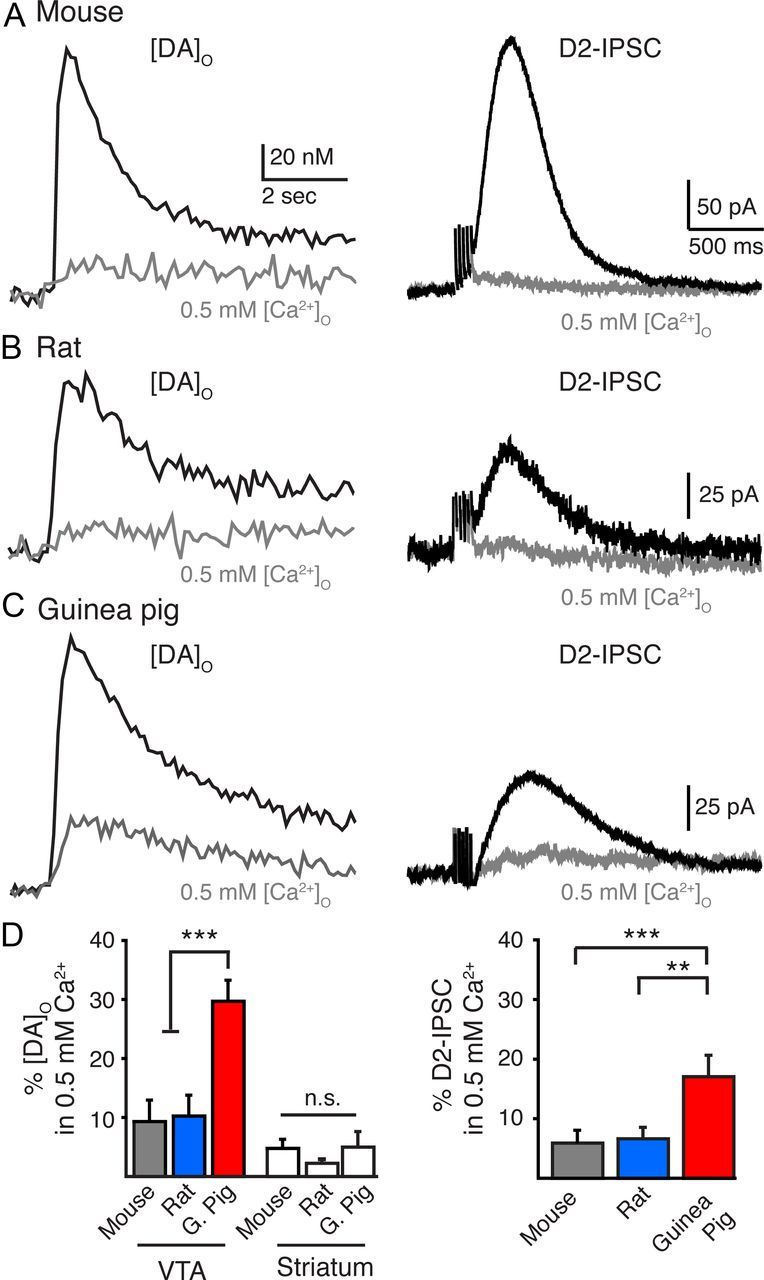
Dopamine release in the VTA shows a weaker dependence on extracellular calcium in guinea pigs than in rats or mice. A–C, Averaged [DA]o (n = 7–10) and representative traces of D2-IPSCs from the mouse, rat, and guinea pig under control conditions (2.5 mm [Ca2+]o; black) and low calcium (0.5 mm [Ca2+]o; gray). D, Summary data illustrating the [DA]o remaining in 0.5 mm [Ca2+]o in the VTA and striatum and the amplitude of D2-IPSCs in the VTA in 0.5 mm Ca2+compared with control (2.5 mm [Ca2+]o). **p < 0.01; ***p < 0.001.
In the guinea pig VTA, a significant amount of extracellular dopamine remained when calcium levels were lowered (Fig. 2C,D). Lowering [Ca2+]o to 0.5 mm led to a reduction of only 70 ± 4% (n = 8) in the [DA]o. D2-IPSCs in the VTA were also less sensitive to lowering the [Ca2+]o because, in the presence of 0.5 mm [Ca2+]o, D2-IPSCs were reduced by 83 ± 3% of control levels (n = 9) (p < 0.01) (Fig. 2C,D). These results support previous findings that somatodendritic dopamine release in the guinea pig shows weak dependence on extracellular calcium (Chen et al., 2011). The calcium dependence of somatodendritic release of dopamine therefore also varies between guinea pigs and other species.
