

Abstract
Multiple lines of evidence support the idea that osteocytes act as mechanosenors in bone and that they control bone formation, in part, by expressing the Wnt antagonist sclerostin. However, the role of osteocytes in the control of bone resorption has been less clear. Recent studies have demonstrated that osteocytes are the major source of the cytokine RANKL involved in osteoclast formation in cancellous bone. The goal of this review is to discuss these and other studies that reveal mechanisms whereby osteocytes control osteoclast formation and thus bone resorption.
Keywords: osteocyte, osteoclast, resorption, RANKL, apoptosis
Introduction
Bone resorption serves many different physiological functions, including removal of calcified cartilage during bone growth, modeling of bones during growth or adaptation, maintenance of mineral homeostasis, the removal of damaged bone matrix, and the creation of a pathway for tooth eruption. Excessive or pathological bone resorption, on the other hand, is associated with a variety of conditions, such as sex steroid deficiency, inflammation, and malignancies. In each case, bone resorption is accomplished by osteoclasts. Because osteoclasts are relatively short-lived cells, they must be continuously generated at or near the site of bone resorption. The generation of osteoclasts at specific sites, in turn, is governed by the production of differentiation and survival factors provided by support cells, whose identity may vary depending on the location and cause of bone resorption.
Regardless of the identity of the cell types that support osteoclast differentiation, such cells do so by expressing the cytokine receptor activator of NFκB ligand (RANKL), which is essential for osteoclast differentiation, function, and survival [1]. RANKL function is inhibited by osteoprotegerin (OPG), which binds to RANKL and prevents its binding to receptor activator of NFκB (RANK) on osteoclast progenitors [2]. Since OPG can be produced in the same vicinity as RANKL, possibly by the same cell types that produce RANKL, the ratio of RANKL and OPG at a particular location is a major determinant of the magnitude of osteoclast formation and bone resorption at that site.
The osteocyte lacuna-canalicular network is contained within the mineralized matrix of bone and is therefore ideally suited to both sense the need for and then orchestrate bone resorption. However, until recently there was little evidence that osteocytes directly participate in the control of either bone resorption or bone formation. The identification the Wnt antagonist sclerostin, together with the demonstration that expression of this protein in bone is restricted primarily to osteocytes, provided the first functional evidence that osteocytes directly control bone formation [3–7]. More recently, genetic studies in mice have revealed that osteocytes provide the majority of the RANKL that controls osteoclast formation in cancellous bone [8, 9]. The goal of this review is to discuss how this recent progress has enhanced our view of osteocyte biology as well at some of the new questions raised by these studies.
Control of osteoclast differentiation and function
Subsequent to the discovery that osteoclasts differentiate from hematopoietic progenitors, it was determined that support from non-hematopoietic cells is also required for osteoclast differentiation [10]. Eventually, the mechanistic basis for this requirement was revealed to be production of M-CSF and RANKL by the supporting cells [1, 11]. Although both cytokines are essential for osteoclastogenesis, M-CSF is primarily involved in stimulating the replication of osteoclast progenitors while RANKL is responsible for stimulating their differentiation [12]. Because changes in the amount of RANKL are sufficient to change osteoclast numbers, and because RANKL is essential for osteoclastogenesis, this cytokine has been viewed as a genetic marker of osteoclast support cells [13]. Many different cell types express RANKL and some have been shown to support osteoclast differentiation, at least in vitro. These cell types include osteoblasts at different stages of differentiation, osteocytes, stromal cells of undefined origin, B and T lymphocytes, synovial fibroblasts, hypertrophic chondrocytes, and even osteoclasts themselves [14–21]. Changes in RANKL expression in each of these cell types has been purported to control osteoclast formation during physiological or pathophysiological bone resorption. However, the functional significance of RANKL produced by a particular cell type for bone resorption was, until recently, unclear.
Once RANK signaling has been initiated in osteoclast progenitors, the differentiation and function of osteoclasts can be further influenced by modulation of the signaling pathways downstream from RANK activation. RANK lacks intrinsic enzymatic activity in its intracellular domain and transduces signals by recruiting adaptor molecules such as the TNF receptor-associated factor (TRAF) family of proteins [22, 23]. TRAF6 functions as the main adaptor molecule that links RANK to both the differentiation and function of osteoclasts, although additional RANK-specific adaptor molecules might exist which link RANK signaling to other pathways [22, 24, 25]. RANKL binding to RANK induces the trimerization of RANK and TRAF6, which leads to the activation of NF-κB and the mitogen-activated kinases (MAPKs) [22, 23]. The essential role of NF-κB and the activator protein 1 (AP-1) transcription factor complex in osteoclastogenesis has been demonstrated genetically [26–28]. RANK activates AP-1 through the induction of c-Fos, which is dependent on the activation of calcium/calmodulin-dependent protein kinase type IV (CaMKIV) and cyclic AMP-responsive element-binding protein (CREB) [29] as well as NF-κB. AP-1 activity is also dependent on the phosphorylation of c-Fos partners including c-Jun by MAPKs [26, 30].
