

Abstract
Patients with platelet α or dense δ-granule defects have bleeding problems. Although several proteins are known to be required for δ-granule development, less is known about α-granule biogenesis. Our previous work showed that the BEACH protein NBEAL2 and the Sec1/Munc18 protein VPS33B are required for α-granule biogenesis. Using a yeast two-hybrid screen, mass spectrometry, coimmunoprecipitation, and bioinformatics studies, we identified VPS16B as a VPS33B-binding protein. Immunoblotting confirmed VPS16B expression in various human tissues and cells including megakaryocytes and platelets, and also in megakaryocytic Dami cells. Characterization of platelets from a patient with arthrogryposis, renal dysfunction, and cholestasis (ARC) syndrome containing mutations in C14orf133 encoding VPS16B revealed pale-appearing platelets in blood films and electron microscopy revealed a complete absence of α-granules, whereas δ-granules were observed. Soluble and membrane-bound α-granule proteins were reduced or undetectable, suggesting that both releasable and membrane-bound α-granule constituents were absent. Immunofluorescence microscopy of Dami cells stably expressing GFP-VPS16B revealed that similar to VPS33B, GFP-VPS16B colocalized with markers of the trans-Golgi network, late endosomes and α-granules. We conclude that VPS16B, similar to its binding partner VPS33B, is essential for megakaryocyte and platelet α-granule biogenesis.
Introduction
Inherited platelet disorders are important causes of abnormal bleeding. Studies of congenital platelet disorders have made major contributions to platelet biology,1,2 and key insights into platelet granule formation and function have come from investigating patients with deficient or absent α and/or δ-granules. Most inherited defects involve platelets lacking α-granules or δ-granules but not both, suggesting the existence of distinct granule development pathways.
Platelets arise from bone marrow-resident megakaryocytes (MKs), which differentiate from hematopoietic stem cells and produce masses of platelets via the formation of microtubule-dependent proplatelet extensions.3,4 Budding vesicles in the megakaryocyte trans-Golgi network mature into multivesicular bodies (MVBs) and granules5–7 that are transported into proplatelets8 to produce mature platelets containing 50 to 80 α-granules, 3 to 8 δ-granules, and a few lysosomes. α-granules contain a wide range of proteins that are both endogenously synthesized as well as endocytosed.5
Several insights into MK and platelet δ-granule development have come from studying patients with Hermansky-Pudlak syndrome (HPS), and Chediak-Higashi syndrome (CHS; MIM214500) for which mouse models exist.9–12 In both of these genetic diseases platelets lack δ-granules. Several genes/proteins linked to the regulation of vesicle trafficking have been implicated in δ-granule formation, including components of BLOC (biogenesis of lysosome-related organelles complex) protein complexes (BLOC-1, 2, 3), known vesicle-trafficking proteins (VPS33A, and the β3A and δ subunit of AP-3), and the BEACH domain-containing protein LYST.9,10,12,13
Less is known about α-granule development, which is affected in 3 inherited human disorders: Quebec platelet disorder, gray platelet syndrome (GPS; MIM139090), and ARC syndrome (arthrogryposis, renal dysfunction, and cholestasis). The α-granule defect in the Quebec platelet disorder is because of the abnormal presence of urokinase in platelets resulting in the proteolytic degradation of multiple α-granule proteins,14,15 arising from tandem duplication of the PLAU gene.16 GPS is characterized by variable thrombocytopenia and large gray-appearing platelets on blood smears with markedly decreased to absent α-granules and α-granule proteins,17,18 recently shown by us and others to be caused by mutations in NBEAL2 encoding a BEACH protein.19–21 Earlier we identified VPS33B as a key component in the formation of platelet α-granules based on their absence in platelets from ARC syndrome patients with mutations in VPS33B (VPS33B-ARC; MIM608552).22,23 Absence of platelet α-granules has subsequently been observed by others in VPS33B-ARC patients.24,25 The observation that GPS and VPS33B-ARC platelets lack α-granules but contain δ-granules, whereas HPS platelets are devoid of δ-granules but contain α-granules, suggests there are distinct pathways for δ-granule and α-granule biogenesis in maturing MKs.
VPS33B is a member of the Sec1/Munc18 (SM) protein family, known to be important for intracellular vesicle trafficking.26 SM proteins are known to interact with membrane-associated soluble N-ethylmaleimide-sensitive fusion (NSF)–attachment protein receptors (SNAREs) of the syntaxin subfamily. Although VPS33B has yet to be characterized in detail, its metazoan homolog VPS33A is known to be part of a multiprotein complex involved in intracellular vesicle trafficking27 with mutations causing δ-granule deficiency in a mouse model (buff) of HPS.28 Saccharomyces contains a single Vps33p homologue that is a component of 2 multiprotein complexes: the HOPS (homotypic fusion and protein vacuole sorting) and CORVET (class C core vacuole/endosome tethering) complexes.29 CORVET is present at endosomes and is required for endosomal trafficking, whereas HOPS is present at vacuoles and is required for vacuolar trafficking.29 In Drosophila, mutations in dVps33A result in the carnation eye color pigmentation defect, and dVps33A exists as part of a complex.30 Drosophila HOPS complex subunits are required for lysosomal delivery of different cargos and for fusion of lysosomes with autophagosomes.30–33 That dVps33A and dVps33B are part of distinct complexes with nonredundant functions is demonstrated by the specific interactions between dVps33A and dVps16A, as well as between dVps33B and dVps16B.32 Drosophila dVps33A is required for late endosome-to-lysosome fusion, whereas early endosomes are unaffected.34 Loss of dVps33A function was not restored by expression of dVps33B, confirming the distinct roles of these proteins during vesicular trafficking. Vps16B was also recently shown to be required for phagosome maturation in Drosophila.35 These observations suggest that mammalian VPS33B also interacts with other proteins to facilitate vesicular trafficking.
In this report, we investigated whether there are potential VPS33B-binding proteins that facilitate MK and platelet α-granule biogenesis. Using a yeast two-hybrid screen with VPS33B as bait against a human bone marrow library, we found binding to an uncharacterized gene product of chromosome 14 open reading frame 133 (C14orf133). The interaction was confirmed through coimmunoprecipitation (Co-IP) experiments, as well as mass spectrometry. Bioinformatics sequence analysis revealed that C14orf133 encodes human VPS16B. Immunoblot analysis confirmed the presence of VPS16B in various human tissues including MKs, platelets, and megakaryocytic Dami cells. While we were conducting these experiments, an interaction between VPS33B and VPS16B (SPE-39, VIPAR) was shown36,37 and mutations in C14orf133 were associated with ARC syndrome (MIM613401).36 Blood from an ARC patient containing a homozygous nonsense mutation in exon 8 of the VPS16B gene (C14orf133, VIPAR) revealed gray-appearing platelets on blood films. Whole-mount and transmission electron microscopy (TEM) of platelets confirmed the presence of δ-granules but a complete absence of α-granules. As with VPS33B-ARC platelets, soluble and membrane-bound α-granule proteins were significantly decreased to absent in VPS16B-ARC platelets. Dami cells stably expressing GFP-VPS16B costained with various intracellular markers revealed that VPS16B, similar to VPS33B, traffics between the trans-Golgi network late endosomes and α-granules. Thus our studies confirm that VPS16B, similar to its binding partner VPS33B, is required in megakaryocyte and platelet α-granule biogenesis.