Recurrent axon collaterals are also present within the VTA (Deutch et al., 1988; Bayer and Pickel, 1990). These terminals may also contribute to dopamine release in the VTA (Chen et al., 2011). We next compared release in the VTA to release in the dorsal striatum, where release is purely axonal. Lowering [Ca2+]o to 0.5 mm reduced the amount of dopamine detected in the striatum by 95 ± 1% (n = 5, Fig. 2D) in the mouse and likewise by 98 ± 4% (n = 5, Fig. 2D) in the rat, indicating that, in the rat and mouse, dopamine exhibits a strong dependence on [Ca2+]o at both axonal and dendritic release sites. In the guinea pig striatum, dopamine release was also strongly reduced in 0.5 mm [Ca2+]o (96 ± 4%, n = 5, Fig. 2D). Thus, the weak dependence of dopamine release on extracellular calcium entry was selective for only somatodendritic terminals of the guinea pig.
Dopamine D2-autoreceptor currents vary among species
The variation in D2-IPSCs among species and the lack of correlation with the measured [DA]o may reflect differences in D2-receptor/GIRK channel density, D2-receptor signaling efficiency, and/or the concentration of dopamine at postsynaptic D2-receptors. To examine these possibilities, we first examined D2-receptor-mediated outward potassium currents evoked by the exogenous iontophoretic application of dopamine. Application of a saturating concentration of dopamine by iontophoresis evoked a maximal outward current of 309 ± 26 pA in mouse VTA cells (n = 22), which was greater than the outward currents observed in rat (221 ± 18 pA, n = 18) or guinea pig dopamine neurons (143 ± 12, n = 46; p < 0.05) (Fig. 3A). Likewise, a similar difference among species was present when cell size was taken into account by normalizing the outward currents to the input capacitance (mouse: 7 ± 0.6 pA/pF, n = 22; rat: 5.3 ± 0.4 pA/pF, n = 18; guinea pig: 3.7 ± 0.3 pA/pF, n = 46; p < 0.05, Fig. 3B). Thus, D2-receptor-mediated GIRK-mediated currents are largest in mouse VTA dopamine cells and smaller in rat and guinea pig neurons. Different levels of receptor expression among species (Werkman et al., 2011) may explain why the density of D2-receptors in the midbrain is greater in the mouse than in the rat or guinea pig (Camps et al., 1990).
Figure 3.

Dopamine-mediated D2-autoreceptor currents are smallest in guinea pig VTA dopamine neurons. A, Representative traces of outward currents evoked by the exogenous iontophoretic application of dopamine (160 nA, 1 s). B, Current density (pA/pF) showing that a maximal application of dopamine evoked the largest currents in the mouse and the smallest currents in the guinea pig. *p < 0.05; ***p < 0.001.
Dopamine transporter regulation of extracellular dopamine in the VTA
Dopamine transporters regulate the amplitude and duration of D2-IPSCs following evoked release. Blocking transporters enhances spillover and prolongs the time course of transmission (Isaacson et al., 1993; Beckstead et al., 2004; Christie and Jahr, 2006; Ford et al., 2009). Because the duration of [DA]o and D2-IPSCs were longer in the guinea pig VTA than in the VTA of rats or mice (Fig. 1A,C), we next asked whether transport function differed within the VTA. Blocking dopamine reuptake with the monoamine transport blocker cocaine (1 μm) led to a similar increase in the amount and duration of [DA]o within the VTA of all species. Cocaine (1 μm) increased the peak [DA]o detected by 225 ± 19% (n = 6) in the mouse, by 227 ± 32% in the rat (n = 6), and by 171 ± 8% (n = 4) in the guinea pig (p > 0.2; one-way ANOVA). Blocking reuptake, however, increased the amplitude of D2-IPSCs in the rat and mouse VTA more than in the guinea pig VTA. Cocaine (1 μm) enhanced the peak of the D2-IPSC in the mouse and rat by 264 ± 31% (n = 7) and 336 ± 29% (n = 8), respectively, which was greater than the increase in the D2-IPSCs in guinea pig (167 ± 9%; n = 7; p < 0.05) (Fig. 4A,B). The smaller effect on D2-IPSCs from the guinea pig suggests that dopamine transporters are less effective in regulating the time course of dopamine at somatodendritic synapses in guinea pig VTA dopamine neurons than in the VTA dopamine neurons of rat or mouse. These observations indicate that D2-autoreceptor-mediated transmission varies in both strength and transporter-dependent regulation among species.