RANKL specifically and potently induces nuclear factor of activated T-cells cytoplasmic 1 (NFATc1), the master regulator of osteoclast differentiation, and this induction is dependent on both the TRAF6-NF-κB and c-Fos pathways [31–33]. The activation of NFAT is mediated by a specific phosphatase, calcineurin, which is activated by calcium-calmodulin signaling. The Nfatc1 promoter contains NFAT binding sites and NFATc1 specifically autoregulates its own promoter during osteoclastogenesis, resulting in a robust induction of NFATc1 [32]. AP-1 containing c-Fos, together with continuously activated calcium signaling, is crucial for this autoamplification [31]. NFATc1 regulates a number of osteoclast-specific genes, such as osteoclast-associated receptor (OSCAR), dendritic cell-specific transmembrane protein (DC-STAMP) and β3-integrin in cooperation with other transcription factors such as AP-1, PU.1 and MITF [22, 23].
Phospholipase Cγ (PLCγ), which mediates Ca2+ release from intracellular stores, is crucially important for the activation of the key transcription factor NFATc1 via calcineurin [31]. The activation of PLCγ by RANK requires the protein tyrosine kinases Btk/Tec and Syk, along with immunoreceptor tyrosine-based activation motif (ITAM)-bearing molecules, such as DNAX-activating protein (DAP12) and the Fc receptor common gamma chain (FcRγ) [34, 35]. In the osteoclast lineage, the immunoglobulin-like receptors (IgLR) associated with DAP12 include the triggering receptor expressed in myeloid cells 2 (TREM-2) and signal-regulatory protein β1 (SIRPβ1); those associated with FcRγ include OSCAR and paired immunoglobulin-like receptor A (PIR-A). Although the ligands for the IgLR remain largely unknown, a recent finding suggests that OSCAR binds to specific motifs within collagens in the extracellular matrix that become uncovered on the nonquiescent bone surface while osteoclasts are undergoing differentiation [36]. As ITAM signals are essential for osteoclastogenesis, but by themselves cannot induce osteoclastogenesis, these signals should properly be considered co-stimulatory signals for RANK. Recent studies showed that NFATc1 choreographs the determination of cell fate in the osteoclast lineage by inducing the repression of negative regulators as well as through its effect on positive regulators [23].
Osteocyte Control of Osteoclastogenesis
The idea that osteocytes influence bone remodeling has existed for decades [37]. However, until recently there has been little functional evidence that osteocytes can directly control the differentiation and activity of either osteoclasts or osteoblasts. The discovery that osteocytes are the primary source of the Wnt antagonist sclerostin provided the first functional evidence that osteocytes regulate any aspect of bone remodeling, in this case bone formation [3–7]. The Wnt signaling that is controlled by sclerostin appears to regulate the proliferation of osteoblast progenitors, as well as osteoblast differentiation and survival but does not appear to alter osteoclastogenesis or bone resorption [38–40].
The first functional evidence that osteocytes control bone resorption was provided by Tatsumi et al., who utilized the dentine matrix protein 1 (DMP1) promoter to express a diphtheria toxin receptor transgene specifically in osteocytes [41]. Administration of diphtheria toxin to these mice induced massive osteocyte apoptosis which was followed temporally by a dramatic but transient increase in bone resorption. Osteocyte death was accompanied by large increases in RANKL expression in bone, which likely played a role in the increased bone resorption. However, the cellular source of RANKL in this model or the mechanisms by which osteocyte apoptosis induced RANKL production, remain unknown.
Additional evidence that osteocytes control bone resorption was provided by studies of transgenic mice expressing a constitutively-active form of the parathyroid hormone receptor (caPTHR) specifically in osteocytes using the DMP1 promoter [42]. Both osteoclast and osteoblast numbers are elevated in these mice and this is associated with increased RANKL and decreased sclerostin expression in bone. Whether the elevated RANKL levels are due to RANKL production by osteocytes has not been determined. Nonetheless, these studies demonstrate that hormone action directly on osteocytes is sufficient to control both bone resorption and bone formation. Consistent with other work demonstrating that the Wnt signaling controlled by Lrp5 or sclerostin does not influence bone resorption, deletion of Lrp5 or over-expression of sclerostin in the DMP1-caPTHR mice had no impact on bone resorption even though it reduced bone formation [42, 43]. Together these results suggest that osteocytes can independently control bone resorption and bone formation via modulation of different pathways.
While modulation of Wnt signaling by Lrp5 does not alter bone resorption, genetic deletion of sclerostin and inhibition of sclerostin action with antibodies have variable effects on bone resorption; genetic deletion in mice does not alter osteoclastogenesis [40] while anti-sclerostin antibodies cause a transient suppression of bone resorption, at least in some studies [44]. Nonetheless, osteocyte-specific deletion of beta-catenin, which would be expected to inhibit canonical Wnt signaling in these cells, led to reduced production of OPG by osteocytes resulting in elevated bone resorption [45]. The same study demonstrated that cell preparations enriched in osteocytes express higher levels of OPG, as well as RANKL, than osteoblast-enriched preparations. Based on this, these authors concluded that OPG may be a factor produced directly by osteocytes that allows them to modulate the rate of bone resorption.
