

Abstract
Mitochondria are primarily responsible for providing the contracting cardiac myocyte with a continuous supply of ATP. However, mitochondria can rapidly change into death-promoting organelles. In response to changes in the intracellular environment, mitochondria become producers of excessive reactive oxygen species and release pro-death proteins, resulting in disrupted ATP synthesis and activation of cell death pathways. Interestingly, cells have developed a defense mechanism against aberrant mitochondria that can cause harm to the cell. This mechanism involves selective sequestration and subsequent degradation of the dysfunctional mitochondrion before it causes activation of cell death. Induction of mitochondrial autophagy, or mitophagy, results in selective clearance of damaged mitochondria in cells. In response to stress such as ischemia/reperfusion, pro-survival and pro-death pathways are concomitantly activated in cardiac myocytes. Thus, there is a delicate balance between life and death in the myocytes during stress, and the final outcome depends on the complex crosstalk between these pathways. Mitophagy functions as an early cardioprotective response, favoring adaptation to stress by removing damaged mitochondria. In contrast, increased oxidative stress and apoptotic proteases can inactivate mitophagy, allowing for the execution of cell death. Herein, we discuss the importance of mitochondria and mitophagy in cardiovascular health and disease, and provide a review of our current understanding of how these processes are regulated in the myocardium.
Keywords: mitochondria, autophagy, apoptosis, p53, Parkin, PINK1
Introduction
Mitochondria are primarily responsible for producing ATP via oxidative phosphorylation in the inner mitochondrial membrane. Due to the high energy demand of the heart, mitochondria make up at least 30% of the myocyte volume 1. Mitochondria also play central roles in both necrosis and apoptosis, and can quickly change from a source of energy for sustaining contraction to an organelle that promotes cell death. In response to changes in the environment, ATP synthesis is disrupted, and mitochondria can become producers of excessive reactive oxygen species (ROS) and release proteins that participate in cell death pathways. Interestingly, cells have developed a defense mechanism against aberrant mitochondria, which can cause harm to the cell. This mechanism involves selective sequestration and subsequent degradation of the dysfunctional mitochondrion before it causes activation of cell death. This occurs through a process known as mitochondrial autophagy, or mitophagy.
Autophagy is an evolutionarily conserved process that is responsible for the degradation of components in the cytoplasm via the lysosomal pathway 2. When the cell receives a signal to initiate autophagy, a membrane called the phagophore is formed (Figure 1). Initial phagophore formation, or nucleation, requires assembly of a complex consisting of BECLIN1, vacuolar protein sorting 34 (VPS34), and VPS15 3. Subsequent expansion of the membrane is mediated by two ubiquitin-like conjugation systems, LC3 and ATG12-ATG5, that promote assembly of the ATG16L complex and the conjugation of LC3 with phosphatidylethanolamine 4, 5. The phagophore expands until its edges fuse around its target(s), forming a double-membrane structure called the autophagosome. Next, the autophagosome fuses with a lysosome and the contents are degraded by lysosomal enzymes 2.
Figure 1.
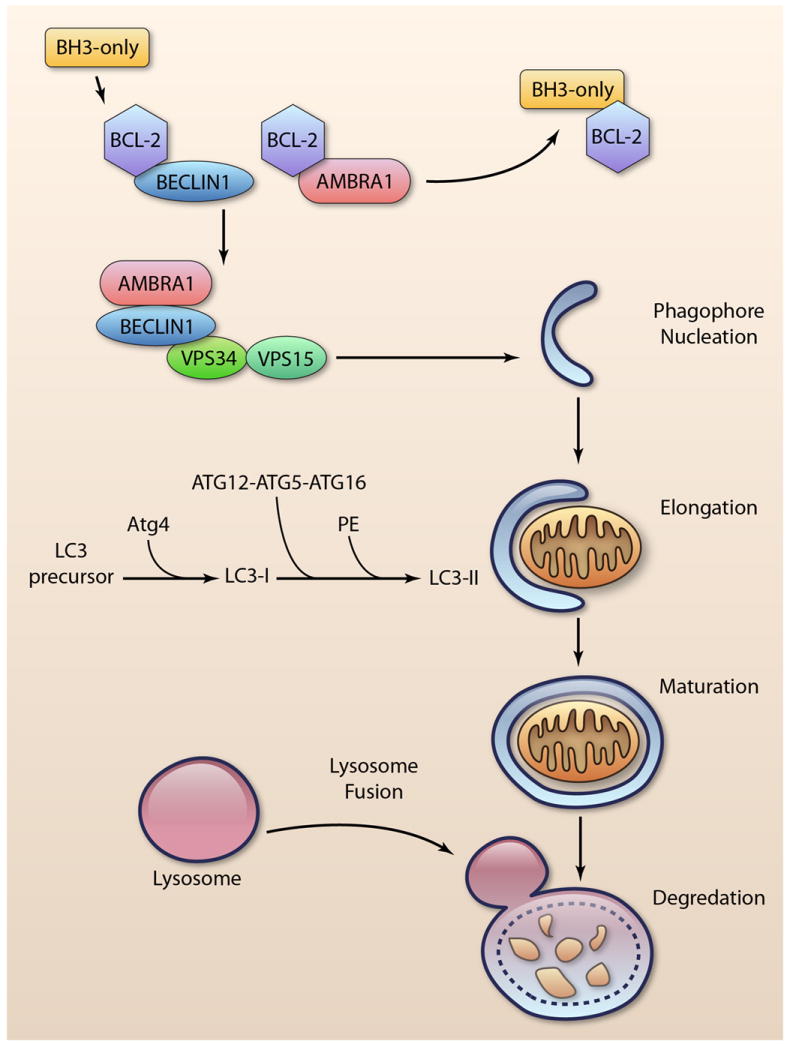
Induction of autophagy. BCL-2/BCL-XL prevents induction of autophagy by binding BECLIN1 and AMBRA1. Displacement by BH3-only proteins leads to activation of the BECLIN1-VPS34-VPS15 complex and phagophore nucleation. Elongation of the membrane requires two ubiquitin-like conjugation systems: ATG12-ATG5-ATG16L, and conjugation of LC3-I with phosphatidylethanolamine (PE) to form LC3-II. After maturation of the autophagosome, it fuses with the lysosome to degrade the cargo. (Illustration Credit: Ben Smith).
Autophagy was initially believed to be a non-selective process whereby autophagosomes randomly engulf cytosolic material. However, it is now clear that autophagy specifically targets invading bacteria 6, protein aggregates 7, and organelles such as mitochondria 8 and ER 9. Thus, autophagy constitutes a very important quality control mechanism in cells, particularly in post-mitotic cells such as cardiac myocytes.
Interestingly, mitochondria can activate both cell death and mitophagy, two opposing forces in the cell, in response to the same stimulus. These are highly regulated processes, and the delicate balance between the two determines whether a cell will live or die. Defining the role of mitochondria and mitophagy in various cardiovascular pathologies is currently an area of great interest. The importance of understanding how dysfunctional mitochondria and mitophagy contribute to cell survival and death in the myocardium during ischemia/reperfusion and heart failure is becoming increasingly apparent. This review highlights the role of mitochondria in cell death and mitophagy, and discusses our current understanding of how these processes are regulated in cells.
Mitochondria and Cell Death
Mitochondria are important regulators of cell death and respond to many different stress signals, including loss of growth factors, hypoxia, oxidative stress, and DNA damage. The switch to a cell death program is mediated by permeabilization of the outer mitochondrial membrane via BAX and/or BAK, or by opening of the mitochondrial permeability transition pore (mPTP) in the inner mitochondrial membrane (Figure 2). Permeabilization of the outer membrane by BAX/BAK results in the release of proapoptotic proteins such as cytochrome c, SMAC/DIABLO, apoptosis-inducing factor (AIF), and endonuclease G (EndoG) to activate apoptosis 10. In contrast, opening of the mPTP causes rapid influx of solutes and water into the mitochondrial matrix, collapse of the proton gradient, disruption of ATP synthesis. This influx causes swelling of the inner membrane and eventual rupture of the outer membrane, culminating in necrotic cell death. Both forms of cell death are highly regulated by mitochondria, and have been implicated in loss of myocardial cells in pathologies such as I/R, cardiomyopathy, and congestive heart failure 11-14.
Figure 2.