Figure 4.

Dopamine uptake in the VTA regulates D2-IPSCs to a greater extent in mice and rats than in guinea pigs. A, Representative traces illustrating evoked D2-IPSCs under control conditions (black) and in the presence of cocaine (1 μm, gray) from VTA dopamine neurons in mice, rats, and guinea pigs. Note the greater increase in amplitude of D2-IPSCs recorded from rats and mice than from guinea pigs in the presence of cocaine. D2-IPSCs under control conditions have been sized to the same initial amplitude to illustrate the relative increase induced by blocking reuptake. B, Summarized data illustrating the increase in amplitude of D2-IPSCs in the presence of cocaine (1 μm). *p < 0.05; ***p < 0.001.
Extent of somatodendritic dopamine transmission correlates with evoked pauses in pacemaker firing
We next asked what functional consequences somatodendritic dopamine transmission would have on the firing patterns of dopamine neurons. Evoked somatodendritic release was hypothesized to have the greatest effect on mouse dopaminergic activity and less of an effect on firing in guinea pig or rat neurons. Initially, the predicted change in voltage in VTA dopamine neurons was calculated based upon the observed amplitude of D2-IPSCs from all cells recorded (including failures) and the species-specific cellular input resistance of VTA dopamine neurons (VD2-IPSP = ID2-IPSC × Rinput). The estimated hyperpolarization in mouse VTA cells was ∼14.8 mV (115 pA * 129 MΩ), which was greater than that predicted for rat or guinea pig VTA neurons (rat: ∼2.1 mV = 21 pA * 101 MΩ; guinea pig: ∼0.7 mV = 11 pA * 68 MΩ).
To test whether the D2-IPSC could regulate the firing patterns of VTA neurons, whole-cell current-clamp recordings were made from dopamine cells in the mouse, rat, and guinea pig. Dopamine neurons typically fire in vitro in a tonic pacemaker pattern at ∼1–5 Hz. A train of stimuli, similar to that used to evoke D2-IPSCs (5 pulses at 40 Hz) evoked large hyperpolarizations in mouse VTA dopamine neurons, sufficient to induce a pause in pacemaker firing (Fig. 5A). The hyperpolarization-induced pause lasted 1.5 ± 0.1 s (n = 18), which was similar to duration of D2-IPSCs recorded in voltage-clamp (Fig. 5A). The pause in pacemaker firing was blocked by bath application of the D2-receptor antagonist sulpiride (200 nm) (0.5 ± 0.1 s; n = 13) (Fig. 5A). When normalized to the baseline pacemaker firing frequency of each neuron, stimulation led to a 364 ± 23% inhibition in firing frequency (n = 18). Sulpiride (200 nm) reduced the pause in firing to 134 ± 11% of control levels (n = 13; p < 0.0001). In contrast, trains of stimuli failed to evoke large dopamine-mediated hyperpolarizations in dopamine cells from rat or guinea pig VTA slices (Fig. 5B,C). Thus the stimulation-induced change in firing rate was not different in rat or guinea pig VTA dopamine neurons between control slices and slices treated with sulpiride (200 nm) (rat control: 164 ± 16%, n = 19; sulpiride: 145 ± 11%, n = 12; guinea pig control: 164 ± 22%, n = 18; sulpiride: 177 ± 25%, n = 16%; p > 0.7; one-way ANOVA) (Fig. 5C). Together these data suggest that the extent of somatodendritic transmission determines the role that D2-autoreceptors play in regulating the phasic actions of dopamine on VTA pacemaker firing.
Figure 5.