Osteoclast formation is induced by the cell-cell contact which occurs between osteoclast precursor cells of the monocyte/macrophage lineage and anchorage-dependent mesenchymal cells in bone [46, 47]. Osteoblast linage cells and bone marrow stromal cells (BMSCs) are thought to be the major cell types that express RANKL in support of osteoclastogenesis [22, 47]. However, since RANKL is expressed by other cell types in both bone and bone marrow, including osteocytes and lymphocytes, the actual major source of RANKL in vivo is as yet unclear. Osteocyte support of osteoclastogenesis was previously suggested by the observation that both isolated avian osteocytes and the osteocyte-like cell line MLO-Y4 induced osteoclastogenesis [16, 48]. Using a mouse genetic approach, targeted ablation of osteoblasts in mice did not affect the number of osteoclasts and bone resorption, and also had no effect on expression levels of RANKL in bone tissues [49, 50]. Thus, it appears that osteoblasts are not required for osteoclastogenesis and osteocytes may play an important role in the initiation of bone remodelling through their role in the differentiation and activation of osteoclasts.
To identify the most physiologically relevant osteoclastogenesis-supporting cells among the mesenchymal lineage cells in bone, we explored the expression of RANKL in osteoblasts and osteocytes. We initially followed the conventional method for isolation and purification by the enzymatic digestion of bone to obtain an osteocyte-rich fraction [51] in which cells exhibited a high expression level of osteocyte marker genes such as Dmp1 (encoding dentin matrix protein 1) and Sost (encoding sclerostin), but not the osteoblast marker Kera (encoding keratocan) [52]. Interestingly, Tnfsf11, which encodes RANKL, was more highly expressed in the osteocyte-rich fraction than the osteoblast-rich fraction, although the percentage of osteocytes is reported to be only around 60 % using this conventional method [51]. To unambiguously isolate high purity osteocytes from bone tissue, we generated osteocyte-specific enhanced green fluorescence protein (EGFP) reporter mice in which osteocytes could be identified by the expression of EGFP driven by the Dmp1 promoter. Using fluorescence activated cell sorting, we established a method for the isolation of high purity osteocytes from the fractions obtained by enzymatic digestion of the neonatal calvariae or adult long bones [8]. A similar approach was successfully utilized in a previous report in which osteocytes and osteoblasts express different fluorescent proteins [52].
The isolated osteocytes (over 99% EGFP-positive cells) morphologically exhibited dendritic processes and exclusively expressed Dmp1 and Sost, which are well-known osteocyte-specific genes. In contrast, osteoblast-specific genes such as Kera and Fmod (encoding fibromodulin) were strongly expressed in isolated osteoblasts (EGFP-negative cells). Most importantly, osteocytes express a much higher amount of RANKL and have a much greater capacity to support osteoclastogenesis than either osteoblasts or BMSCs [8]. These results revealed that primary osteocytes produce relatively large amounts of RANKL and efficiently support osteoclast differentiation in vitro. To address the functional importance of RANKL produced by osteocytes in vivo, our laboratories independently developed mice harboring a conditional allele for RANKL [8, 9]. In both models, exons 3 and 4 of the Tnfsf11 gene were flanked by loxP sites such that deletion of these exons results in complete loss of RANKL function. To delete the Tnfsf11 gene specifically in osteocytes, these mice were crossed with transgenic mice expressing the Cre recombinase under the control of Dmp1 regulatory elements [53]. Specific deletion in osteocytes was confirmed by comparison with isolated osteoblasts and soft tissues that express RANKL such as spleen and thymus [8, 9].
Mice lacking RANKL in osteocytes developed normally and displayed no changes in skeletal development when analyzed at birth and had only a mild increase in bone mass by 5 weeks of age [8, 9]. In contrast, mice with germline deletion of Tnfsf11 displayed obvious osteopetrosis at birth [1]. In addition, tooth eruption was normal in the conditional knockout mice whereas it was completely blocked in mice with germline Tnfsf11 deletion. Despite normal development and early growth, mice lacking RANKL in osteocytes had increased bone mass when analyzed at 3 or 6 months of age. This high bone mass was associated a dramatic reduction in osteoclast numbers in cancellous bone and reduced circulating levels of CTX, a marker of bone resorption. Moreover, bone formation rate and the levels of a circulating marker of bone formation were low in the conditional knockout mice, most likely due to the coupling of bone formation to bone resorption. Thus, these results demonstrated for the first time that osteocytes are an essential source of the RANKL that stimulates osteoclast formation and remodeling in cancellous bone.
The presence of a normal skeleton during development and early growth in mice lacking RANKL in osteocytes indicates that the osteoclast formation required for processes such as tooth eruption and calcified cartilage resorption was not affected in these mice. Therefore the RANKL required for these processes must be supplied by cell types other than osteocytes. In one of the studies mentioned above [9], the RANKL conditional allele was also deleted using osterix 1-Cre (Osx1-Cre) [54] and osteocalcin-Cre (OCN-Cre) [55] transgenic mice, which display Cre-mediated deletion in the chondrocytic as well as the osteoblastic lineage [9] (table 1). Deletion of Tnfsf11 using either the Osx1-Cre or OCN-Cre models resulted in a failure of tooth eruption as well as a failure to resorb the calcified cartilage beneath the growth plate, suggesting that chondrocytes or osteoblasts provide the RANKL necessary for these processes. Importantly, RANKL immunostaining in hypertrophic chondrocytes was abolished in both models, strongly implicating this cell type as the source of RANKL for resorption of calcified cartilage. To directly address this latter possibility, the RANKL conditional allele was deleted using a collagen X-Cre transgene, which is active predominantly in hypertrophic chondrocytes [56]. This maneuver also prevented resorption of calcified cartilage but had no effect on tooth eruption. Together these results suggest that RANKL produced by hypertrophic chondrocytes is responsible for the osteoclast formation necessary to remove the mineralized cartilage matrix produced during endochondral bone growth. Although the cells expressing the RANKL required for tooth eruption must express the Osx1-Cre and OCN-Cre transgenes, the precise identification of these cells will require additional studies.