Methods
Patients
This study was performed according to the guidelines of the Research Ethics Board at The Hospital for Sick Children and the Internal Review Board of the Pitt County Memorial Hospital. Informed consent was provided according to the Declaration of Helsinki. A full-term female patient was born to healthy nonconsanguineous parents. The findings of arthrogryposis, conjugated hyperbilirubinemia, and renal tubular acidosis led to a diagnosis of ARC syndrome. Frequent episodes of epistaxis suggested a bleeding diathesis in this patient. Molecular studies confirmed that the neonate has a homozygous mutation in exon 8 of C14orf133, (VIPAR; c.484C > T) leading to a premature stop codon (p.Arg162Stop). Although mutations in C14orf133 have been shown to cause ARC syndrome (https://grenada.lumc.nl/LOVD2/ARC),36 the c.484C > T mutation has not been previously described. Limited blood sample availability precluded platelet function testing.
Yeast two-hybrid analysis and cDNA constructs
The yeast two-hybrid library screen against premade human bone marrow library (Clontech) with VPS33B as bait was performed as per the manufacturer's specifications. The coding sequence of human VPS16B was PCR amplified from the vector pCMV6-XL5, which contained the library inserts with 5′-TAGTAGAATTCATGAATCGGACAAAGGGTGA-3′ and 5′-ATCTAGGTACCAAGGTAGTCAATGCCACTTA-3′. The PCR product was then inserted into pEGFP-C2 (Clontech). Using 5′-GACACGAATTCAGATGAATCGGACAAAGGGT-3′ and 5′-CGTCCGGTACCTTAATTCTTCCATCGAATTT-3′ human VPS16B was inserted into pCMV-Myc (Clontech). GFP-hVPS16B was then subcloned from pEGFP-C2 using AseI and BamHI restriction sites into pIRES2-DsRed-Express vector (Clontech) for the Dami stable cell line. For bacterial expression of S-tagged hVPS16B the coding sequence was PCR amplified and inserted into the pETDuet-1 vector (Novagen, EMD Millipore) with 5′-ATACGGTACCATGAATCGGACAAAGGGTGATGAGG-3′ and 5′-GTGAGG-TACCATTCTTCCATCGAATTTGCGAGCTG-3′.
Cell cultures
Human embryonic kidney (HEK293) cells were grown in DMEM (with 4.5 g/L glucose, 4mM l-glutamine, 100 μg/mL penicillin/streptomycin; Wisent Bioproducts), supplemented with 10% fetal bovine serum (FBS). Transfection of plasmids in HEK293 cells was performed with Lipofectamine 2000 (Invitrogen; Life Technologies), using one-half the amount of reagent and a quarter of the DNA suggested by the manufacturer. Dami cells were the generous gift of Dr David Wilcox. Cells were grown in Iscove Modification of DMEM supplemented with 10% horse serum (HS; Wisent Bioproducts). Dami cell induction was achieved by supplementing growth media for 3 to 5 days with 1μM phorbol 12-myristate 13-acetate (PMA; Invitrogen) and 50 ng/μL thrombopoietin (TPO; Kirin). Plasmids were introduced into Dami cells through electroporation using Amaxa Nucleofector kits and devices, as per manufacturer specifications (Lonza). Dami cells stably expressing GFP-hVPS16B were selected with 2 mg/mL G418 (Wisent Bioproducts), and individual clones assayed through immunoblotting with anti-GFP antibody. Human megakaryocytes were cultured from CD34+ cells as previously described.23 Human platelets were isolated and prepared for immunostaining as previously described.38
Immunoblot and coimmunoprecipitation analysis
Immunoblot analysis of platelet and cell lysates was done as previously described.23 For VPS16B protein expression in different human tissues we used the human tissue INSTA-Blot (Imgenex) containing 11 human tissue lysates. For Co-IP experiments, HEK293 cells were grown in 10-cm dishes and transiently transfected with GFP-VPS33B and Myc-hVPS16B constructs at 80% confluence. Cells were harvested 24 hours later, washed in ice-cold PBS and lysed in 1 mL 1% TX-100 buffer (100mM NaCl, 30mM HEPES, 20mM NaF, 1mM EGTA; pH 7.5) with 1 × protease inhibitor cocktail without EDTA (Roche) at 4°C. Cell debris was removed by centrifugation at 16 000g for 10 minutes at 4°C. The supernatant was then divided in 2 for preclearing with 100 μL slurry of protein A sepharose (Sigma-Aldrich) and with protein G sepharose (GE Healthcare) for 1 hour at 4°C. The precleared lysates were incubated with 2 μg anti-GFP and 1 μg anti-Myc antibodies, respectively, with the appropriate IgG controls (Dako Canada) for 1 hour at 4°C. Immune complexes were incubated with 50 μL protein A or G slurry for 1 hour at 4°C, washed 3 times in 1 mL 1% TX-100 buffer, and eluted in 25 μL Laemmli buffer. Eluates were subjected to SDS-PAGE followed by immunoblotting with anti-Myc and anti-GFP antibodies. Co-IP from Dami cells was performed similarly, with 10 μL anti-VPS33B serum, and 10 μL of the preimmune rabbit serum as a control. Human megakaryocytes were collected from a 50 mL culture at 400g for 5 minutes, washed twice in ice-cold PBS, and resuspended in nondenaturing lysis buffer (1% TX-100, 50mM Tris-chloride, 300mM NaCl, 5mM EDTA, 0.02% NaN3, pH 7.4) with 1 × protease inhibitor cocktail without EDTA (Roche) for 30 minutes at 4°C. Cell debris was removed through centrifugation at 16 000g for 15 minutes at 4°C. The lysate was precleared with 30 μL protein G sepharose slurry (GE Healthcare Biosciences) at 4°C for 30 minutes with end-over-end rotation. Ten milliliters of preimmune serum or anti-VPS33B serum were preincubated with 50% protein G sepharose beads for 1 hour at 4°C, washed 4 times in 1 mL nondenaturing lysis buffer, and subsequently incubated with the precleared lysate with 10% BSA overnight at 4°C. Sepharose bead-immune complexes were washed 3 times in 1 mL wash buffer (lysis buffer with 0.1% TX-100), once in 1 mL PBS, and flushed with 40 μL Laemmli buffer.