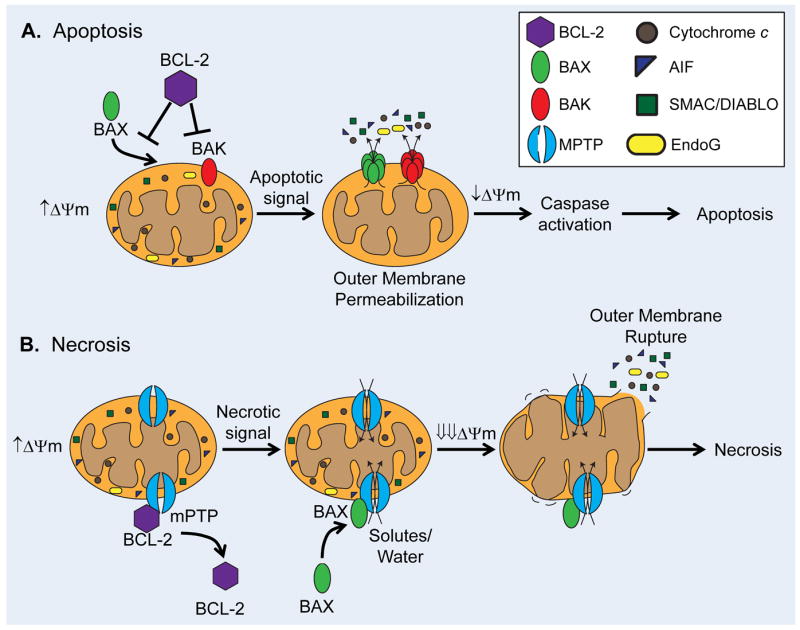
Mechanisms of mitochondrial membrane permeabilization. A. Anti-apoptotic BCL-2 family proteins, such as BCL-2 and BCL-XL, inhibit BAX/BAK-mediated outer mitochondrial membrane permeabilization. BAX/BAK activation causes release of cytotoxic proteins, slow loss of DYm, and culminates in apoptosis. B. Opening of the mPTP results in rapid influx of solutes and water which causes dissipation of the Dym, inner membrane swelling and subsequent outer membrane rupture. Destruction of the outer membrane releases cytotoxic proteins into the cytosol and leads to necrotic cell death. Anti-apoptotic BCL-2 proteins also prevent mPTP opening, while pro-apoptotic proteins such as Bax can enhance mPTP opening.
Regulation of Mitochondrial Permeabilization by BCL-2 Family Proteins
The BCL-2 family of proteins is at the center of mitochondrial apoptosis regulation. Anti-apoptotic members such as BCL-2 and BCL-XL promote survival by inhibiting the function of the pro-apoptotic BCL-2 proteins. The pro-apoptotic members can be separated into two distinct subfamilies: the BH3-only proteins, which include BID, BNIP3, NIX/BNIP3L, and PUMA; and the effector proteins BAX and BAK 11. The BH3-only proteins transduce stress signals from the cytosol to the mitochondria to initiate cell death by binding to and neutralizing the anti-apoptotic BCL-2 proteins, thereby activating BAX and BAK 15, 16. Some of the BH3-only proteins, such as tBID, can interact directly with BAX and BAK to induce their activation. Upon activation, BAX and BAK form pores in the outer mitochondrial membrane large enough to allow passage of cytotoxic proteins from the intermembrane space to the cytosol (Figure 2A) 17. The regulation of apoptosis in the heart by BCL-2 proteins has been implicated in the pathogenesis of myocardial hypertrophy, infarction and heart failure 11. Numerous studies have demonstrated a cardioprotective role for the anti-apoptotic BCL-2 proteins in response to various stressors, and transgenic mice overexpressing BCL-2 in the heart are resistant to I/R injury 18, 19. Moreover, BAX is activated in myocytes in response to oxidative stress 20 and during ischemia 21. Hearts from BAX-deficient mice have reduced mitochondrial damage and decreased infarct size after I/R compared to wild type mice, implicating BAX as a major player of mitochondrial dysfunction in I/R 22. Among the BH3-only proteins, BID, PUMA, BNIP3, and NIX/BNIP3L have been implicated in cardiac myocyte death 23-26.
Mitochondria and the mPTP
The mPTP is a channel in the inner mitochondrial membrane that allows for passage of molecules up to 1.5 kDa 12. Opening of the pore causes a collapse of the proton gradient and electrical potential across the inner mitochondrial membrane, leading to disruption of oxidative phosphorylation (Figure 2B). Opening of the mPTP also causes influx of water and subsequent swelling of the inner membrane. The outer mitochondrial membrane is unable to expand, resulting in rupture and release of pro-apoptotic proteins into the cytosol. The composition of the mPTP has remained elusive, and to date the only protein identified to be an essential component is cyclophilin D (CypD) 27, 28. The mPTP is a major contributor of myocardial I/R injury, and inhibitors of the mPTP reduce infarct size in ex vivo I/R 29, 30. Moreover, CypD-deficient mice are resistant to I/R injury 27, 28. Interestingly, cells lacking CypD are still sensitive to apoptotic stimuli, suggesting that mPTP opening is not required for induction of apoptosis via the mitochondrial pathway 27.
Studies have also suggested the voltage-dependent anion channel (VDAC) and the adenine nucleotide transporter (ANT) as potential components of the mPTP. Interestingly, experiments in mice deficient for multiple isoforms of VDAC demonstrate that pore opening occurs in the absence of VDAC 31. Similarly, mitochondria lacking ANT1/2 still undergo permeability transition, albeit at higher Ca2+ thresholds 32. However, considering the critical role of the ANTs in ATP/ADP exchange, it is likely that a functional compensation by other mitochondrial carrier proteins counterbalances the lack of ANT. There’s strong evidence that ANT and VDAC are important regulators of cell death. Studies have found that modulation of the levels of different ANTs can result in either cytoprotection or exacerbated cell death. For instance, overexpression of ANT1 causes apoptosis 33, whereas ANT2 inhibits mPTP opening 34. Differential regulation of cell survival and death has also been reported for the different isoforms of VDAC. Tajeddine et al. reported that VDAC1 contributes to mitochondrial membrane permeabilization via activation of BAX 35. In contrast, VDAC2 has been reported to inhibit apoptosis by sequestering BAK 36.
There exists substantial cross talk between the BCL-2 proteins and the mPTP. In the heart, BCL-2 has been reported to increase the calcium threshold for mPTP opening by blocking opening of the pore 37. Additionally, Kitsis and colleagues recently made the discovery that Bax/Bak/CypD triple knockout mice fail to show further reduction in infarct size compared to Bax/Bak double knockout mice 38. Moreover, the authors found that cells deficient only for Bax/Bak are also resistant to mPTP opening and necrotic cell death, 38 and that BAX can promote mPTP opening via a mechanism that is distinct from its ability to induce outer mitochondrial membrane permeabilization. Thus, while apoptosis and necrosis are distinct pathways, there is substantial overlap of the two in both regulation and mechanism of action.
Regulation of ER Ca2+ Flux and Metabolism by BCL-2 Proteins
Release of Ca2+ from the ER via inositol triphosphate (IP3) receptors is another critical event for the initiation of apoptosis. Mitochondria are closely associated with ER, and a major portion of the Ca2+ that is released into the cytosol is absorbed by mitochondria 39. This mitochondrial Ca2+ buffering both protects cells from the cytotoxic effects of Ca2+ and results in activation of several key enzymes in the mitochondrial matrix to enhance ATP production 39. However, excess Ca2+ uptake by mitochondria leads to Ca2+ overload and opening of the mPTP 27. The level of Ca2+ stored in the ER, and by extension released during stress, determines how much is subsequently taken up by mitochondria. BCL-2 proteins also localize to the ER and regulate Ca2+ homeostasis. BCL-2 and BCL-XL repress pro-apoptotic Ca2+ signals from the ER by reducing ER Ca2+ stores, consequently reducing Ca2+ release during stress 40, 41. Alternatively, BCL-2 can directly interact with IP3 receptors to inhibit IP3-mediated ER Ca2+ release 42. In contrast, ER-localized BAX and BAK elevate the resting ER Ca2+ stores and trigger release of ER Ca2+ into the cytosol during stress 40, 43. Moreover, the BH3-only protein NIX is localized to the ER/SR and is implicated with increasing Ca2+ release from ER/SR, leading to opening of the mPTP in cardiac myocytes 44. Thus, pro-apoptotic BCL-2 proteins promote cell death both by directly causing mitochondrial permeability and indirectly by increasing ER calcium release for subsequent uptake by mitochondria.