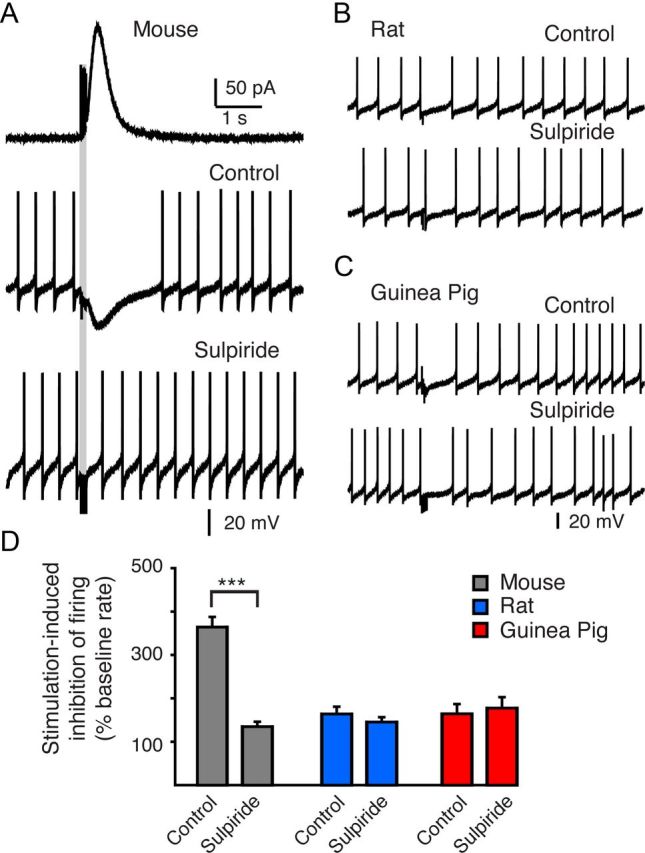
Evoking D2-IPSCs does not induce a pause in the firing of VTA dopamine neurons from the rat or guinea pig. A, Voltage-clamp recording of a D2-IPSC (top trace) from a mouse VTA dopamine neuron and current-clamp recordings of tonic pacemaker firing of the same neuron (lower traces). The time course of the evoked D2-IPSCs was similar to that of the evoked hyperpolarization and the pause in firing when recorded in current clamp. The D2-receptor antagonist, sulpiride (200 nm), blocks the evoked pause in firing, suggesting that D2-receptor activation during phasic release underlies the pause in firing. B, C, Current-clamp recordings of tonic pacemaker firing of rat and guinea pig VTA dopamine neurons under control conditions and in the presence of sulpiride (200 nm). Stimulation does not induce an extended pause in firing. Sulpiride does not change the firing rate following stimulation, suggesting that D2-receptor activation by evoked release in the rat and guinea pig is not sufficient to induce a phasic pause in firing. D, Summary data of the above conditions. Data are illustrated as the time from the start of stimulation to the first action potential following the stimulation as a percentage of the average interspike interval recorded from each cell during 3 s of baseline tonic firing. ***p < 0.001.
Discussion
The results show that the mechanisms controlling release, time course, and strength of somatodendritic dopamine transmission vary widely across the VTA of different species. While much of the past work examining somatodendritic release has been done in rats and guinea pigs (Cragg and Greenfield, 1997; Rice et al., 1997; Hoffman and Gerhardt, 1999; Chen and Rice, 2001; Chen et al., 2011), the physiological actions of dopamine as a synaptic transmitter have been examined only in mice (Beckstead et al., 2004; Ford et al., 2009, 2010; Bello et al., 2011). In contrast to somatodendritic dopamine transmission in the guinea pig, somatodendritic dopamine transmission in rats and mice exhibited a strong dependence on extracellular calcium. The time course of dopamine within somatodendritic synapses was also more strongly regulated by dopamine transporters in rats and mice than in guinea pigs. This limited dopamine spillover and prevented prolonged activation of postsynaptic D2-receptors. Thus, somatodendritic dopamine transmission in the guinea pig varied from that in other species in time course and the dependence of release on extracellular calcium entry. This work suggests that species-specific differences may account for at least some of the differences in the mechanisms underlying somatodendritic release and transmission reported in previous publications.