Cell fractionation
Dami cells were counted and a suspension of 40 × 106 cells was collected by centrifugation at 500g for 5 minutes, washed in ice-cold PBS, and resuspended in 300 μL CF buffer (25mM HEPES, 150mM NaCl, 1mM EGTA, 1mM DTT, 0.5mM MgCl2, pH 7.4). Cells were lysed by passage through a 25-G needle 50 times, and centrifuged at 16 000g for 10 minutes at 4°C. The supernatant was centrifuged at 100 000g for 1 hour at 4°C in a Beckman-Coulter Optima Max-XP ultracentrifuge with the TLA-110 rotor. The resulting supernatant constituted the cytosolic fraction and was saved, while the pellet was resuspended in CF Buffer with 1% TX-100, and centrifuged at 10 000g for 1 hour at 4°C. Both supernatants were then subjected to SDS-PAGE followed by immunoblotting with anti-VPS16B and anti-VPS33B antibodies.
Mass spectrometry, antibodies, molecular evolutionary analysis of C14orf133, bright field microscopy, electron microscopy, and confocal immunofluorescence microscopy
The experimental details are described in supplemental Methods (available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Results
VPS33B interacts with chromosome 14 open reading frame 133 (C14orf133)
To identify potential VPS33B binding proteins, we performed a yeast two-hybrid library screen with VPS33B as bait against a human bone marrow library. More than 52% of the positive interactions were with an uncharacterized gene product of chromosome 14 open reading frame 133 (C14orf133; data not shown). To confirm this interaction we performed Co-IP experiments of transiently transfected Myc-C14orf133 and GFP-VPS33B tagged constructs in HEK293 cells. Anti-Myc immunoprecipitation (IP) followed by anti-GFP immunoblotting (IB) showed specific binding of C14orf133 to VPS33B but not to VPS33A (Figure 1A). The interaction between C14orf133 and VPS33B was further confirmed using a stable HEK293 cell line expressing C-terminal tagged VPS33B-FLAG combined with mass spectrometry (supplemental Figure 2A-B). That this interaction is also present in vivo in MKs and Dami cells was shown using anti-VPS33B serum to Co-IP VPS33B with C14orf133 as shown in Figure 1B. Taken together our results show that VPS33B interacts with C14orf133 both in vitro and in vivo.
Figure 1.
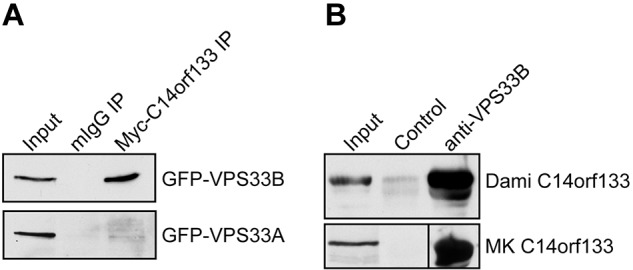
VPS33B binds to C14orf133 in HEK 293 cells, Dami cells, and megakaryocytes. (A) Myc-tagged C14orf133 (Myc-C14orf133) was coimmunoprecipitated (IP) with GFP-tagged VPS33B (GFP-VPS33B) or VPS33A (GFP-VPS33A) after cotransfecting HEK293 cells. The IP antibody was anti-Myc whereas a nonspecific mouse IgG (mIgG) was used as a control. The immunoblot (IB) signals were detected using an anti-GFP antibody. Myc-C14orf133 coimmunoprecipitated with GFP-VPS33B but not GFP-VPS33A. (B) Using anti-VPS33B serum and preimmune serum as control, endogenous VPS33B was immunoprecipitated from Dami and human megakaryocyte cells. The immunoblot signals were detected with anti-C14orf133 mouse serum. VPS33B binds to C14orf133 endogenously in Dami and megakaryocyte cells.
Sequence analysis suggests that C14orf133 is mammalian VPS16B
From multiple sequence alignments, molecular evolutionary analysis reveals C14orf133 to be the human ortholog of VPS16B as shown in Figure 2 and supplemental Figure 3A and B. Secondary structure predictions to identify positions of α-helices demonstrate a well-conserved secondary structure between VPS16A and VPS16B, despite sharing only 20% sequence identity (supplemental Figure 3A-B).
Figure 2.

Phylogenetic reconstruction of the VPS16 domain from selected VPS16A and VPS16B proteins. The multiple sequence alignment was generated using structural information (see supplemental Methods). Only nodes with at least 50% support (from 10 000 bootstrap replicates) are resolved. Under the assumption that the shared region from VPS16A and VPS16B has a common ancestor, VPS16A_YEAST represent the outgroup for this phylogenetic tree. Branch lengths indicate average amino acid substitutions per site (see scale bar).
Presence of VPS16B in human tissues, megakaryocytes, Dami cells, and platelets
Using a polyclonal mouse anti-VPS16B we determined the presence of VPS16B in various human tissues (Figure 3A) as well as MKs and platelets (Figure 3B) via immunoblotting. VPS16B was seen to be present in most tissues, although the relative amount varied considerably compared with the GAPDH loading control. Different sized bands in the tissue blot may represent different isoforms, proteolytic degradation, or nonspecific antibody binding. VPS16B is present in human MKs and platelets (Figure 3B). Because we did not observe VPS33B in platelets using a polyclonal antibody generated against the N-terminal of VPS33B previously,23 or on repeat testing (Figure 3B bottom blot, N-terminal) we were surprised to detect VPS33B in platelets using a newly generated rabbit anti-VPS33B (internal amino acids 450-478) antibody (Figure 3 middle panel, internal). Cellular fractionation studies indicated that in Dami cells the majority of VPS16B and VPS33B are present within the cytosolic fraction, with a small proportion in the membrane compartment (Figure 3C).
Figure 3.