Studies have demonstrated that anti-apoptotic BCL-2 family proteins can also influence cell survival by regulating mitochondrial bioenergetics. A pool of BCL-XL localizes to the mitochondrial matrix, where it interacts with the β-subunit of the F1F0 ATP synthase and prevents proton leakage 45, 46. Since proton leakage reduces available protons for the ATPase, decreasing this leak enhances the efficiency of mitochondrial ATP synthesis. This positive energetic change may increase resistance to stressors such as hypoxia. Similarly, Perciavalle et al. reported that Mcl-1 is localized to distinct mitochondrial locations where it exhibits differing functions 47. Mcl-1 on the outer mitochondrial membrane has anti-apoptotic activity and maintains mitochondrial integrity, whereas Mcl-1 in the matrix is important in assembly of F1F0 ATP synthase oligomers.
Regulation of Autophagy by BCL-2 Family Proteins
BCL-2 and BCL-XL are also negative regulators of autophagy via interaction with BECLIN1 48 (Figure 1). Although BECLIN1 contains a BH3 domain, it does not function as a pro-apoptotic protein 49. Instead, it is involved in autophagosome nucleation. He et al. recently reported that both fasting- and exercise-induced autophagy in the myocardium requires disruption of the BCL2 BECLIN1 complex, and knock-in mice expressing a mutant BCL-2 that is unable to dissociate from BECLIN1 fail to induce autophagy 50. BCL-2 also inhibits autophagy by interacting with AMBRA1 (activating molecule in BECLIN1-regulated autophagy) 51. AMBRA1 activates the BECLIN1-VPS34-VPS15 complex that is required for nucleation of phagophores52. During autophagy induction, BECLIN1 and AMBRA1 are released from BCL-2 to initiate the formation of the phagophore. The interaction between BECLIN1 and BCL-2/BCL-XL can be disrupted by BH3-only proteins to activate autophagy 53, 54. However, it is currently unknown what mechanism triggers the dissociation of AMBRA1 from BCL-2.
BCL-2 family proteins can also regulate autophagy via ER Ca2+ stores. Treatment of cells with intracellular Ca2+-mobilizing agents results in increased autophagy, which is inhibited by ER-targeted BCL-2 55. Similarly, Brady et al. demonstrated that ER/SR-targeted BCL-2 negatively regulates starvation-mediated autophagy by depleting ER/SR Ca2+ content rather than via direct interaction with BECLIN1 in HL-1 myocytes 56.
BNIP3 and NIX/BNIP3L were originally identified as pro-apoptotic BH3-only proteins that cause cell death via permeabilization of the outer mitochondrial membrane 57-59. Although it is now understood that they can activate both apoptosis and mitophagy, it is also evident that these are two distinct processes activated independently and in differing contexts. For instance, NIX/BNIP3L is essential for removal of mitochondria in reticulocytes by mitophagy 60, 61. Similarly, BNIP3 is a potent inducer of mitophagy in cells including cardiac myocytes 8, 24. In cells lacking BAX and BAK, overexpression of BNIP3 results in increased mitophagy in the absence of outer mitochondrial membrane permeabilization 62. Interestingly, we found that disrupting BNIP3-mediated mitophagy has no effect on its pro-apoptotic activity 9. This suggests that BNIP3 and NIX have the dual function of regulating both mitophagy 24, 60, 63 and cell death 57-59. It is still unclear exactly under what conditions BNIP3 and NIX switch from mitophagy regulators to mitochondrial pro-death proteins.
Regulation of Cell Death and Autophagy by p53
The p53 tumor suppressor protein regulates both apoptosis and autophagy. The induction of cell death by p53 occurs via regulation of gene expression and by direct action at the mitochondria. Under normal conditions, p53 is maintained at low levels by the E3 ubiquitin ligase MDM2, which targets p53 for degradation by the proteasome 64. In response to stress such as hypoxia or DNA damage, the levels of p53 increase in the cell. Nuclear p53 transactivates several pro-apoptotic genes, including Bax65, Bak65, 66, Noxa67 and Puma68, while it represses the transcription of anti-apoptotic Bcl-2, Bcl-XL, and Mcl-166, 69, 70. A portion of p53 also translocates to mitochondria, where it promotes mitochondrial membrane permeabilization by interacting with BCL-2 family proteins. p53 has been reported to directly bind to and activate BAX 71 and BAK 72, and to inhibit BCL-2/BCL-XL73. Accumulating evidence indicates that p53 plays an important role in stress-induced apoptosis in the heart. Studies have found that p53 is upregulated in the heart in response to ischemia 74, oxidative stress 75, and anthracycline exposure 76. Moreover, p53-deficient mice have reduced susceptibility to anthracycline-induced myocardial apoptosis and heart failure 77, and cardiac-specific deletion of Mdm4, a homolog of Mdm2, results in elevated levels of p53 and development of dilated cardiomyopathy 78.
p53 plays dual roles in regulating autophagy depending on the context and subcellular localization. Under normal conditions, cytoplasmic p53 suppresses the induction of autophagy. Tasdemir et al. initially reported that genetic deletion, knockdown, or inhibition of p53 induces autophagy in cells, suggesting that cytoplasmic p53 functions as an autophagy repressor 79. In response to metabolic stress such as nutrient deprivation, nuclear p53 binds to the promoter region of multiple genes involved in inducing autophagy. Many products of these target genes, such as AMPK, tuberous sclerosis protein 2 and sestrin1/2 converge on the mTOR pathway 80, 81. mTOR is a negative regulator of autophagy, and inhibition of this pathway results in the induction of autophagy. Zhang et al. initially discovered that p53 activates autophagy during glucose deprivation via inhibition of the mTOR pathway 82. In contrast, during hypoxia, nuclear p53 reduces autophagy by repressing expression of BNIP3 83. Additionally, Hoshino et al. found that nuclear p53 attenuates autophagy and mitophagy in the ischemic heart, which results in accumulation of damaged mitochondria and increased myocardial damage 84. Collectively, these studies suggest that p53 suppress autophagy under normal conditions, and that in response to stress, p53 can either induce or inhibit autophagy.
Regulation of Mitophagy by the PINK1/Parkin Pathway
Damage to mitochondria often results in activation of both mitophagy and mitochondrial apoptosis/permeabilization in the same cell. In an effort to prevent cell death, damaged mitochondria are sequestered by autophagosomes and degraded before apoptosis or necrosis can be triggered. Electron microscopy (EM) analysis of the infarct border zone reveals that 10% of autophagosomes contain mitochondria 8 hours after the myocardial infarction, while no mitochondria are found in autophagosomes in control mice 84. EM also shows many autophagosomes containing mitochondria following ex vivo I/R 24. The PTEN-induced putative kinase 1 (PINK1)/Parkin pathway is important in regulating mitophagy in cells. Mitophagy allows for the selective removal of only dysfunctional mitochondria by autophagosomes. The E3 ubiquitin ligase Parkin is predominantly cytosolic under basal conditions, but rapidly translocates to mitochondria upon loss of mitochondrial membrane potential (Δψm) (Figure 3) 85, 86. Parkin then promotes ubiquitination of mitochondrial proteins, which serves as a signal for mitophagy 87. To date, only four Parkin substrates have been identified on mitochondria; VDAC1 87, Mitofusins1/2 88, 89, and MIRO 90.
Figure 3.