Table 1.
Specificity of Cre-driver strains
| Transgene | Deletes in: | Reference | |||
|---|---|---|---|---|---|
| Osx1-Cre | chondrocyte | pre-osteoblast | osteoblast | osteocyte | [9, 54] |
| OCN-Cre | chondrocyte | osteoblast | osteocyte | [9, 55] | |
| Dmp1-Cre | osteoblast* | osteocyte | [8, 9, 53] | ||
| ColX-Cre | chondrocyte | osteoblast* | osteocyte* | [9, 56] |
deletion in a small percentage of cells
Signals controlling osteocyte RANKL
Parathyroidectomy or inactivation of the gene encoding parathyroid hormone (PTH) significantly reduces cancellous bone remodeling without altering endochondral bone growth [57, 58]. Moreover, these manipulations also reduce the expression of RANKL in bone, suggesting that PTH controls bone remodeling in part by stimulating RANKL expression. Consistent with this, PTH is a potent stimulator of RANKL transcription in osteoblastic cell lines in vitro [59–61]. The finding that transgenic expression of a constitutively active PTH receptor specifically in osteocytes is sufficient to increase both RANKL expression and osteoclast formation led to the idea that PTH controls osteoclast formation by promoting RANKL expression by osteocytes. This concept has been further supported by preliminary studies demonstrating that deletion of the PTHR specifically in osteocytes decreases osteoclast formation in cancellous bone [62, 63].
In vitro studies in stromal/osteoblastic cell lines have identified a transcriptional enhancer, designated the distal control region (DCR), that mediates stimulation of RANKL expression by PTH [64]. This and additional enhancers mediate the stimulation of RANKL expression by 1,25-dihydroxyvitamin D3 [64–66]. Consistent with these observations, deletion of the DCR in mice reduced expression of RANKL in bone and consequently reduced the rate of bone remodeling [67]. Based on the results mentioned earlier, it is likely that deletion of the DCR reduced RANKL expression in osteocytes. Overall, these findings suggest a model whereby basal levels of PTH maintain bone remodeling by stimulating RANKL expression in osteocytes via the DCR. Surprisingly, bone loss induced by hyperparathyoidism was normal in mice lacking the DCR, even though the increase in RANKL expression in bone was prevented [68]. These results suggest that PTH controls physiological bone remodeling via stimulation of RANKL expression by osteocytes, but that additional or alternative mechanisms are involved in the bone resorption stimulated by high levels of PTH, such as suppression of OPG expression by osteocytes and other cell types [60, 61, 69–71].
In contrast to PTH, estrogen suppresses the rate of bone remodeling [72]. Recently, estrogen receptor gene deletion studies have shown that estrogens act directly on osteoclasts and their progenitors to suppress bone resorption [73, 74]. However, the increase in bone resorption observed in these mice does not fully replicate that seen with estrogen deficiency, suggesting that additional mechanisms are involved. Studies in estrogen-deficient women demonstrated that RANKL abundance is increased on the surface of bone marrow lymphocytes and stromal cells, compared to estrogen-replete women [75]. It remains unclear whether estrogens alter the expression of RANKL mRNA or protein by osteocytes. However, soluble RANKL levels are elevated in the bone marrow of rats after orchidectomy, a condition that causes an increase in bone resorption similar to that observed after ovariectomy [76, 77]. The finding that osteocytes are a major source of RANKL in bone implicates this cell type as a potential source of the soluble RANKL induced by loss of androgens, and possibly loss of estrogens as well.
Loss of estrogen also stimulates osteocytes to undergo apoptotic cell death [72, 78]. Schaffler and colleagues have demonstrated that intra-cortical bone resorption is spatially associated with apoptotic osteocytes in estrogen-deficient rats [79]. Moreover, inhibition of osteocyte death via administration of a caspase inhibitor concomitantly suppressed intra-cortical bone resorption in these animals [79]. These results suggest that estrogen suppresses cortical bone resorption by promoting osteocyte viability. The role of RANKL expressed by osteocytes in this process is unknown, although the osteocyte ablation model mentioned earlier demonstrates that osteocyte death is sufficient to increase RANKL expression in bone [41]. Gonadectomy of mice lacking RANKL in osteocytes, as well as in mice lacking RANKL in other cell types, should clarify the role of RANKL production by various cell types in the elevated bone resorption associated with sex steroid deficiency.