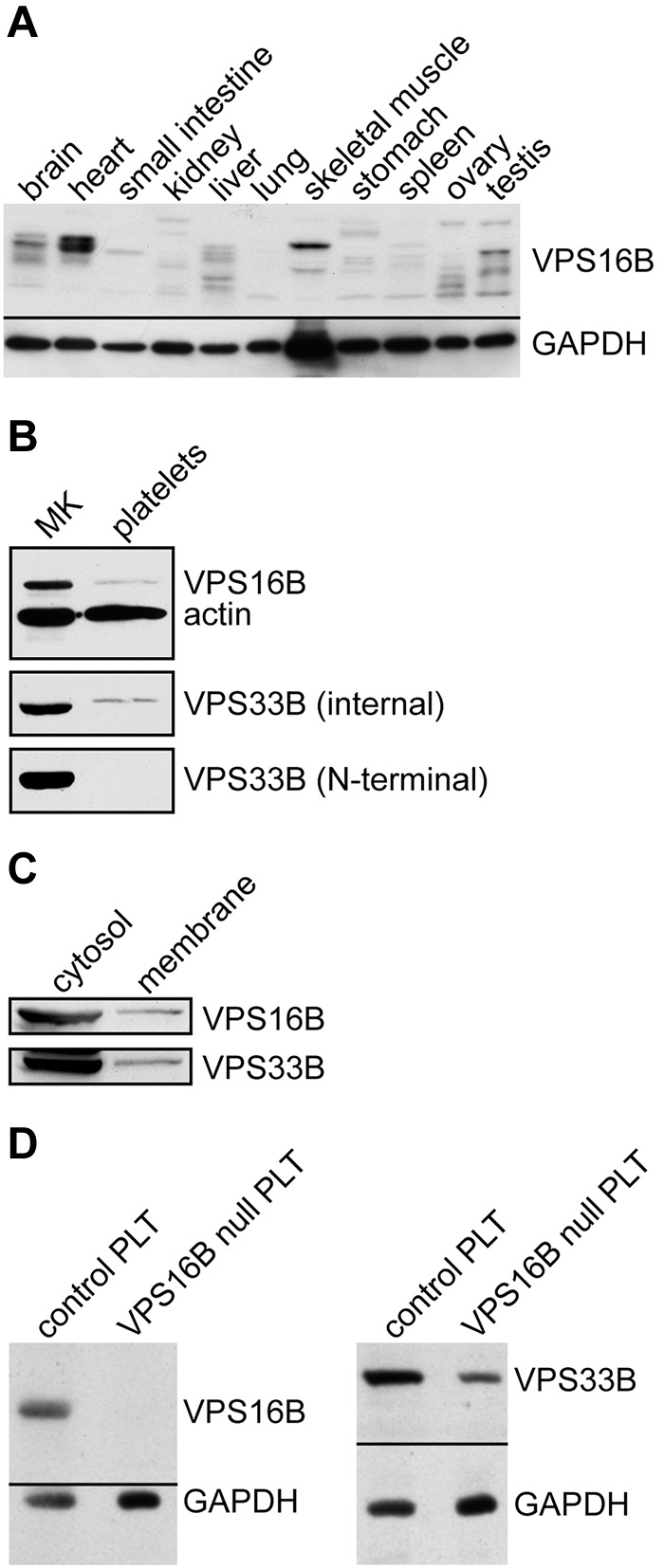
VPS16B and VPS33B distribution in tissue and cells lysates. (A) VPS16B is found in various tissues. A ready-to-use PVDF membrane with 11 human tissue lysates (Imgenex IMB-120) was probed with anti-VPS16B mouse serum, and anti-GAPDH antibody as a loading control. (B) VPS16B and VPS33B are present in both megakaryocyte (MK) and platelet lysates (reduced 10% SDS-PAGE). The latter were probed with anti-VPS16B mouse serum, anti-VPS33B internal as well as N-terminal antibodies, along with actin as a loading control. VPS16B and actin were probed and developed simultaneously (top panel). VPS33B is detected in platelets using the internal but not N-terminal antibody. (C) VPS16B and VPS33B are primarily found in the cytosol rather than the membrane in fractionated Dami cell lysates when probed with mouse anti-VPS16B and rabbit anti-VPS33B serum, respectively. (D) Platelets from a patient with ARC syndrome containing a homozygous nonsense mutation in exon 8 of C14orf133 (VIPAR) are devoid of VPS16B (left panel; VPS16B null PLT) and contain significantly reduced VPS33B (right panel; VPS16B null PLT), as shown by immunoblotting.
Structural abnormalities of platelets from an ARC patient with loss of VPS16B
We analyzed platelets from an ARC patient containing a homozygous nonsense mutation in exon 8 of C14orf133, (VIPAR) encoding VPS16B (VPS16B-ARC; VPS16B null) resulting in a complete loss of VPS16B and a partial loss of VPS33B protein expression in platelets (Figure 3D). The platelet count was 280 × 109/L with a mean platelet volume (MPV) of 10.9 fL. The platelet size distribution revealed some very large platelets with a MPV of 15 to 25 fL. The platelets appeared pale and agranular in Wright-Giemsa stained blood films (Figure 4A) compared with control neonatal platelets (Figure 4B).
Figure 4.
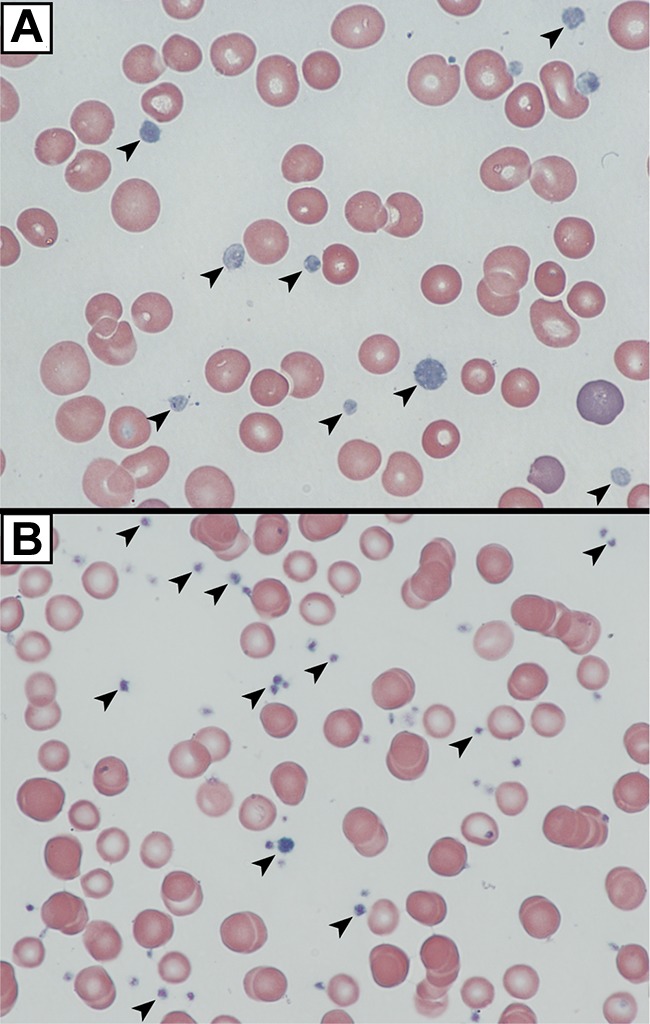
VPS16B null platelets appear abnormal in blood films. Romanowsky (Wright-Giemsa)–stained blood films from a VPS16B-deficient ARC patient compared with a neonatal control visualized using oil-immersion 63×/1.4 objective lenses. (A) VPS16B null platelets appeared large, homogeneous, pale and nongranular (arrows). (B) Normal neonatal control platelets are typically smaller, and granular (arrows).