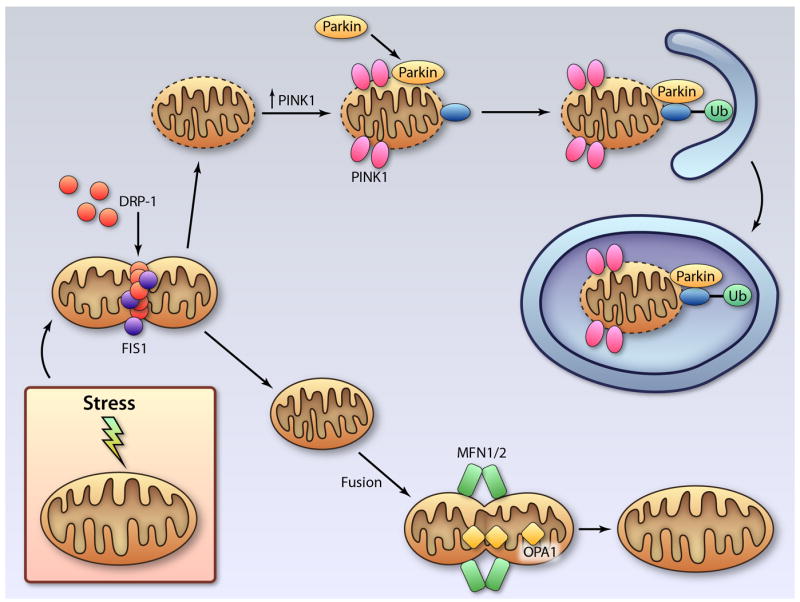
Regulation of mitophagy. Damaged mitochondria undergo DRP1-mediated fission prior to mitophagy. Reduced mitochondrial membrane potential leads to accumulation of PINK1 and subsequent recruitment of the E3 ubiquitin ligase Parkin to mitochondria. Parkin promotes ubiquitination of proteins in the mitochondrial membrane, which targets the damaged mitochondrion for removal by an autophagosome. The healthy mitochondrial fragment will undergo fusion mediated by MFN1/2 and OPA1. (Illustration Credit: Ben Smith).
The serine/threonine kinase PINK1 plays a central role in communicating the collapse of the Δψm to Parkin. PINK1 is found at very low levels on mitochondria with intact Δψm because it is rapidly imported and cleaved by mitochondrial proteases, and then is degraded by the proteasome 91-93. Upon collapse of the Δψm, the import and degradation of PINK1 is blocked, and PINK1 accumulates on the outer mitochondrial membrane. Studies have demonstrated that expression of PINK1 is necessary for the recruitment of Parkin to depolarized mitochondria 92, 94. However, exactly how accumulation of PINK1 on the outer mitochondrial membrane results in recruitment and activation of Parkin is currently unclear. Based on experimental findings, three different models have been proposed. First, it has been suggested that PINK1 directly interacts with Parkin, thereby anchoring it to the mitochondria 95, 96. Another model suggests that PINK1 directly phosphorylates Parkin, resulting in its activation 95, 97. Alternatively, PINK1 may phosphorylate Parkin substrates on the mitochondria, thereby increasing their affinity for Parkin90. In contrast, other studies have found that Parkin does not require PINK1 for its function 98-100. Additional studies are needed to elucidate the relationship between PINK1 and Parkin in mitophagy.
To date, very few PINK1 substrates have been identified, and only one of them is associated with the mitochondria. The atypical Rho GTPase MIRO plays an important role in the movement of mitochondria within cells by connecting mitochondria to microtubules 101. Wang et al. reported that phosphorylation of MIRO by PINK1 activates proteasomal degradation of MIRO in a Parkin-dependent manner 90. This supports the notion that the PINK1/Parkin pathway disrupts movement of damaged mitochondria in cells, thereby separating them from the pool of healthy mitochondria for mitophagy. Unlike neuronal cells, whose survival depends on efficient transport of mitochondria from the cell body to the axon terminal, mitochondria in cardiac myocytes do not display significant motility 102. Interestingly, MIRO is highly expressed in heart and skeletal muscle 103, and it will be greatly beneficial to determine what role MIRO plays in PINK1/Parkin-mediated mitophagy in the myocardium.
PINK1 and Parkin are highly expressed in the heart, 104, 105 and recent studies have provided some insights into their functional role in the myocardium. Billia et al. reported that PINK1 protein levels are markedly reduced in end-stage human heart failure 104, and that PINK1-deficient mitochondria have reduced oxidative capacity, which correlates with the development of cardiac dysfunction and hypertrophy by two months of age 104. Moreover, Parkin has been found to play an important role in clearing mitochondria in ischemic preconditioning (IPC), and mice deficient in Parkin are resistant to IPC 106. We have also found that BNIP3-mediated mitophagy is associated with the translocation of Parkin to mitochondria 107.
Selective Mitophagy is mediated via Autophagy Adaptors/Receptors
Previously, autophagy was thought to be an essentially non-selective pathway that accidentally sequestered material in the cytosol including mitochondria, but recent evidence suggests that autophagosomes carry selective cargoes. Until recently, it was unclear what signals or labels were used to target specific mitochondria for removal by the autophagy pathway. The recent identification of autophagy adaptor proteins and autophagy receptors on mitochondria has provided important new insights into this process. Currently, there is experimental evidence of at least two different mechanisms for selective mitophagy in mammalian cells (Figure 4).
Figure 4.
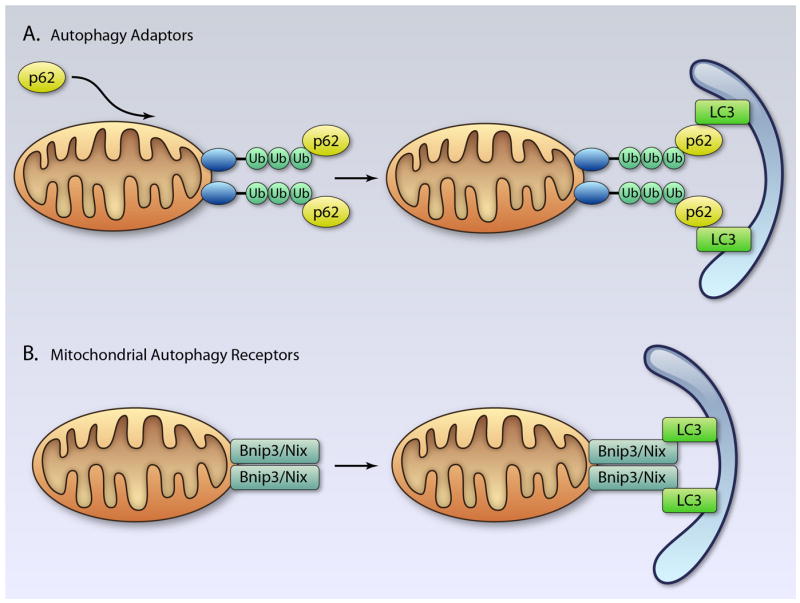
Removal of mitochondria via autophagy adaptors or autophagy receptors. A. The p62 protein interacts with ubiquitinated proteins on the mitochondrion. The complex is then selectively sequestered by an autophagosome through the interaction between p62 and LC3. B. Autophagy receptors on the mitochondria such as Bnip3 and Nix interact directly with LC3 on the autophagosome. (Illustration Credit: Ben Smith).
Mitophagy via Autophagy Adaptors
One mechanism of mitochondrial clearance involves autophagy adaptors and ubiquitin, in which the ubiquitin is used as a signal for autophagic degradation of mitochondria 108, 109. In this model, the dysfunctional mitochondrion is recognized by Parkin, which ubiquitinates specific protein substrates on the mitochondrion. The identification of the autophagy adaptor protein p62/SQSTM1 (hereafter referred to as p62) has provided important insights into the process of mitophagy 109. The p62 protein binds to ubiquitinated proteins via its ubiquitin-associated (UBA) domain 110, and to LC3 on the phagophore via its LC3 Interacting Region (LIR) 109. Thus, the recruitment and binding of p62 to ubiquitinated mitochondrial proteins docks the mitochondrion to the LC3-positive phagophore for engulfment (Figure 4A). In support of this model, it has been reported that p62 is recruited to mitochondria in a Parkin-dependent manner, and that knockdown of p62 substantially inhibits mitophagy 87, 111. In contrast, another study found that depletion of p62 by RNAi has little effect on mitophagy in HeLa cells. The authors also found that p62 can be recruited to mitochondria by a mitochondrion-anchored ubiquitin fusion protein, but this does not induce substantial mitophagy. This suggests that p62 recruitment to mitochondria alone is not sufficient for mitophagy to ensue 112. Therefore, p62 may not be responsible for Parkin-induced mitophagy, or there may be redundancy in autophagy adaptor proteins. NBR1 (neighbor of BRCA1 gene) contains both LIR and UBA motifs, and has been identified to act as an autophagy adaptor protein 108. However, it is currently unknown if NBR1 plays a role in mitophagy.