Calcium dependence of dopamine release in the VTA
Dopamine release from dendritic terminals has often been reported to persist in low extracellular calcium, leading to the conclusion that somatodendritic release exhibits a weak dependence on calcium entry not seen at conventional CNS synapses (Hoffman and Gerhardt, 1999; Chen and Rice, 2001; Fortin et al., 2006; Chen et al., 2011). Here we found that, in the rat and the mouse, both axonal and dendritic dopamine release showed an equally strong dependence on extracellular calcium. Lowering calcium to 0.5 mm led to a ≥90% reduction in dopamine release in the VTA and dorsal striatum. The VTA contains both axon collaterals and somatodendritic terminals (Deutch et al., 1988; Bayer and Pickel, 1990) and the release of dopamine from both sets of terminals can contribute to the total detected increase in [DA]o with FSCV (Ford et al., 2010; Chen et al., 2011). However, because D2-IPSCs from VTA and SNc dopamine cells are similar in amplitude, kinetics, duration, and pharmacological regulation (Beckstead et al., 2004; Ford et al., 2007, 2010), the physiological activation of D2-receptors during phasic release may occur primarily at somatodendritic synapses. Like the reduction in the [DA]o, D2-IPSCs in the VTA were also similarly reduced by >90% in the presence of 0.5 mm calcium in rats and mice. Thus, whether measured electrochemically or electrophysiologically, somatodendritic dopamine release in both the rat and mouse shows a strong dependence on calcium entry from the extracellular space.
Dopamine transmission in the VTA of the guinea pig, however, showed a marked difference in regulation. As previously reported, lowering extracellular calcium (0.5 mm) did not inhibit dopamine release to the same extent in the VTA compared with the striatum (Chen et al., 2011). In 0.5 mm extracellular calcium, ∼30% of total dopamine release remained in the VTA. Likewise, D2-IPSCs in VTA dopamine neurons from the guinea pig were also less sensitive to reductions in extracellular calcium. Because dopamine neurons of the guinea pig do not express many of the vesicular fusion release proteins commonly expressed at fast synapses (Witkovsky et al., 2009), the results suggest that dopamine transmission in the guinea pig may occur via a different set of mechanisms than at other central synapses.
Time course of transmission
The duration of synaptic transmission is dependent upon both the concentration time course of neurotransmitter and the kinetics of the neurotransmitter-receptor interactions (Katz and Miledi, 1973; Lester et al., 1990; Barbour et al., 1994). Many G-protein-coupled receptors that mediate slow metabotropic transmission, such as GABAB receptors, are located at extrasynaptic sites and are activated by transmitter spillover away from the synaptic cleft (Otis and Mody, 1992; Isaacson et al., 1993; Scanziani, 2000; Kulik et al., 2002; Beenhakker and Huguenard, 2010). The long-lasting presence of dopamine detected in the extracellular space of rats and guinea pigs has often thus been interpreted to indicate that dopamine transmission also occurs by a paracrine spillover-type mechanism (volume transmission). In the mouse VTA, somatodendritic dopamine transmission is thought to be mediated by a high concentration of dopamine that activates a pool of receptors located within 1 μm from the site of release (Ford et al., 2009). Somatodendritic dopamine transmission may therefore be more localized than previously assumed.
Dopamine D2-IPSCs were significantly larger in the mouse than in the rat. The iontophoresis of exogenous dopamine evoked smaller maximal outward currents in rat dopamine cells, suggesting that D2-receptor/GIRK channel density, number of release sites, and/or the number of points of synaptic contact may be reduced in the rat compared with the mouse. However, despite the smaller amplitude, the kinetics and duration of synaptic transmission were similar. Because the concentration of dopamine determines the rate of activation of the GIRK conductance (Ford et al., 2009), the similarity in the rate of rise of D2-IPSCs and time to peak of synaptic currents suggests that, during transmission, D2-autoreceptors are exposed to similar concentrations of dopamine in both the rat and mouse.
Dopamine D2-IPSCs were significantly slower in the guinea pig. In guinea pig dopamine cells, IPSCs had a slower rise time, time to peak, and decay kinetics than either the rat or mouse. Because blocking dopamine reuptake with cocaine had a limited effect on the amplitude of D2-IPSCs in the guinea pig, the reuptake of dopamine in the VTA of the guinea pig may be less efficient. The slow kinetics of D2-IPSCs in the guinea pig most likely results from this decreased rate of dopamine uptake. The effect of this in the guinea pig would be an increase in dopamine spillover to extrasynaptic sites and a prolonged activation of postsynaptic receptors.