To determine the α-granule content and analyze the ultrastructure of VPS16B-ARC platelets, thin section transmission EM was performed as shown in Figure 5. Platelet α-granule deficiency was clearly evident in micrographs (Figure 5B) compared with control neonatal platelets (Figure 5A). A complete absence of α-granules was noted in VPS16B-ARC platelets (n = 50 platelets) compared with an average of 5.5 α-granules per platelet section in controls (n = 25 platelets).23 Dense granule quantification using whole-mount EM revealed an average count of 10.3 dense granules per VPS16B-ARC platelet (n = 25 platelets) compared with 1.4 per platelet in neonatal controls.23 Thus ultrastructural studies confirmed a complete absence of α-granules and increased δ-granules in VPS16B-ARC platelets, similar to observations in VPS33B-ARC platelets.23
Figure 5.

Absent α-granules in platelets from VPS16B null patients. Thin-section transmission electron micrographs of representative platelets from control neonatal and VPS16B null neonatal blood. Magnification is ×30 000 and scale bar represents 500 nm. (A) Control platelets contain multiple α-granules (white arrowheads), which are absent in VPS16B null platelets (B). The dark-staining structures seen in the VPS16B null platelet (B) contain distinct internal membranes that probably represent mitochondria.
α-granule proteins are deficient in VPS16B null platelets
To demonstrate that the observed α-granule deficiency in VPS16B null platelets was also accompanied by a deficit of α-granule proteins, we analyzed several α-granule proteins using immunoblots (Figure 6). Equivalent sample loading was performed by loading lysates from similar numbers of platelets, measuring total protein concentrations and probing for actin or GAPDH. The results shown in Figure 6 indicate that the MK-synthesized proteins PF4, β-TG, osteonectin, TSP1, and VWF are markedly reduced to absent in VPS16B null platelets compared with controls (Figure 6A-E). Although VWF was markedly decreased in VPS16B null platelets (Figure 6E), it was present in normal amounts in VPS16B-ARC patient plasma (Figure 6F), suggesting that endothelial VWF synthesis was unaffected. Fibrinogen was also reduced in VPS16B null platelets compared with controls (Figure 6G) but present in normal amounts in patient plasma (not shown), suggesting that exogenous α-granule protein uptake is diminished in the absence of α-granules. The membrane protein P-selectin is absent in VPS16B null platelets (Figure 6H), suggesting an absence of α-granule membrane structures.
Figure 6.

α-granule protein deficiencies in VPS16B null platelets. Immunoblots comparing platelet (PLT) whole-cell lysates or plasma from VPS16B null and normal (N) neonatal sources as indicated for each lane. Significantly reduced PF4 (A), β-TG (B), TSP1 (D), VWF (E), and fibrinogen (G) content in VPS16B null platelets (lane 2) compared with control neonatal platelets (lane 1) were observed. Platelet osteonectin (C) and P-selectin (H) were undetectable in VPS16B null platelets (lane 2) compared with normal neonatal (lanes 1). (F) In contrast, VWF was present at roughly equivalent quantities (0.5 μL plasma/lane) in VPS16B null plasma (lane 2) compared with normal neonatal plasma (lanes 1). Lysate from equivalent numbers of platelets was loaded on each gel. Equivalent protein loading is indicated by visualizing actin or GAPDH on the same immunoblot. Panels A through C were probed and developed simultaneously using primary rabbit antibodies (PF4, β-TG, osteonectin) and mouse antibodies (actin, GAPDH). (A-D) Reduced 15% SDS-PAGE. (E-F,H) Reduced 8% SDS-PAGE. (G) Nonreduced 8% SDS-PAGE.
VPS16B localizes with trans-Golgi network, late endosomal and α-granule markers
Studies in our laboratory have demonstrated the presence of VPS33B in the proximity of late endosomes and α-granules in human megakaryocytes.23 Because our anti-VPS16B antibody was not adequate for IF microscopy, we generated a Dami cell line expressing stable levels of GFP-VPS16B to determine its intracellular localization relative to different vesicular markers in this megakaryocytic cell (Figure 7, supplemental Figure 4). These studies were done in triplicate where multiple cells were scored (supplemental Table 1) and representative images were taken. The distribution of GFP-VPS16B is in a granular pattern throughout the cytoplasm of Dami cells (green in Figure 7, supplemental Figure 4). GFP-VPS16B partially colocalizes with the trans-Golgi marker AP-1 (Figure 7A-C), the late endosome/lysosomal marker LAMP1 (Figure 7D-F), the late endosome marker Rab7 (Figure 7G-I), the α-granule marker VWF (Figure 7J-L), but not the δ-granule/lysosome marker CD63 (Figure 7M-O). The relative colocalization with these vesicular markers is indicated by the Pearson correlation coefficients (supplemental Table 1). There is little or no colocalization between GFP-VPS16B and the cis-Golgi marker GM130 (supplemental Figure 4A-C), the endosome/trans-Golgi marker M6PR (supplemental Figure 4D-F), the early endosome marker EEA1 (supplemental Figure 4G-I), and the recycling endosome marker TfR (supplemental Figure 4J-L). These studies suggest that VPS16B interacts with the trans-Golgi network, late endosomes and associates with α-granules as observed with VPS33B.23
Figure 7.