AMBRA1 and Mitophagy
Recent evidence indicates that AMBRA1 plays a role in mitophagy. Upon induction of autophagy, AMBRA1 translocates to the ER and mitochondria, where it interacts with BECLIN1 to initiate nucleation. Strappazzon et al. reported that a pool of AMBRA1 is already docked at mitochondria by BCL-2 under normal conditions. This reserve of AMBRA1 is released from BCL-2 when autophagy is induced, allowing it to bind to BECLIN1 and initiate formation of the phagophore at the mitochondrion 51. PARKIN also interacts with AMBRA1 at mitochondria to promote mitochondrial clearance, and depolarization of mitochondria increases the interaction between Parkin and AMBRA1 113. Interestingly, AMBRA1 is required for the clearance of mitochondria, but not for the translocation of Parkin. In contrast, AMBRA1-mediated mitophagy is dependent on Parkin, and overexpression of AMBRA1 in Parkin-/- cells fails to restore mitophagy levels. It has been hypothesized that local activation of the PI3K complex by AMBRA1 at the mitochondria allows for formation of new phagophores near or even directly around depolarized mitochondria. AMBRA1 recruitment to damaged mitochondria may therefore contribute to the efficient, spatially restricted, selective nature of mitophagy.
Currently, it is unclear whether these two different models of Parkin-mediated mitophagy are different or complimentary. Because LC3 only associates with phagophores after they have started to form 114, p62 can only recruit pre-existing phagophores to mitochondria and cannot induce the formation of new autophagosomes. In contrast, AMBRA1 stimulates new phagophore formation 52. Thus, we believe that the two mechanisms have complimentary effects to allow for efficient mitophagy; AMBRA1 induces formation of new phagophores at the mitochondria, which, after incorporation of LC3, may be tethered to ubiquitinated mitochondria via p62.
NIX and BNIP3 as Autophagy Receptors on Mitochondria
There is also evidence that mitochondria can be cleared via an ubiquitin-independent pathway involving direct binding of ATG8 family proteins to autophagy receptors on the mitochondria (Figure 4B). In yeast, mitophagy is regulated by a single, ubiquitin-independent pathway that requires the autophagy receptor ATG32 115, 116. The ATG32 protein is an outer mitochondrial membrane protein that interacts with ATG8 via its LIR 115. In mammalian cells, mitochondrial NIX/BNIP3L and BNIP3 have been identified as autophagy receptors for the selective clearance of mitochondria in cells 9, 117, 118. We have found that BNIP3 promotes mitophagy in cells including cardiac myocytes 24, 119, 120. NIX and BNIP3 target mitochondria for autophagy by directly binding to LC3/GABARAP on the autophagosome via their conserved LIR motifs 9, 117. Interestingly, disrupting the interaction between LC3 and BNIP3 significantly decreases, but does not completely abrogate mitophagy 9, suggesting that BNIP3 is not the only autophagy receptor on the mitochondria. Similarly, disrupting the interaction between NIX and LC3/GABARAP causes only a partial reduction in mitophagy 117. A study from Dorn’s group confirmed that BNIP3 and NIX have overlapping functions as regulators of mitophagy in the adult myocardium. Long-term studies of nix and bnip3 knockout mice revealed that these mice accumulate dysfunctional mitochondria in the heart with age. Interestingly, the accumulation occurs faster in nix/bnip3 DKO mice 121, suggesting that BNIP3 and NIX have overlapping functions in regulating mitochondrial clearance in the adult heart. To date, only NIX and BNIP3 have been identified as mitophagy receptors in mammalian cells. However, it is likely that there exists a redundancy in the mitophagy pathway, and that multiple proteins can act as autophagy receptors to ensure the removal of aberrant mitochondria. On the autophagosome, only ATG8 family proteins have been identified to participate in the clearance of mitochondria and protein aggregates. However, it is possible that other autophagosomal proteins can participate in binding proteins on mitochondria to ensure docking and removal.
An important question is whether BNIP3/NIX and PINK1/Parkin participate in the same pathway. On this matter, studies have produced contradictory evidence. Parkin translocation is dependent on loss of Δψm, yet we have found that BNIP3 promotes mitophagy in BAX/BAK-deficient MEFs even when mitochondria retain their Δψm 62. In contrast, we have also found that Parkin translocates to mitochondria in response to BNIP3 overexpression in cardiac myocytes, and that Parkin-deficient myocytes have reduced autophagy in response to BNIP3 107. Furthermore, Ding et al. reported that CCCP-induced Parkin translocation is significantly reduced in NIX-deficient MEFs 111. This suggests that NIX and BNIP3 may be important in recruiting Parkin to mitochondria.
Interplay Between Mitophagy and Mitochondrial Biogenesis
Not surprisingly, mitophagy is closely coupled to mitochondrial biogenesis. Studies have demonstrated that mitophagy has the capacity to clear most of the mitochondria in cells. In fact, cells overexpressing PINK1 or Parkin can degrade all their mitochondria within 24-96 h in response to treatment with mitochondrial uncouplers 86, 92. It is therefore important for the cell to quickly replace the mitochondria that have been removed by mitophagy. Mitochondria have a significant reserve capacity that can be utilized upon demand 122. In response to modest mitophagy, myocytes can utilize their mitochondrial reserve to maintain energy production without affecting contractility. However, excessive mitophagy in the absence of mitochondrial biogenesis will result in the depletion of the bioenergetic reserve in the remaining mitochondria and subsequent cell death.
The transcriptional coactivator peroxisome-proliferator-activated receptor γ coactivator 1α (PGC-1α) is a master regulator of mitochondrial biogenesis 123. PGC-1α is induced at birth in the mouse heart when there is a striking increase in mitochondrial biogenesis 123, 124. Studies have found that fasting is associated with both increased mitophagy 125 and rapid upregulation of PGC-1α in heart tissues 123. Additionally, Parkin has also been shown to be a central regulator of both mitophagy and mitochondrial biogenesis. Shin et al. recently identified PARIS (ZNF746), a KRAB and zinc finger protein, to be a Parkin substrate 126. PARIS represses the expression of PGC-1α by binding to insulin response sequences in the PGC-1α promoter. Activation of Parkin promotes degradation of PARIS and subsequent activation of PGC-1α transcription. Although it has been shown that Parkin regulates mitophagy in myocytes 106, 107, it remains to be determined whether Parkin regulates mitochondrial biogenesis via PARIS in the heart.
Mitochondrial Dynamics are Integrated with Cell Death and Mitophagy
Mitochondrial fission and fusion are regulated by several different GTPases. Mitofusins 1 and 2 (MFN1 and MFN2) regulate fusion of the outer mitochondrial membrane, whereas optic atrophy protein 1 (OPA1) promotes fusion of the inner membrane 127, 128. Mitochondrial fission is regulated by dynamin-related protein 1 (DRP1) and fission protein 1 (FIS1) 129, 130. These proteins are highly expressed in the heart 131-134. Deletion of Drp1 is embryonic lethal, and myocytes in Drp1 null embryos have reduced contractility 135. Defects in DRP1 function are also associated with early infant mortality and cardiomyopathy 136, 137. Knockdown of fusion proteins mitochondrial assembly regulatory factor (MARF) or OPA1 leads to the development of cardiomyopathy in Drosophila 138. These studies indicate that functional mitochondrial dynamics are important for normal heart and mitochondrial function.
Importantly, mitochondrial dynamics influence cell death by mechanisms that are not fully understood, and it is unclear whether mitochondrial fragmentation is the cause or a consequence of the pathogenesis. Inhibition of mitochondrial fission attenuates disease progression in models of neurodegenerative and cardiovascular diseases 132, 139, 140. Studies have found that excessive DRP1-mediated mitochondrial fission contributes to apoptotic cell death 141-143. Mitochondrial fission occurs within the same time frame as activation of pro-apoptotic BAX and permeabilization of the mitochondrial outer membrane. Additionally, DRP1 co-localizes with BAX on the mitochondrial membrane at the onset of apoptosis 141. However, fission is not required for BAX/BAK-dependent apoptosis, and inhibiting DRP1 only delays cell death 143, 144. It has been found that inhibiting the fission machinery partially prevents cytochrome c release from mitochondria 143, and that cells with fragmented mitochondria are more sensitive to apoptosis 144. This suggests that although DRP1-mediated fission is not required for apoptosis to proceed, it increases sensitivity to apoptotic stimuli possibly by enhancing cytochrome c release.