Functional implications
The functional consequence of D2-IPSCs in dopamine cells from the VTA of the mouse was a pause in pacemaker firing. In rats and guinea pigs, electrical stimulation often led to slower firing. However, the effect was not altered in the presence of the D2-receptor antagonist, sulpiride. Multiple metabotropic neurotransmitter receptors, including GABAB, mGluR, and α1-noradrenergic receptors, inhibit dopamine cells (Fiorillo and Williams, 1998; Paladini and Tepper, 1999; Morikawa et al., 2003; Paladini and Williams, 2004; Morikawa and Paladini, 2011). The pause was unlikely to be mediated by GABAB and mGluR receptors because receptor signaling was prevented by antagonists (CGP 55845) or blockers of the receptor-effector signaling cascade (BAPTA), suggesting that activation or inactivation of other conductances may have been engaged as a result of the train of stimuli. This species difference in ability of D2 autoreceptors to induce a pause in firing may result from a higher level of expression of D2 receptors in the mouse than in either the rat or guinea pig (Camps et al., 1990; Werkman et al., 2011). Lower coupling efficiency or GIRK channel expression in rats or guinea pigs may also have contributed to the observed differences. However, this possibility seems less likely because robust GABAB-mediated synaptic potentials that couple via GIRK channels have been reported previously in guinea pig dopamine neurons (Cameron and Williams, 1993; Bonci and Williams, 1996).
The activation of D2 autoreceptors has long been known to regulate dopaminergic neuron firing (Aghajanian and Bunney, 1977). The present results indicate that strong activation of these receptors during robust somatodendritic synaptic events can be an efficient form of lateral inhibition that regulates the firing patterns of VTA dopamine neurons. The species-dependent variations in the strength of D2-IPSCs that we observed would therefore be expected to differentially control the firing pattern of dopamine neurons in vivo and subsequently the forebrain terminal release of dopamine during behaviorally relevant tasks. Because multiple mechanisms can underlie dopamine transmission, subtle alterations in any of these mechanisms would be expected to have significant effects on the efficiency of transmission. Recent work in humans has shown that reduced midbrain D2-autoreceptor levels correlated with highly impulsive individuals (Buckholtz et al., 2010). Disruptions in somatodendritic transmission may therefore lead to widespread dysfunctions in this system at the behavioral and clinical level. Differences in this system highlight the need to consider species-specific aspects of dopamine signaling in the future when assessing the preclinical efficacy of pharmacological treatments for a variety of psychiatric diseases.
Footnotes
This work was supported by National Institutes of Health Grant DA026417 (C.P.F.) and a NARSAD (National Alliance for Research on Schizophrenia and Depression) Young Investigator Grant from the Brain & Behavior Research Foundation (C.P.F.). C.P.F. was the Francoeur Investigator of the Brain & Behavior Research Foundation and a Mt. Sinai Health Care Foundation Scholar.
The authors declare no competing financial interests.
References
- Aghajanian GK, Bunney BS. Dopamine“autoreceptors”: pharmacological characterization by microiontophoretic single cell recording studies. Naunyn Schmiedebergs Arch Pharmacol. 1977;297:1–7. doi: 10.1007/BF00508803. [DOI] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Bayer VE, Pickel VM. Ultrastructural localization of tyrosine hydroxylase in the rat ventral tegmental area: relationship between immunolabeling density and neuronal associations. J Neurosci. 1990;10:2996–3013. doi: 10.1523/JNEUROSCI.10-09-02996.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci. 2010;30:15262–15276. doi: 10.1523/JNEUROSCI.3243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, Rubinstein M. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci. 2011;14:1033–1038. doi: 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J Neurosci. 2001;21:9134–9141. doi: 10.1523/JNEUROSCI.21-23-09134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Hikosaka O. Midbrain dopamine neurons signal preference for advance information about upcoming rewards. Neuron. 2009;63:119–126. doi: 10.1016/j.neuron.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckholtz JW, Treadway MT, Cowan RL, Woodward ND, Li R, Ansari MS, Baldwin RM, Schwartzman AN, Shelby ES, Smith CE, Kessler RM, Zald DH. Dopaminergic network differences in human impulsivity. Science. 2010;329:532. doi: 10.1126/science.1185778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- Camps M, Kelly PH, Palacios JM. Autoradiographic localization of dopamine D 1 and D 2 receptors in the brain of several mammalian species. J Neural Transmission General Section. 1990;80:105–127. doi: 10.1007/BF01257077. [DOI] [PubMed] [Google Scholar]