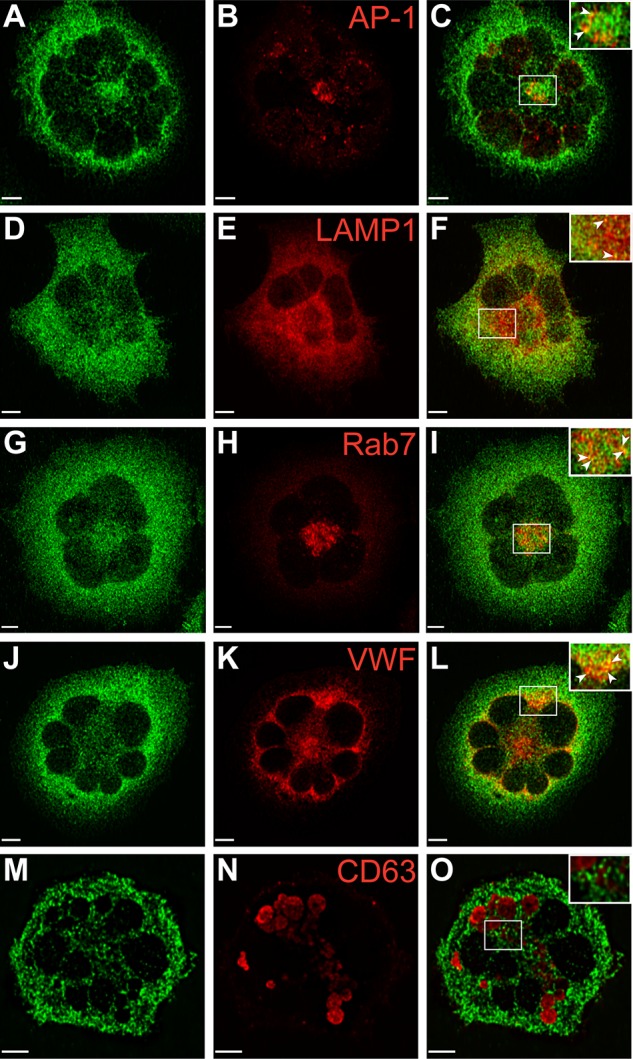
Localization of GFP-VPS16B within stable megakaryocytic Dami cells. Dami cells stably transfected with GFP-VPS16B fusion protein (green) were costained with different intracellular markers (red) as indicated. Merged images are shown on the right of each row (C,F,I,L,O). Partial colocalization (merge) of GFP-VPS16B is observed with the trans-Golgi network (AP-1, A-C), late endosomes/lysosomes (LAMP1, D-F), late endosomes (Rab7, G-I), α-granules (VWF, J-L) but not δ-granules (CD63, M-O). Insets with arrows highlight areas of colocalization. Multilobed nuclei are readily observed in Dami cells stimulated with phorbol ester and TPO. Representative z-stack images are shown. The white bar represents 5 μm. Images were obtained using a Leica DMIRE2 inverted fluorescence microscope equipped with a Hamamatsu C9100-12 back-thinned EM-CCD camera and Yokogawa CSU 10 spinning disk confocal scan head (with Spectral Aurora Borealis upgrade), with 63×/1.4 objective lenses. Data were acquired and processed using the PerkinElmer Volocity Verison 5.1.1 software.
Discussion
Platelet secretory granules develop within maturing bone marrow megakaryocytes, where α-granules, δ-granules, and lysosomes are transported to extending proplatelets8 and undergo further maturation after platelets are released into the circulation. Immunogold electron microscopy studies of megakaryoblastic CHRF-288 and MK cells suggest that α-granules develop from budding vesicles of the Golgi complex, from where they mature into multivesicular bodies and α-granules.5,6 Morphologically, 2 types of multivesicular bodies can be distinguished: immature type I MVBs contain internal vesicles only, and type II MVBs contain internal vesicles and an electron dense matrix.6 Observations that the dense granule/lysosomal transmembrane CD63 protein is localized to MVBs6 and deficient in HPS patients39 suggest that MVBs are also intermediate stages during δ-granule biogenesis. Although MVBs provide common sorting compartments for α and δ-granule formation, the mechanisms by which these vesicles develop into distinct granules is unknown.
Several molecules have been implicated in δ-granule development, whereas only 2 proteins have been identified as being required for α-granule biogenesis: the BEACH domain containing protein neurobeachin-like 2 (NBEAL2; deficient in GPS)19–21 and the Sec1/Munc18 protein VPS33B (deficient in VPS33B-ARC syndrome).23 The molecular details of how NBEAL2 functions during vesicular trafficking are unknown. As for VPS33B, the known functions of other Sec1/Munc18 proteins, the Drosophila homolog Vps33B and the yeast homolog Vps33p suggest that mammalian VPS33B interacts with SNAREs and other proteins to facilitate membrane docking and fusion during vesicular trafficking and α-granule formation.23
We identified C14orf133 as a VPS33B binding protein using a yeast two-hybrid screen, and confirmed the interaction by Co-IP (Figure 1A) and mass spectrometry (supplemental Figure 2) experiments in mammalian cells. That the interaction occurs in human MKs and Dami cells was shown by native IP experiments using a polyclonal anti-VPS33B antibody (Figure 1B). Other groups have also identified C14orf133 as a VPS33B interacting protein using Caenorhabditis elegans,37 mammalian36,37 and Drosophila protein binding assays.32 The protein encoded by C14orf133 has been variably termed SPE-39,37 VIPAR (VPS33B-interacting protein, apical-basolateral polarity regulator),36 and dVps16B.32
Previous work in Drosophila identified dVps16B, the product of the CG18112 gene, as a homolog of dVps16A, based on shared sequence similarity in their C-terminal domains.32 Here, we show that the C-terminal regions of the human homolog of CG18112 and VPS16A are likewise conserved. Despite the relatively low sequence identity associated with these sequences, they are nonetheless predicted to have a conserved secondary structure (supplemental Figure 3B). A multiple sequence alignment of the conserved C-terminal region of the VPS16 family guided by secondary structure predictions allowed us to improve on previous attempts to align this family,36,37 extend the taxonomic range and construct a robust phylogeny, which supports the hypothesis that the ancestral form of VPS16A duplicated early in the Metazoan lineage (Figure 2, supplemental File 1). In light of this relationship and consistent with the earlier study,32 we propose the divergent family should be named VPS16B. Intriguingly, whereas the C-terminal domains of VPS16A and VPS16B (termed Vps16_C; Pfam ID: PF04840) are well conserved, VPS16B appears to have lost the relatively well conserved Vps16_N domain (Pfam ID: PF04841) associated with VPS16A. Although the N-terminus of VPS16B is more variable (supplemental Figure 3B), this region still represents a common domain (Golgin_A5; Pfam ID: PF09787.2). Golgin_A5 has also been identified in plants, but its combination with Vps16_C represents a metazoan innovation and is only present as a single copy in any species. Together these data suggest that through duplication and subsequent replacement of Vps16_N domains with Golgin_A5, VPS16B provides metazoans with the prospect of either expanding (neofunctionalization) or specializing (subfunctionalization) the function of the Vps-C complex.
The binding interaction of VPS16B to VPS33B suggests that VPS16B, similar to VPS33B, may play a role during α-granule biogenesis. Previous studies in Drosophila have suggested that there are 2 functionally distinct class C Vps complexes.32,34 Importantly, dVps33B was shown to interact only with dVps16B. Furthermore, the 2 dVps33 homologues appear to have nonredundant functions, as dVps33B was unable to rescue the effect of a dVps33A deletion.34 Based on these findings, we performed Co-IP experiments with tagged human VPS16B and VPS33A (Figure 1A, top immunoblot). Although there is a strong interaction between Myc-VPS16B (Myc-C14orf133) and GFP-VPS33B, we were unable to observe any binding between Myc-VPS16B and GFP-VPS33A (Figure 1A, bottom immunoblot). In contrast to these findings and those of Pulipparacharuvil et al,32 interactions between tagged VPS16B and VPS33A were observed by another group.37 Although the discrepant results are not explained, it should be noted that, similar to Drosophila, VPS33A and VPS33B have nonredundant functions in platelet granule biogenesis, because mutations in VPS33A (missense mutation in buff mouse) cause δ-granule defects,28 whereas mutations in VPS33B (VPS33B-ARC) cause α-granule defects.23 Taken together our results suggest that human VPS33A and VPS33B form components of functionally distinct complexes involved in different aspects of platelet granule biogenesis.