In addition, Piquereau et al. recently reported that cardiac mitochondria of Opa1+/− mice accumulate more calcium and present a delay in calcium-induced mPTP opening 145. Opa1 down-regulation induces clear changes in mitochondrial morphology and alterations of the mitochondrial cristae. Unexpectedly, mitochondria appear larger in Opa1+/− myocytes. Similar findings have been reported in Mfn2-/- mice, in which MFN2 deficiency leads to accumulation of large mitochondria in the myocardium 133. In contrast, MFN1-deficient myocytes accumulate small spherical mitochondria, as has been previously reported in Mfn1 null cell lines 146. Although mitochondria in Mfn1-/- and Mfn2-/- hearts have opposing morphologies, they are both more resistant to mPTP opening 133,146, suggesting that the process of fusion influences mPTP more than mitochondrial size.
Mitochondrial dynamics are closely integrated with mitophagy (Figure 3). Consistently, mitophagy is attenuated in cells with reduced mitochondrial fission, suggesting that fission is a prerequisite for mitophagy to occur 107, 147-149. For instance, DRP1-deficient MEFs have significantly reduced Parkin-mediated mitophagy compared with wild-type MEFs 150. We found that DRP1-mediated fission is a prerequisite for mitophagy by BNIP3 in cardiac myocytes 107. It is currently unclear why mitochondrial fission must occur prior to mitophagy. One possibility could be that fission produces smaller mitochondrial fragments that can more easily be engulfed by autophagosomes. Mitochondria usually have an elongated shape and can be up to 5 μm long 151, whereas autophagosomes are spherical in shape with a diameter of about 1 μm 152. Recent studies have connected the PINK1/Parkin mitophagy pathway with mitochondrial dynamics. MFN1 and MFN2 are substrates for Parkin 88, 150, although their ubiquitination does not serve as a signal for mitophagy. Instead, MFN1/2 ubiquitination leads to their proteasomal degradation prior to mitophagy 88, 150, indicating that ubiquitination of MFNs does not constitute a signal for mitophagy. Tanaka et al. also identified that p97 is required for proteasomal elimination of MFN1 and MFN2 150. p97 is an AAA+ ATPase that is involved in the retrotranslocation of ER membrane-spanning proteins after their ubiquitination 153. Thus, the data suggest that degradation of MFN1/2 might serve to switch the balance of mitochondrial dynamics towards fission to facilitate mitophagy. The loss of MFNs may prevent damaged mitochondria from fusing with healthy mitochondria in the cell. Additional evidence that mitochondrial size plays a role in mitophagy came from studies published by Lippincott-Schwartz’s and Scorrano’s groups. They found that nutrient deprivation induces formation of hyperfused mitochondrial networks that protect mitochondria from elimination by autophagosomes 154, 155. An alternative possibility is that fission segregates dysfunctional mitochondria prior to removal by autophagy. Shirinai and colleagues provided evidence that mitochondrial fission gives rise to two fragments with different Δψm 149. Mitochondrial fragments with low Δψm are more likely to be targeted by autophagosomes, whereas fragments with high Δψm have a higher probability of undergoing mitochondrial fusion.
Regulation of Cell Death and Autophagy by Reactive Oxygen Species (ROS)
Mitochondria are both a major source of and targets of ROS, which are by-products of mitochondrial electron transport activity 156. Although significant amounts of ROS are generated by cardiac mitochondria, myocytes have developed a highly efficient antioxidant system that can neutralize ROS under normal conditions. However, damaged mitochondria can produce ROS at levels that exceed the capacity of the antioxidant system and can result in cell death. ROS can cause the oxidative modification of mitochondrial proteins, lipids, and mtDNA, resulting in mitochondrial dysfunction. ROS can also contribute to opening of the mPTP 12, 27. Oxidative stress has been associated with loss of cells in both I/R injury 157, 158 and anthracycline-induced cardiomyopathy 159.
In addition, ROS play an important role in inducing autophagy in cells and during myocardial I/R 160. Exactly how ROS regulate autophagy is unknown, although it is very possible that oxidative modification of transcription factors affects the levels of autophagy proteins. ROS can also directly regulate the formation of autophagosomes. ATG4 enables the conversion of LC3-I to lipidated LC3-II, its insertion into the autophagosome, and the recycling of LC3-II after autophagosome-lysosome fusion. ATG4 is subject to oxidation and subsequent inactivation, which leads to accumulation of LC3-II and increased formation of autophagosomes 161. Interestingly, increased oxidative/nitrosative stress can inhibit mitophagy by modification and inactivation of Parkin. Parkin contains multiple conserved cysteine residues that are important for maintaining its solubility 162, but are also susceptible to modification by oxidative and nitrosative stress that lead to Parkin inactivation and aggregation 163, 164. Since cardiac mitochondria are a major source of ROS/RNS during stress, it is likely that aberrant modification of Parkin and subsequent inhibition of mitophagy will result.
Cross Talk Between Apoptosis and Mitophagy
During stress, pro-survival and pro-death processes are concomitantly activated in cells. The final outcome (life vs. death) depends on the balance between these pathways (Figure 5). Although it might be surprising that the cell is activating two opposing pathways, it is prudent to have multiple strategies in place for dealing with stress. The ability of a cell to repair itself and prevent unnecessary death is particularly important in a post-mitotic cell such as a myocyte that cannot be easily replaced. If exposure to modest stress results in damage to only a few mitochondria, the cell can easily clear those mitochondria via mitophagy. However, if the number of damaged mitochondria exceeds the capacity of mitophagy, or if mitophagy becomes inactivated, the cell is beyond rescue and apoptosis will become the dominant pathway to minimize extraneous tissue damage upon cell death.
Figure 5.
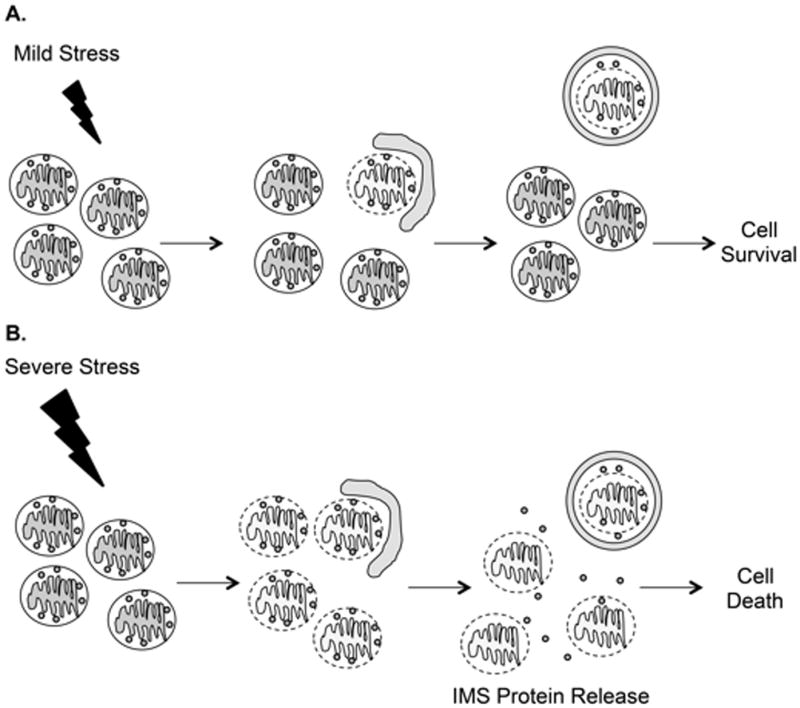
Balance between life and death during stress. A. Mild stress causes damage to a few mitochondria, which are rapidly sequestered by autophagosomes. B. In response to severe stress, there is overwhelming mitochondrial damage that autophagosomes are unable to efficiently clear. These mitochondria will release pro-death proteins from the intermembrane space (IMS), such as cytochrome c, AIF, and SMAC/Diablo, that will activate cell death pathways.
Although there is overwhelming evidence that dysfunctional mitochondria activate mitophagy, several studies have found that mitochondrial stress and apoptosis inhibit induction of autophagy in cells. For instance, caspases have been reported to cleave BECLIN1 during apoptosis, thereby preventing the initiation of autophagy 165. The resulting BECLIN1 fragment is unable to form a complex with VPS34, as is required for autophagy 166. Instead, the BECLIN1 fragment translocates to mitochondria and enhances apoptosis. Atg5 has also been reported to be cleaved by calpain in response to apoptotic stimuli 167. Similar to the BECLIN1 fragment, truncated Atg5 translocates from the cytosol to mitochondria, where it associates with BCL-XL and triggers cytochrome c release and caspase activation.