Osteocyte apoptosis also increases in response to changes in biomechanical loading of the skeleton. Interestingly, both over-loading and unloading of the skeleton stimulate osteocyte apoptosis and both conditions result in increased bone resorption [80, 81]. Thus, induction of osteocyte death appears to be a common mechanism for initiating targeted bone resorption [82]. However, when considering the observation that osteocytes are an important source of RANKL, the question arises of how osteocytes can simultaneously undergo apoptosis and also supply RANKL to initiate bone resorption. A recent set of experiments using fatigue-loaded rat ulnae performed by the Schaffler laboratory provide one potential answer. They demonstrated that osteocyte apoptosis localizes to areas containing fatigue damage whereas expression of factors that support focal bone resorption, such as RANKL and VEGF, is elevated in osteocytes surrounding the apoptotic osteocytes [83]. Thus dying osteocytes may signal to neighboring healthy osteocytes to control bone remodeling. Consistent with this idea, apoptotic bodies produced by dying osteocyte-like cells in vitro are able to promote osteoclast formation in vitro and in vivo [84]. It is also possible that changes in load control expression of osteocyte RANKL independent of cell death. Sclerostin abundance in osteocytes is elevated by unloading and suppressed by anabolic loading [39, 85]. Activation of the same signaling pathways that control sclerostin production may also control RANKL expression.
In the pathogenesis of bone destruction associated with rheumatoid arthritis (RA), synovium is the active site for the interplay between immune and bone cells. These pathological findings led us to hypothesize that osteoclasts play an important role in bone resorption in RA and that osteoclasts are formed in the synovium [86, 87]. Importantly, inflammatory cytokines such as IL-1, IL-6 and TNF-α, which are abundant in the synovial fluid and synovium of RA patients, have a potent capacity to induce RANKL on synovial fibroblasts and thus accelerate RANKL signaling, thereby directly contributing to the bone destruction process [22]. Several groups, including our own, have demonstrated a high level of RANKL expression in the synovium of RA patients [14, 88]. RANKL was found to be expressed by synovial and T cells, both of which are found in inflamed synovium [14, 19, 88], but at that point it was unclear which cell types are the major RANKL-expressing cells in joint destruction. Since then, a series of reports has established that the pathological bone damage associated with inflammation is caused by an abnormal expression of RANKL. Although physiological bone resorption in bone remodelling is mainly regulated by osteocyte RANKL, the contribution of osteocytes in bone destruction associated with inflammation is not well understood. A few in vitro reports suggest that the expression of RANKL on the osteocyte-like cell line MLO-Y4 is increased by IL-1, resulting in enhanced MLO-Y4-mediated osteoclastogenesis [89], and IL-1 also stimulates apoptosis of MLO-Y4 cells [90]. These findings suggest that osteocytes, via inflammatory cytokines such as IL-1, might contribute to the pathological regulation of osteoclastogenesis and bone destruction in autoimmune diseases such as RA. However, in vivo functions of osteocyte RANKL under inflammatory conditions remain unclear. The relative contribution of synoviocytes, T cells, and osteocytes to RANKL expression in pathological bone destruction should be elucidated in the future. A summary of pathways potentially controlling RANKL production by osteocytes is depicted in figure 1.
Fig. 1.
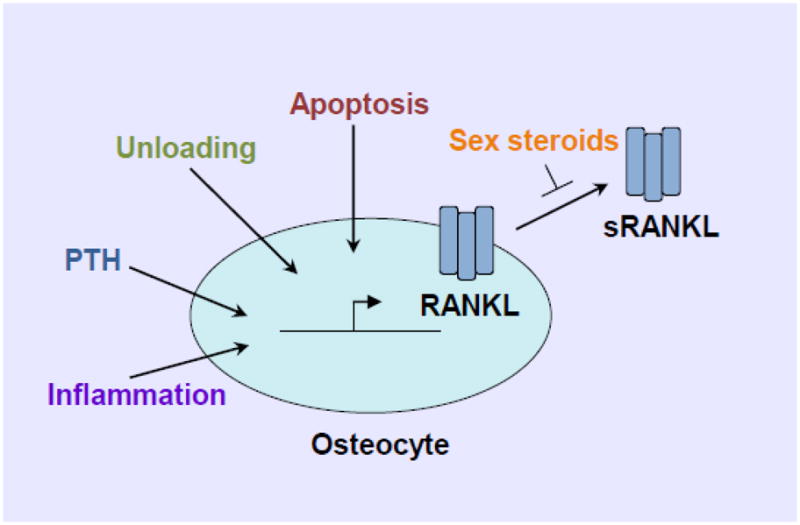
Signals controlling RANKL production by osteocytes.
Conclusions
Although osteocytes have long been thought to play a role in bone resorption, the advent of tools allowing manipulation of gene expression specifically in this cell type has created new opportunities to address this idea. The studies discussed here have revealed a surprisingly central role for osteocytes as orchestrators of bone resorption due to their expression of a rate-limiting factor required for osteoclast formation and function. Future work will be required to identify the signaling pathways and transcriptional programs that control RANKL expression in osteocytes and to determine whether RANKL produced by osteocytes plays a role in various pathological conditions that lead to increased bone resorption. In addition, considering the physical isolation of this cell type, the relative importance of membrane-bound and soluble forms of RANKL will need to be addressed. Finally, it is possible that osteocytes regulate bone resorption via mechanisms in addition to the production of RANKL and OPG. If recent experience is any indication, it seems likely that further surprises are in store.