To detect VPS16B protein in human cells we set out to make a monoclonal antibody directed against VPS16B. Although we were unsuccessful in getting a monoclonal antibody, sufficient polyclonal mouse serum was available showing immunoblot specificity in MK and Dami cells (supplemental Figure 1). The protein expression of VPS16B appears fairly widespread in human tissues (Figure 3A) and importantly was present in human platelets (Figure 3B).This was unexpected because we did not previously identify its binding partner, VPS33B, in human platelets.23 However, using a newly generated antibody against an internal region of VPS33B (see supplemental Methods), we also confirmed the presence of VPS33B in platelets (Figure 3B). The absence of a signal, using the antibody generated against the 16 N-terminal amino acids of VPS33B, could reflect posttranslational modification or proteolysis of this region in VPS33B in platelets. The presence of VPS33B and VPS16B in platelets raises the possibility that these proteins function in circulating platelets for new granule biogenesis during platelet progeny formation40 and/or release processes.
Because VPS33B-VPS16B is predicted to function in vesicular docking and fusion events during α-granule development, we wanted to assess its relative distribution in the cytosolic and membrane compartments of megakaryocytic Dami cells. As can be seen from cell-fractionation experiments the majority of VBS16B and VPS33B is found in the cytosolic compartment with smaller amounts associated with membrane fractions (Figure 3C). Because the majority of VPS33B-VPS16B is cytosolic, the recruitment to membranes for facilitating vesicular fusion is probably a dynamic process. It is possible that after membrane fusion is completed, VPS33B-VPS16B disengage from the membranes of the fused vesicles and return to the cytosol of the cell.
Based on the complex formation between VPS33B and VPS16B, it was not surprising to observe, that mutations in VPS16B (C14orf133, VIPAR) similar to VPS33B, cause ARC syndrome in humans.22,36 In zebrafish and mouse cells, deficiency of VPS16B causes biliary secretion and tight junction defects of the medullary collecting ducts, respectively, confirming the role of VPS16B in regulating apical-basolateral polarity in the liver and kidney.36 In C elegans, VPS16B is involved in vesicular biogenesis during spermatogenesis and processing of endocytosed proteins in oocytes and coelomocytes.37 The Ras-related GTP-binding proteins Rab5, Rab7, and Rab11 that are known regulators of vesicular trafficking have been associated with VPS16B.36,37
Because we previously showed that VPS33B-ARC patients have abnormal platelets that lack α-granules,23 we predicted that platelets from patients with mutations in C14orf133 have similar abnormalities. We characterized platelets from a patient with ARC syndrome (VPS16B-ARC) containing a homozygous nonsense mutation in exon 8 of C14orf133, (c.484C > T) leading to a premature stop codon at amino acid 162. This mutation has not been previously described compared with 7 other subjects with C14orf133 (VIPAR) mutations.36 In contrast to normal control platelets, VPS16B protein is completely absent and VPS33B is markedly reduced in platelets from this patient determined by immunoblotting (Figure 3D, compare lanes 1 and 2 in left and right panels, respectively). Decreased VPS33B in the absence of VPS16B suggests that these proteins have a stabilizing effect on each other within the VPS33B-VPS16B complex.
The dark blue staining pattern of normal platelets in blood films is largely because of the basophilic protein content of platelet α-granules. The abnormal, pale and agranular appearance of VPS16B-ARC (VPS16B null) platelets in blood films (Figure 4A-B) suggested α-granule deficiency, as was also noted in VPS33B-ARC platelets.23 Evidence that VPS16B null platelets contain no recognizable α-granules was confirmed by thin-section transmission EM (Figure 5B) compared with an average number of 5.5 α-granules per platelet thin section, seen in controls (Figure 5A).23 Whole-mount EM determined that δ-granules were present at increased numbers (10.3 per platelet) compared with neonatal controls (1.4 per platelet) as was also observed in VPS33B-ARC platelets.23 By comparison, normal human adults contain 3 to 8 δ-granules per platelet.41 Taken together, the ultrastructural studies confirm a complete absence of α-granules in all VPS16B-ARC platelets examined but the presence of δ-granules that are increased in some VPS16B null platelets, findings that were similar to those in VPS33B-ARC platelets.23 These data confirm that the VPS33B-VPS16B complex has a distinct functional role during platelet α-granule biogenesis whereas the VPS33A-containing complex (mutated in HPS buff mouse)28 has a specific role in δ-granule formation. The distinct roles of the VPS33B-VPS16B and VPS33A-VPS16A containing complexes is further supported by their nonredundant functions during lysosomal trafficking and pigment granule formation in Drosophila.32,34
Soluble proteins contained within platelet α-granules originate from endogenous megakaryocyte5 (eg PF4, β-TG, osteonectin, TSP1, VWF) or platelet42 synthesis or are plasma derived via processes, such as endocytosis through the αIIbβ3 integrin receptor (eg, fibrinogen).5 The proteins appear to be either packed into discrete α-granule subpopulations43,44 or are spatially packaged into distinct compartments within the same α-granule.45,46 We observed that both endogenously synthesized (eg, PF4, β-TG [NAP-2/CXCL7], osteonectin, TSP1, VWF) as well as plasma-derived (eg, fibrinogen) soluble α-granule proteins were reduced or absent in VPS16B null platelets (Figure 6A-E,G). Because VWF is also expressed in endothelial cells where it is stored in Weibel-Palade bodies and actively secreted,47 the presence of normal amounts of VWF in VPS16B null plasma (Figure 6F) suggests that the packaging defect of VWF is confined to VPS16B-ARC MKs, as was observed in VPS33B-ARC patients.23
Absent α-granule membrane structures could explain the inability of VPS16B-ARC MKs and/or platelets to capture proteins into α-granule vesicles. This possibility is supported by the complete absence of the α-granule membrane protein P-selectin in VPS16B null platelets (Figure 6H). P-selectin is a type I integral membrane protein residing within α-granule membranes of resting platelets that is translocated to the plasma membrane of stimulated platelets.48 The P-selectin deficit in VPS16B null platelets per immunoblot was also observed in VPS33B-ARC platelets,23 therefore probably reflects absent α-granule membrane compartments. This is in contrast to gray platelet syndrome platelets (lacking NBEAL2)19–21 where EM immunolocalization studies and immunoblots have detected P-selectin in platelets,49,50 albeit in reduced amounts in some patients,17 confirming the presence of α-granule membrane structures in GPS platelets.