AMBRA1 is also an important target during apoptosis. Pagliarini et al. found that AMBRA1 is subjected to cleavage by caspases and calpains, which results in inactivation of autophagy and promotion of cell death168. Interestingly, knockdown of AMBRA1 results in increased sensitivity to cell death, whereas overexpression of a caspase cleavage resistant form of AMBRA1 prolongs autophagy and reduces cell death. These findings suggest that when there is vast mitochondrial damage, such as that which occurs during a myocardial infarction, activation of apoptotic proteases will shut down autophagy/mitophagy and activate apoptosis to ensure cell death. Whether or not proteolytic cleavage of BECLIN1, Atg5, or AMBRA1 occurs in the myocardium in response to stress such as I/R is currently unknown and remains to be explored.
Conclusion
Understanding the interface between adaptation to stress and cell death is important for understanding the pathogenesis of cardiovascular disease. Basal levels of mitophagy are important for maintaining cellular homeostasis and protecting cells against accumulation of dysfunctional mitochondria. There is also cross talk between the autophagy and apoptotic pathways. During stress, there is concomitant activation of autophagy/mitophagy and apoptosis, where enhanced mitophagy is an early response to promote survival by removing damaged mitochondria. With overwhelming mitochondrial damage, apoptosis becomes dominant, and inactivation of critical proteins of the autophagy pathway allows for cell death. Interestingly, enhanced levels of mitophagy can lead to excessive removal of mitochondria, loss of cardiac myocytes, and development of heart failure. It is important to clarify the differences between mitophagy pathways utilized in the degradation of damaged versus merely superfluous mitochondria. The manipulation of proteins that regulate mitochondrial integrity and mitophagy represents future therapeutic targets to preserve myocyte viability and prevent the development of heart disease. Therefore, it is important to gain insights into the mechanisms regulating the balance between survival and death, both under normal conditions and in the diseased myocardium.
Acknowledgments
Sources of Funding
ABG is supported by NIH grants R01HL087023, R01HL101217, and R01HL092136.
Non-standard Abbreviations and Acronyms
- Ambra1
Activating molecule in beclin 1-regulated autophagy
- AIF
Apoptosis Inducing Factor
- ANT
Adenine nucleotide transporter
- Atg5/7
Autophagy protein 5/7
- Bnip3
BCL2/adenovirus E1B nineteen kDa protein-interacting protein 3
- Bnip3L
BCL2/adenovirus E1B nineteen kDa protein-interacting protein 3-like
- CypD
Cyclophilin D
- Drp1
Dynamin-related protein 1
- EndoG
Endonuclease G
- Fis1
Mitochondrial fission 1
- IP3
Inositol triphosphate
- LAMP2
Lysosome-associated membrane protein 2
- LC3
Microtubule-associated protein 1 light chain 3
- Mfn1/2
Mitofusin-1/2
- mPTP
Mitochondrial permeability transition
- Opa1
Optic atrophy 1
- PI3K
Phosphoinositide 3-kinase
- PINK1
PTEN-induced putative kinase 1
- ROS
Reactive oxygen species
- VDAC
voltage-dependent anion channel
Footnotes
Disclosures
None
References
- 1.Schaper J, Meiser E, Stammler G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res. 1985;56:377–391. doi: 10.1161/01.res.56.3.377. [DOI] [PubMed] [Google Scholar]
- 2.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999;18:3888–3896. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 6.Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. 2011;13:1319–1327. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105:9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy. 2010;6:17–24. doi: 10.4161/auto.6.7.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-Associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gustafsson AB, Gottlieb RA. Mechanisms of apoptosis in the heart. J Clin Immunol. 2003;23:447–459. doi: 10.1023/b:joci.0000010421.56035.60. [DOI] [PubMed] [Google Scholar]
- 11.Gustafsson AB, Gottlieb RA. Bcl-2 Family Members and Apoptosis, Taken to Heart. Am J Physiol Cell Physiol. 2006 doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 12.Baines CP. The cardiac mitochondrion: nexus of stress. Annu Rev Physiol. 2010;72:61–80. doi: 10.1146/annurev-physiol-021909-135929. [DOI] [PubMed] [Google Scholar]
- 13.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 14.Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, Criollo A, Nemchenko A, Hill JA, Lavandero S. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2011;2:e244. doi: 10.1038/cddis.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 18.Imahashi K, Schneider MD, Steenbergen C, Murphy E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res. 2004;95:734–741. doi: 10.1161/01.RES.0000143898.67182.4c. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313–2320. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 20.Gustafsson AB, Tsai JG, Logue SE, Crow MT, Gottlieb RA. Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J Biol Chem. 2004;279:21233–21238. doi: 10.1074/jbc.M400695200. [DOI] [PubMed] [Google Scholar]
- 21.Capano M, Crompton M. Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem J. 2006;395:57–64. doi: 10.1042/BJ20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hochhauser E, Kivity S, Offen D, Maulik N, Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub V, Tobar A, Vidne BA. Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2003;284:H2351–2359. doi: 10.1152/ajpheart.00783.2002. [DOI] [PubMed] [Google Scholar]
- 23.Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW., 2nd Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation. 2008;117:396–404. doi: 10.1161/CIRCULATIONAHA.107.727073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 25.Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem. 2001;276:30724–30728. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- 26.Toth A, Jeffers JR, Nickson P, Min JY, Morgan JP, Zambetti GP, Erhardt P. Targeted Deletion of Puma Attenuates Cardiomyocyte Death and Improves Cardiac Function during Ischemia/reperfusion. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.01046.2005. [DOI] [PubMed] [Google Scholar]
- 27.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 29.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 30.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 31.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007 doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bauer MK, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol. 1999;147:1493–1502. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Bras M, Borgne-Sanchez A, Touat Z, El Dein OS, Deniaud A, Maillier E, Lecellier G, Rebouillat D, Lemaire C, Kroemer G, Jacotot E, Brenner C. Chemosensitization by knockdown of adenine nucleotide translocase-2. Cancer Res. 2006;66:9143–9152. doi: 10.1158/0008-5472.CAN-05-4407. [DOI] [PubMed] [Google Scholar]
- 35.Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L, Araujo N, Pinna G, Larochette N, Zamzami N, Modjtahedi N, Harel-Bellan A, Kroemer G. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene. 2008;27:4221–4232. doi: 10.1038/onc.2008.63. [DOI] [PubMed] [Google Scholar]
- 36.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 37.Zhu L, Yu Y, Chua BH, Ho YS, Kuo TH. Regulation of sodium-calcium exchange and mitochondrial energetics by Bcl-2 in the heart of transgenic mice. J Mol Cell Cardiol. 2001;33:2135–2144. doi: 10.1006/jmcc.2001.1476. [DOI] [PubMed] [Google Scholar]
- 38.Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, Crow MT, Gavathiotis E, Dorn GW, 2nd, O’Rourke B, Kitsis RN. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dedkova EN, Blatter LA. Mitochondrial Ca2+ and the heart. Cell Calcium. 2008;44:77–91. doi: 10.1016/j.ceca.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, Rizzuto R. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem. 2004;279:54581–54589. doi: 10.1074/jbc.M409663200. [DOI] [PubMed] [Google Scholar]
- 41.White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 44.Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, Molkentin JD, Kranias EG, Dorn GW., 2nd Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest. 2009;119:203–212. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, Hardwick JM. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 49.Ciechomska IA, Goemans GC, Skepper JN, Tolkovsky AM. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene. 2009;28:2128–2141. doi: 10.1038/onc.2009.60. [DOI] [PubMed] [Google Scholar]
- 50.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, Piacentini M, Levine B, Cecconi F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. Embo J. 2011;30:1195–1208. doi: 10.1038/emboj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 53.Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 54.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. Embo J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 56.Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. Febs J. 2007;274:3184–3197. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 57.Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J. 2007;405:407–415. doi: 10.1042/BJ20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–231. doi: 10.1161/01.res.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- 59.Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, Dorn GW., 2nd Distinct Pathways Regulate Proapoptotic Nix and BNip3 in Cardiac Stress. J Biol Chem. 2006;281:1442–1448. doi: 10.1074/jbc.M509056200. [DOI] [PubMed] [Google Scholar]
- 60.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson AB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18:721–731. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang X, Jiang X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012;586:1390–1396. doi: 10.1016/j.febslet.2012.02.049. [DOI] [PubMed] [Google Scholar]