Acknowledgments
The authors would like to thank S.C. Manolagas for critical review of the manuscript. This work was supported by the Central Arkansas Veteran’s Healthcare System (Merit Review), the United States National Institutes of Health (AR049794), the University of Arkansas for Medical Sciences (UAMS) Translational Research Institute (UL1 RR029884), UAMS tobacco settlement funds, and by a grant for ERATO, the Takayanagi Osteonetwork Project from the Japan Science and Technology Agency; Grant-in-Aids for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS); Grant-in-Aid for Challenging Exploratory Research from the JSPS; grants for Global Center of Excellence Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT); and grants from Astellas Foundation for Research on Metabolic Disorders and BMKK RA Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 2.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 3.Brunkow ME, Gardner JC, Van NJ, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–89. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balemans W, Ebeling M, Patel N, Van HE, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–43. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 5.Balemans W, Patel N, Ebeling M, Van HE, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–52. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- 7.Van Bezooijen RL, Roelen BAJ, Visser A, Wee-Pals L, de Wilt E, Karperien M, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–14. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–4. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 9.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley JM, et al. Osteoblastic cells are involved in osteoclast formation. Endocrinology. 1988;123:2600–2. doi: 10.1210/endo-123-5-2600. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–4. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 12.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien CA. Control of RANKL gene expression. Bone. 2010;46:911–9. doi: 10.1016/j.bone.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, et al. Involvement of receptor activator of nuclear factor κ B ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000;43:259–69. doi: 10.1002/1529-0131(200002)43:2<259::AID-ANR4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 15.Gori F, Hofbauer LC, Dunstan CR, Spelsberg TC, Khosla S, Riggs BL. The expression of osteoprotegerin and RANK ligand and the support of osteoclast formation by stromal-osteoblast lineage cells is developmentally regulated. Endocrinology. 2000;141:4768–76. doi: 10.1210/endo.141.12.7840. [DOI] [PubMed] [Google Scholar]
- 16.Zhao S, Kato Y, Zhang Y, Harris S, Ahuja SS, Bonewald LF. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J Bone Miner Res. 2002;17:2068–79. doi: 10.1359/jbmr.2002.17.11.2068. [DOI] [PubMed] [Google Scholar]
- 17.O’Brien CA, Gubrij I, Lin SC, Saylors RL, Manolagas SC. STAT3 activation in stromal osteoblastic cells is required for induction of the receptor activator of NF-κB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D-3 or parathyroid hormone. J Biol Chem. 1999;274:19301–8. doi: 10.1074/jbc.274.27.19301. [DOI] [PubMed] [Google Scholar]
- 18.Kanematsu M, Sato T, Takai H, Watanabe K, Ikeda K, Yamada Y. Prostaglandin E-2 induces expression of receptor activator of nuclear factor-κB ligand/osteoprotegrin ligand on pre-B cells: Implications for accelerated osteoclastogenesis in estrogen deficiency. J Bone Miner Res. 2000;15:1321–9. doi: 10.1359/jbmr.2000.15.7.1321. [DOI] [PubMed] [Google Scholar]
- 19.Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–9. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 20.Usui M, Xing L, Drissi H, Zuscik M, O’Keefe R, Chen D, et al. Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J Bone Miner Res. 2008;23:314–25. doi: 10.1359/JBMR.071025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kartsogiannis V, Zhou H, Horwood NJ, Thomas RJ, Hards DK, Quinn JMW, et al. Localization of RANKL (Receptor activator of NFκB ligand) mRNA and protein in skeletal and extraskeletal tissues. Bone. 1999;25:525–34. doi: 10.1016/s8756-3282(99)00214-8. [DOI] [PubMed] [Google Scholar]
- 22.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 23.Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab. 2012 doi: 10.1016/j.tem.2012.05.005. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24.Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–24. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes to Cells. 1999;4:353–62. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- 26.Wagner EF, Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunol Rev. 2005;208:126–40. doi: 10.1111/j.0105-2896.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 27.Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking nf-κ-b1 and nfκb2. Nat Med. 1997;3:1285–9. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 28.Franzoso G, Carlson L, Xing LP, Poljak L, Shores EW, Brown KD, et al. Requirement for nfκb in osteoclast and b-cell development. Genes Dev. 1997;11:3482–96. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, et al. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med. 2006;12:1410–6. doi: 10.1038/nm1515. [DOI] [PubMed] [Google Scholar]
- 30.David JP, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF. JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J Cell Sci. 2002;115:4317–25. doi: 10.1242/jcs.00082. [DOI] [PubMed] [Google Scholar]
- 31.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 32.Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–9. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM, et al. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118:3775–89. doi: 10.1172/JCI35711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–63. doi: 10.1038/nature02444. [DOI] [PubMed] [Google Scholar]