- Chen BT, Rice ME. Novel Ca2+ dependence and time course of somatodendritic dopamine release: substantia nigra versus striatum. J Neurosci. 2001;21:7841–7847. doi: 10.1523/JNEUROSCI.21-19-07841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Patel JC, Moran KA, Rice ME. Differential calcium dependence of axonal versus somatodendritic dopamine release, with characteristics of both in the ventral tegmental area. Front in Syst Neurosci. 2011;5:39. doi: 10.3389/fnsys.2011.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheramy A, Leviel V, Glowinski J. Dendritic release of dopamine in the substantia nigra. Nature. 1981;289:537–542. doi: 10.1038/289537a0. [DOI] [PubMed] [Google Scholar]
- Chieng B, Azriel Y, Mohammadi S, Christie MJ. Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol. 2011;589:3775–3787. doi: 10.1113/jphysiol.2011.210807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodo LA, Bannon MJ, Grace AA, Roth RH, Bunney BS. Evidence for the absence of impulse-regulating somatodendritic and synthesis-modulating nerve terminal autoreceptors on subpopulations of mesocortical dopamine neurons. Neuroscience. 1984;12:1–16. doi: 10.1016/0306-4522(84)90133-7. [DOI] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ, Greenfield SA. Differential autoreceptor control of somatodendritic and axon terminal dopamine release in substantia nigra, ventral tegmental area, and striatum. J Neurosci. 1997;17:5738–5746. doi: 10.1523/JNEUROSCI.17-15-05738.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ, Hawkey CR, Greenfield SA. Comparison of serotonin and dopamine release in substantia nigra and ventral tegmental area: region and species differences. J Neurochem. 1997;69:2378–2386. doi: 10.1046/j.1471-4159.1997.69062378.x. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Goldstein M, Baldino F, Jr, Roth RH. Telencephalic projections of the A8 dopamine cell group. Ann N Y Acad Sci. 1988;537:27–50. doi: 10.1111/j.1749-6632.1988.tb42095.x. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature. 1998;394:78–82. doi: 10.1038/27919. [DOI] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Beckstead MJ, Williams JT. Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J Neurophysiol. 2007;97:883–891. doi: 10.1152/jn.00963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Phillips PE, Williams JT. The time course of dopamine transmission in the ventral tegmental area. J Neurosci. 2009;29:13344–13352. doi: 10.1523/JNEUROSCI.3546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Gantz SC, Phillips PE, Williams JT. Control of extracellular dopamine at dendrite and axon terminals. J Neurosci. 2010;30:6975–6983. doi: 10.1523/JNEUROSCI.1020-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin GD, Desrosiers CC, Yamaguchi N, Trudeau LE. Basal somatodendritic dopamine release requires snare proteins. J Neurochem. 2006;96:1740–1749. doi: 10.1111/j.1471-4159.2006.03699.x. [DOI] [PubMed] [Google Scholar]
- Geffen LB, Jessell TM, Cuello AC, Iversen LL. Release of dopamine from dendrites in rat substantia nigra. Nature. 1976;260:258–260. doi: 10.1038/260258a0. [DOI] [PubMed] [Google Scholar]
- Groves PM, Linder JC. Dendro-dendritic synapses in substantia nigra: descriptions based on analysis of serial sections. Exp Brain Res. 1983;49:209–217. doi: 10.1007/BF00238581. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Gerhardt GA. Differences in pharmacological properties of dopamine release between the substantia nigra and striatum: an in vivo electrochemical study. J Pharmacol Exp Ther. 1999;289:455–463. [PubMed] [Google Scholar]
- Isaacson JS, Solís JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Jaffe EH, Marty A, Schulte A, Chow RH. Extrasynaptic vesicular transmitter release from the somata of substantia nigra neurons in rat midbrain slices. J Neurosci. 1998;18:3548–3553. doi: 10.1523/JNEUROSCI.18-10-03548.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CE, Budygin EA, Mateo Y, Jones SR. Neurochemical characterization of the release and uptake of dopamine in ventral tegmental area and serotonin in substantia nigra of the mouse. J Neurochem. 2006;96:267–282. doi: 10.1111/j.1471-4159.2005.03557.x. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The binding of acetylcholine to receptors and its removal from the synaptic cleft. J Physiol. 1973;231:549–574. doi: 10.1113/jphysiol.1973.sp010248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik A, Nakadate K, Nyíri G, Notomi T, Malitschek B, Bettler B, Shigemoto R. Distinct localization of GABA(B) receptors relative to synaptic sites in the rat cerebellum and ventrobasal thalamus. Eur J Neurosci. 2002;15:291–307. doi: 10.1046/j.0953-816x.2001.01855.x. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–773. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lester RA, Clements JD, Westbrook GL, Jahr CE. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 1990;346:565–567. doi: 10.1038/346565a0. [DOI] [PubMed] [Google Scholar]