- 65.Perfettini JL, Kroemer RT, Kroemer G. Fatal liaisons of p53 with Bax and Bak. Nat Cell Biol. 2004;6:386–388. doi: 10.1038/ncb0504-386. [DOI] [PubMed] [Google Scholar]
- 66.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 67.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 68.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 69.Sugars KL, Budhram-Mahadeo V, Packham G, Latchman DS. A minimal Bcl-x promoter is activated by Brn-3a and repressed by p53. Nucleic Acids Res. 2001;29:4530–4540. doi: 10.1093/nar/29.22.4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pietrzak M, Puzianowska-Kuznicka M. p53-dependent repression of the human MCL-1 gene encoding an anti-apoptotic member of the BCL-2 family: the role of Sp1 and of basic transcription factor binding sites in the MCL-1 promoter. Biol Chem. 2008;389:383–393. doi: 10.1515/BC.2008.039. [DOI] [PubMed] [Google Scholar]
- 71.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 72.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 73.Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, Pan H, Kessler H, Pancoska P, Moll UM. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. 2006;281:8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 74.Long X, Boluyt MO, Hipolito ML, Lundberg MS, Zheng JS, O’Neill L, Cirielli C, Lakatta EG, Crow MT. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. 1997;99:2635–2643. doi: 10.1172/JCI119452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89:279–286. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 76.Liu J, Mao W, Ding B, Liang CS. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;295:H1956–1965. doi: 10.1152/ajpheart.00407.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM. Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol Cell Biochem. 2005;273:25–32. doi: 10.1007/s11010-005-5905-8. [DOI] [PubMed] [Google Scholar]
- 78.Xiong S, Van Pelt CS, Elizondo-Fraire AC, Fernandez-Garcia B, Lozano G. Loss of Mdm4 results in p53-dependent dilated cardiomyopathy. Circulation. 2007;115:2925–2930. doi: 10.1161/CIRCULATIONAHA.107.689901. [DOI] [PubMed] [Google Scholar]
- 79.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 81.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Feng X, Liu X, Zhang W, Xiao W. p53 directly suppresses BNIP3 expression to protect against hypoxia-induced cell death. Embo J. 2011;30:3397–3415. doi: 10.1038/emboj.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, Ikeda K, Ueyama T, Okigaki M, Matsubara H. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol. 2012;52:175–184. doi: 10.1016/j.yjmcc.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 85.Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A. 2010;107:11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 88.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sha D, Chin LS, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet. 2010;19:352–363. doi: 10.1093/hmg/ddp501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X, Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- 98.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 99.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 100.Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rice SE, Gelfand VI. Paradigm lost: milton connects kinesin heavy chain to miro on mitochondria. J Cell Biol. 2006;173:459–461. doi: 10.1083/jcb.200604071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beraud N, Pelloux S, Usson Y, Kuznetsov AV, Ronot X, Tourneur Y, Saks V. Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J Bioenerg Biomembr. 2009;41:195–214. doi: 10.1007/s10863-009-9214-x. [DOI] [PubMed] [Google Scholar]
- 103.Fransson A, Ruusala A, Aspenstrom P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278:6495–6502. doi: 10.1074/jbc.M208609200. [DOI] [PubMed] [Google Scholar]
- 104.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108:9572–9577. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 106.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning Involves Selective Mitophagy Mediated by Parkin and p62/SQSTM1. PLoS ONE. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;301:H1924–1931. doi: 10.1152/ajpheart.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 109.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 110.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci. 2011;31:10249–10261. doi: 10.1523/JNEUROSCI.1917-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 115.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 116.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielmann Y, Stangler T, Hersch N, Hoffmann B, Merkel R, Willbold D. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–698. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- 119.Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial Autophagy by Bnip3 Involves Drp1-Mediated Mitochondrial Fission and Recruitment of Parkin in Cardiac Myocytes. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy. 2010;6:855–862. doi: 10.4161/auto.6.7.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dorn GW., 2nd Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010;3:374–383. doi: 10.1007/s12265-010-9174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem J. 2009;424:99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res. 2010;107:825–838. doi: 10.1161/CIRCRESAHA.110.223818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy. 2010;6:462–472. doi: 10.4161/auto.6.4.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) Repression of PGC-1alpha Contributes to Neurodegeneration in Parkinson’s Disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 132.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 133.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Hartel S, Jaimovich E, Zorzano A, Hidalgo C, Lavandero S. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res. 2008;77:387–397. doi: 10.1093/cvr/cvm029. [DOI] [PubMed] [Google Scholar]
- 135.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, Lygate CA, Hough T, Townsend S, Williams D, Wells S, Norris D, Glyn-Jones S, Land J, Barbaric I, Lalanne Z, Denny P, Szumska D, Bhattacharya S, Griffin JL, Hargreaves I, Fernandez-Fuentes N, Cheeseman M, Watkins H, Dear TN. A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000. doi: 10.1371/journal.pgen.1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 138.Dorn GW, 2nd, Clark CF, Eschenbacher WH, Kang MY, Engelhard JT, Warner SJ, Matkovich SJ, Jowdy CC. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ Res. 2011;108:12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) Ameliorates Pressure Overload Induced Heart Failure. PLoS ONE. 2012;7:e32388. doi: 10.1371/journal.pone.0032388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cui M, Tang X, Christian WV, Yoon Y, Tieu K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J Biol Chem. 2010;285:11740–11752. doi: 10.1074/jbc.M109.066662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 142.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC. Inhibiting the mitochondrial fission machinery does not prevent bax/bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sugioka R, Shimizu S, Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem. 2004;279:52726–52734. doi: 10.1074/jbc.M408910200. [DOI] [PubMed] [Google Scholar]
- 145.Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, Fortin D, Huynh le H, Nicolas V, Alavi MV, Brenner C, Ventura-Clapier R, Veksler V, Joubert F. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. 2012;94:408–417. doi: 10.1093/cvr/cvs117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF, Jr, Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. 2012;302:H167–179. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 148.Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta. 2008;1777:860–866. doi: 10.1016/j.bbabio.2008.05.442. [DOI] [PubMed] [Google Scholar]
- 149.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cereghetti GM, Scorrano L. The many shapes of mitochondrial death. Oncogene. 2006;25:4717–4724. doi: 10.1038/sj.onc.1209605. [DOI] [PubMed] [Google Scholar]
- 152.Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells. 2010;15:923–933. doi: 10.1111/j.1365-2443.2010.01433.x. [DOI] [PubMed] [Google Scholar]
- 153.Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 154.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Becker LB, vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am J Physiol. 1999;277:H2240–2246. doi: 10.1152/ajpheart.1999.277.6.H2240. [DOI] [PubMed] [Google Scholar]
- 158.Vanden Hoek TL, Li C, Shao Z, Schumacker PT, Becker LB. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J Mol Cell Cardiol. 1997;29:2571–2583. doi: 10.1006/jmcc.1997.0497. [DOI] [PubMed] [Google Scholar]
- 159.Sterba M, Popelova O, Vavrova A, Jirkovsky E, Kovarikova P, Gersl V, Simunek T. Oxidative stress, redox signaling and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2012.4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wong ES, Tan JM, Wang C, Zhang Z, Tay SP, Zaiden N, Ko HS, Dawson VL, Dawson TM, Lim KL. Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J Biol Chem. 2007;282:12310–12318. doi: 10.1074/jbc.M609466200. [DOI] [PubMed] [Google Scholar]
- 163.Winklhofer KF, Henn IH, Kay-Jackson PC, Heller U, Tatzelt J. Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J Biol Chem. 2003;278:47199–47208. doi: 10.1074/jbc.M306769200. [DOI] [PubMed] [Google Scholar]
- 164.Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 165.Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–1719. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 166.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–277. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 168.Pagliarini V, Wirawan E, Romagnoli A, Ciccosanti F, Lisi G, Lippens S, Cecconi F, Fimia GM, Vandenabeele P, Corazzari M, Piacentini M. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012 doi: 10.1038/cdd.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]