- 35.Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806. doi: 10.1016/j.cell.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 36.Barrow AD, Raynal N, Andersen TL, Slatter DA, Bihan D, Pugh N, et al. OSCAR is a collagen receptor that costimulates osteoclastogenesis in DAP12-deficient humans and mice. J Clin Invest. 2011;121:3505–16. doi: 10.1172/JCI45913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–38. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sutherland MK, Geoghegan JC, Yu CP, Turcott E, Skonier JE, Winkler DG, et al. Sclerostin promotes the apoptosis of human osteoblastic cells: a novel regulation of bone formation. Bone. 2004;35:828–35. doi: 10.1016/j.bone.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 39.Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/β-catenin signaling. J Bone Miner Res. 2009;24:1651–61. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–9. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 41.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–75. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 42.O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE. 2008;3:e2942. doi: 10.1371/journal.pone.0002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intra-cortical remodeling. J Bone Miner Res. 2011;26:1035–46. doi: 10.1002/jbmr.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf-1 as Therapeutic Targets in Bone Diseases. Endocr Rev. 2012 doi: 10.1210/er.2011-1060. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 45.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, et al. Osteocyte Wnt/β-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–85. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption--a hypothesis. Calcif Tissue Int. 1981;33:349–51. doi: 10.1007/BF02409454. [DOI] [PubMed] [Google Scholar]
- 47.Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–57. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka K, Yamaguchi Y, Hakeda Y. Isolated chick osteocytes stimulate formation and bone-resorbing activity of osteoclast-like cells. J Bone Miner Metab. 1995;13:61–70. [Google Scholar]
- 49.Corral DA, Amling M, Priemel M, Loyer E, Fuchs S, Ducy P, et al. Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc Natl Acad Sci U S A. 1998;95:13835–40. doi: 10.1073/pnas.95.23.13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galli C, Fu Q, Wang W, Olsen BR, Manolagas SC, Jilka RL, et al. Commitment to the osteoblast lineage is not required for RANKL gene expression. J Biol Chem. 2009;284:12654–62. doi: 10.1074/jbc.M806628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gu G, Nars M, Hentunen TA, Metsikko K, Vaananen HK. Isolated primary osteocytes express functional gap junctions in vitro. Cell Tissue Res. 2006;323:263–71. doi: 10.1007/s00441-005-0066-3. [DOI] [PubMed] [Google Scholar]
- 52.Paic F, Igwe JC, Nori R, Kronenberg MS, Franceschetti T, Harrington P, et al. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone. 2009;45:682–92. doi: 10.1016/j.bone.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ. DMP1-targeted Cre expression in odontoblasts and osteocytes. J Dent Res. 2007;86:320–5. doi: 10.1177/154405910708600404. [DOI] [PubMed] [Google Scholar]
- 54.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133:3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 55.Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–12. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- 56.Gebhard S, Hattori T, Bauer E, Schlund B, Bosl MR, de Crombrugghe B, et al. Specific expression of Cre recombinase in hypertrophic cartilage under the control of a BAC-Col10a1 promoter. Matrix Biol. 2008;27:693–9. doi: 10.1016/j.matbio.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ueno Y, Shinki T, Nagai Y, Murayama H, Fujii K, Suda T. In vivo administration of 1,25-dihydroxyvitamin D-3 suppresses the expression of RANKL mRNA in bone of thyroparathyroidectomized rats constantly infused with PTH. J Cell Biochem. 2003;90:267–77. doi: 10.1002/jcb.10623. [DOI] [PubMed] [Google Scholar]
- 58.Miao D, Li J, Xue Y, Su H, Karaplis AC, Goltzman D. Parathyroid hormone-related peptide is required for increased trabecular bone volume in parathyroid hormone-null mice. Endocrinology. 2004;145:3554–62. doi: 10.1210/en.2003-1695. [DOI] [PubMed] [Google Scholar]
- 59.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee SK, Lorenzo JA. Parathyroid hormone stimulates TRANCE and inhibits osteoprotegerin messenger ribonucleic acid expression in murine bone marrow cultures: Correlation with osteoclast-like cell formation. Endocrinology. 1999;140:3552–61. doi: 10.1210/endo.140.8.6887. [DOI] [PubMed] [Google Scholar]
- 61.Fu Q, Jilka RL, Manolagas SC, O’Brien CA. Parathyroid hormone stimulates receptor activator of NFκB ligand and inhibits osteoprotegerin expression via protein kinase A activation of cAMP-response element-binding protein. J Biol Chem. 2002;277:48868–75. doi: 10.1074/jbc.M208494200. [DOI] [PubMed] [Google Scholar]
- 62.Saini V, Barry K, Fulzele K, Feng J, Divieti PP. PTH/PTHrP (PPR) Receptor signaling in osteocytes regulate bone development in temporal manner. J Bone Miner Res. 2011;26:S16. [Google Scholar]
- 63.Tu X, Edwards R, Olivos N, Benson J, Galli C, Pellegrini G, et al. Conditional deletion of the parathyroid hormone (PTH) receptor 1 from osteocytes results in decreased bone resorption and a progressive increase in cancellous bone mass. J Bone Miner Res. 2011;26:S16. [Google Scholar]