- Madras BK, Miller GM, Fischman AJ. The dopamine transporter and attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1397–1409. doi: 10.1016/j.biopsych.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Meisenzahl EM, Schmitt GJ, Scheuerecker J, Möller HJ. The role of dopamine for the pathophysiology of schizophrenia. Int Rev Psychiatry. 2007;19:337–345. doi: 10.1080/09540260701502468. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Paladini CA. Dynamic regulation of midbrain dopamine neuron activity: intrinsic, synaptic, and plasticity mechanisms. Neuroscience. 2011;198:95–111. doi: 10.1016/j.neuroscience.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J Neurosci. 1996;16:4135–4145. doi: 10.1523/JNEUROSCI.16-13-04135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Mody I. Differential activation of GABAA and GABAB receptors by spontaneously released transmitter. J Neurophysiol. 1992;67:227–235. doi: 10.1152/jn.1992.67.1.227. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Tepper JM. GABA(A) and GABA(B) antagonists differentially affect the firing pattern of substantia nigra dopaminergic neurons in vivo. Synapse. 1999;32:165–176. doi: 10.1002/(SICI)1098-2396(19990601)32:3<165::AID-SYN3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Williams JT. Noradrenergic inhibition of midbrain dopamine neurons. J Neurosci. 2004;24:4568–4575. doi: 10.1523/JNEUROSCI.5735-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips PE, Hancock PJ, Stamford JA. Time window of autoreceptor-mediated inhibition of limbic and striatal dopamine release. Synapse. 2002;44:15–22. doi: 10.1002/syn.10049. [DOI] [PubMed] [Google Scholar]
- Phillips PE, Stuber GD, Heien ML, Wightman RM, Carelli RM. Subsecond dopamine release promotes cocaine seeking. Nature. 2003;422:614–618. doi: 10.1038/nature01476. [DOI] [PubMed] [Google Scholar]
- Rice ME, Richards CD, Nedergaard S, Hounsgaard J, Nicholson C, Greenfield SA. Direct monitoring of dopamine and 5-HT release in substantia nigra and ventral tegmental area in vitro. Exp Brain Res. 1994;100:395–406. doi: 10.1007/BF02738400. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ, Greenfield SA. Characteristics of electrically evoked somatodendritic dopamine release in substantia nigra and ventral tegmental area in vitro. J Neurophysiol. 1997;77:853–862. doi: 10.1152/jn.1997.77.2.853. [DOI] [PubMed] [Google Scholar]
- Scanziani M. GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Schmauss C, Sulzer D. Altered dopamine release and uptake kinetics in mice lacking D2 receptors. J Neurosci. 2002;22:8002–8009. doi: 10.1523/JNEUROSCI.22-18-08002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Behavioral dopamine signals. Trends Neurosci. 2007;30:203–210. doi: 10.1016/j.tins.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Tobler PN, Fiorillo CD, Schultz W. Adaptive coding of reward value by dopamine neurons. Science. 2005;307:1642–1645. doi: 10.1126/science.1105370. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine's role in drug abuse and addiction. Neuropharmacology. 2009;56(Suppl 1):3–8. doi: 10.1016/j.neuropharm.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werkman TR, McCreary AC, Kruse CG, Wadman WJ. NK3 receptors mediate an increase in firing rate of midbrain dopamine neurons of the rat and the guinea pig. Synapse. 2011;65:814–826. doi: 10.1002/syn.20908. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Groves PM, Fifkov á E. Monoaminergic synapses, including dendro-dendritic synapses in the rat substantia nigra. Exp Brain Res. 1977;30:161–174. doi: 10.1007/BF00237248. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Witkovsky P, Patel JC, Lee CR, Rice ME. Immunocytochemical identification of proteins involved in dopamine release from the somatodendritic compartment of nigral dopaminergic neurons. Neuroscience. 2009;164:488–496. doi: 10.1016/j.neuroscience.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]