- 64.Fu Q, Manolagas SC, O’Brien CA. Parathyroid Hormone Controls Receptor Activator of NF-κB Ligand Gene Expression via a Distant Transcriptional Enhancer. Mol Cell Biol. 2006;26:6453–68. doi: 10.1128/MCB.00356-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim S, Yamazaki M, Zella LA, Shevde NK, Pike JW. Activation of Receptor Activator of NF-κB Ligand Gene Expression by 1,25-Dihydroxyvitamin D3 Is Mediated through Multiple Long-Range Enhancers. Mol Cell Biol. 2006;26:6469–86. doi: 10.1128/MCB.00353-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martowicz ML, Meyer MB, Pike JW. The mouse RANKL gene locus is defined by a broad pattern of histone H4 acetylation and regulated through distinct distal enhancers. J Cell Biochem. 2011;112:2030–45. doi: 10.1002/jcb.23123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galli C, Zella LA, Fretz JA, Fu Q, Pike JW, Weinstein RS, et al. Targeted deletion of a distant transcriptional enhancer of the receptor activator of nuclear factor-κB ligand gene reduces bone remodeling and increases bone mass. Endocrinology. 2008;149:146–53. doi: 10.1210/en.2007-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Onal M, Galli C, Fu Q, Xiong J, Weinstein RS, Manolagas SC, et al. The RANKL distal control region is required for the increase in RANKL expression, but not the bone loss, associated with hyperparathyroidism or lactation in adult mice. Mol Endocrinol. 2012;26:341–8. doi: 10.1210/me.2011-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Onyia JE, Miles RR, Yang X, Halladay DL, Hale J, Glasebrook A, et al. In vivo demonstration that human parathyroid hormone 1–38 inhibits the expression of osteoprotegerin in bone with the kinetics of an immediate early gene. J Bone Miner Res. 2000;15:863–71. doi: 10.1359/jbmr.2000.15.5.863. [DOI] [PubMed] [Google Scholar]
- 70.Kondo H, Guo J, Bringhurst FR. Cyclic adenosine monophosphate/protein kinase A mediates parathyroid hormone/parathyroid hormone-related protein receptor regulation of osteoclastogenesis and expression of RANKL and osteoprotegerin mRNAs by marrow stromal cells. J Bone Miner Res. 2002;17:1667–79. doi: 10.1359/jbmr.2002.17.9.1667. [DOI] [PubMed] [Google Scholar]
- 71.Huang JC, Sakata T, Pfleger LL, Bencsik M, Halloran BP, Bikle DD, et al. PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res. 2004;19:235–44. doi: 10.1359/JBMR.0301226. [DOI] [PubMed] [Google Scholar]
- 72.Manolagas SC. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–37. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 73.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, et al. Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell. 2007;130:811–23. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 74.Martin-Millan M, Almeida M, Ambrogini E, Han L, Zhao H, Weinstein RS, et al. The estrogen receptor-α in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol Endocrinol. 2010;24:323–34. doi: 10.1210/me.2009-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111:1221–30. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Proell V, Xu H, Schuler C, Weber K, Hofbauer LC, Erben RG. Orchiectomy upregulates free soluble RANKL in bone marrow of aged rats. Bone. 2009;45:677–81. doi: 10.1016/j.bone.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 77.Li X, Ominsky MS, Stolina M, Warmington KS, Geng Z, Niu QT, et al. Increased RANK ligand in bone marrow of orchiectomized rats and prevention of their bone loss by the RANK ligand inhibitor osteoprotegerin. Bone. 2009;45:669–76. doi: 10.1016/j.bone.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 78.Tomkinson A, Reeve J, Shaw RW, Noble BS. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J Clin Endocrinol Metab. 1997;82:3128–35. doi: 10.1210/jcem.82.9.4200. [DOI] [PubMed] [Google Scholar]
- 79.Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2010;46:577–83. doi: 10.1016/j.bone.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res. 2009;24:597–605. doi: 10.1359/JBMR.081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21:605–15. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 82.Parfitt AM. Targeted and nontargeted bone remodeling: relationship to basic multicellular unit origination and progression. Bone. 2002;30:5–7. doi: 10.1016/s8756-3282(01)00642-1. [DOI] [PubMed] [Google Scholar]
- 83.Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50:1115–22. doi: 10.1016/j.bone.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008;23:915–27. doi: 10.1359/jbmr.080207. [DOI] [PubMed] [Google Scholar]
- 85.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–75. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 86.Takayanagi H, Oda H, Yamamoto S, Kawaguchi H, Tanaka S, Nishikawa T, et al. A new mechanism of bone destruction in rheumatoid arthritis: synovial fibroblasts induce osteoclastogenesis. Biochem Biophys Res Commun. 1997;240:279–86. doi: 10.1006/bbrc.1997.7404. [DOI] [PubMed] [Google Scholar]
- 87.Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol. 1998;152:943–51. [PMC free article] [PubMed] [Google Scholar]
- 88.Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, et al. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis & Rheumatism. 2000;43:250–8. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 89.Kulkarni RN, Bakker AD, Everts V, Klein-Nulend J. Mechanical loading prevents the stimulating effect of IL-1β on osteocyte-modulated osteoclastogenesis. Biochem Biophys Res Commun. 2012;420:11–6. doi: 10.1016/j.bbrc.2012.02.099. [DOI] [PubMed] [Google Scholar]
- 90.Bakker AD, Silva VC, Krishnan R, Bacabac RG, Blaauboer ME, Lin YC, et al. Tumor necrosis factor α and interleukin-1β modulate calcium and nitric oxide signaling in mechanically stimulated osteocytes. Arthritis Rheum. 2009;60:3336–45. doi: 10.1002/art.24920. [DOI] [PubMed] [Google Scholar]