VPS33B has been associated with markers of late endosome/lysosomes and α-granules but not with cis-Golgi complexes or δ-granules/lysosomes in human MKs.23 To determine whether VPS16B is present in similar intracellular compartments, colocalization studies using megakaryocytic Dami cells expressing stable GFP-VPS16B were performed (Figure 7, supplemental Figure 4, and supplemental Table 1). Colocalization of GFP-VPS16B to markers of the trans-Golgi network (AP-1, Figure 7A-C), late endosome/lysosomes (LAMP1, Figure 7D-F), late endosomes (Rab7, Figure 7G-I), and α-granules (VWF, Figure 7J-L) was observed. In comparison, the relative colocalization of GFP-VPS16B to markers of δ-granules/lysosomes (CD63, Figure 7M-O), cis-Golgi complexes (GM130, supplemental Figure 4A-C), endosome/trans-Golgi (M6PR, supplemental Figure 4D-F), early endosomes (EEA1, supplemental Figure 4G-I), and recycling endosomes (TfR, supplemental Figure 4J-L) was much reduced. The unavailability of adequate primary anti-VPS16B and anti-VPS33B antibodies for immunofluorescence microscopy studies did not allow us to look at the native distribution of these proteins in human MKs. Taken together our results suggest that VPS16B, together with VPS33B, acts along the trans-Golgi network to late endosome to α-granule vesicular trafficking pathway during formation of α-granules in megakaryocytes.
These results confirm the essential role of VPS16B in α-granule but not δ-granule biogenesis in human megakaryocytes and platelets. It is predicted that VPS16B binds to VPS33B within MKs and Dami cells to facilitate docking and fusion of intracellular vesicles during α-granule formation. After the membrane-containing α-granule vesicles have been established by VPS16B-VPS33B, NBEAL2 somehow promotes further α-granule maturation. Identification of VPS16B as an essential component of human α-granule biogenesis represents an important new insight into a process that has until recently been poorly understood.
Supplementary Material
Acknowledgments
The authors thank the family for participating in this research. They also thank the Kirin Brewery Company for their generous gift of recombinant pegylated human megakaryocyte growth and development factor (thrombopoietin, TPO).
This work was supported by grants from the Canadian Institutes of Health Research (MOP 81208, W.H.A.K.; MOP 84556, J.P.), and the National Institutes of Health (EY10199, H.K.).
Footnotes
There is an Inside Blood commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.U., J.D.W., J.P., and W.H.A.K. designed experiments; D.U., L.L., H.C., F.G.P., S.Z.C, M.P., H.K., J.D.W., J.P., and W.H.A.K. performed research, analyzed data; P.M.G., K.K.L., and J.J.C. facilitated patient recruitment and participation in this study; and D.U., F.G.P., H.K., J.D.W., J.P., and W.H.A.K. wrote and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Walter H. A. Kahr, Departments of Pediatrics and Biochemistry, University of Toronto, Division of Haematology/Oncology, Program in Cell Biology, The Hospital for Sick Children, 555 University Avenue, Toronto, ON, M5G 1X8, Canada; e-mail: walter.kahr@sickkids.ca.
References
- 1.Cox K, Price V, Kahr WH. Inherited platelet disorders: a clinical approach to diagnosis and management. Expert Rev Hematol. 2011;4(4):455–472. doi: 10.1586/ehm.11.41. [DOI] [PubMed] [Google Scholar]
- 2.Nurden A, Nurden P. Advances in our understanding of the molecular basis of disorders of platelet function. J Thromb Haemost. 2011;9(Suppl 1):76–91. doi: 10.1111/j.1538-7836.2011.04274.x. [DOI] [PubMed] [Google Scholar]
- 3.Italiano JE, Jr, Lecine P, Shivdasani RA, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. 1999;147(6):1299–1312. doi: 10.1083/jcb.147.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel SR, Hartwig JH, Italiano JE., Jr The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest. 2005;115(12):3348–3354. doi: 10.1172/JCI26891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009;23(4):177–189. doi: 10.1016/j.blre.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heijnen HF, Debili N, Vainchencker W, Breton-Gorius J, Geuze HJ, Sixma JJ. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood. 1998;91(7):2313–2325. [PubMed] [Google Scholar]
- 7.King SM, Reed GL. Development of platelet secretory granules. Semin Cell Dev Biol. 2002;13(4):293–302. doi: 10.1016/s1084952102000599. [DOI] [PubMed] [Google Scholar]
- 8.Richardson J, Shivdasani R, Boers C, Hartwig J, Italiano JE., Jr Mechanisms of organelle transport and capture along proplatelets during platelet production. Blood. 2005;106:4066–4075. doi: 10.1182/blood-2005-06-2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Rusiniak ME, Chintala S, Gautam R, Novak EK, Swank RT. Murine Hermansky-Pudlak syndrome genes: regulators of lysosome-related organelles. Bioessays. 2004;26(6):616–628. doi: 10.1002/bies.20042. [DOI] [PubMed] [Google Scholar]
- 11.Wei ML. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19(1):19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15(1):22–29. doi: 10.1097/MOH.0b013e3282f2bcce. [DOI] [PubMed] [Google Scholar]
- 13.Raposo G, Marks MS. Melanosomes: dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 2007;8(10):786–797. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahr WH, Zheng S, Sheth PM, et al. Platelets from patients with the Quebec platelet disorder contain and secrete abnormal amounts of urokinase-type plasminogen activator. Blood. 2001;98(2):257–265. doi: 10.1182/blood.v98.2.257. [DOI] [PubMed] [Google Scholar]
- 15.Sheth PM, Kahr WH, Haq MA, Veljkovic DK, Rivard GE, Hayward CP. Intracellular activation of the fibrinolytic cascade in the Quebec platelet disorder. Thromb Haemost. 2003;90(2):293–298. doi: 10.1160/TH02-12-0323. [DOI] [PubMed] [Google Scholar]
- 16.Paterson AD, Rommens JM, Bharaj B, et al. Persons with Quebec platelet disorder have a tandem duplication of PLAU, the urokinase plasminogen activator gene. Blood. 2010;115(6):1264–1266. doi: 10.1182/blood-2009-07-233965. [DOI] [PubMed] [Google Scholar]
- 17.Nurden AT, Nurden P. The gray platelet syndrome: clinical spectrum of the disease. Blood Rev. 2007;21(1):21–36. doi: 10.1016/j.blre.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 18.White JG. Ultrastructural studies of the gray platelet syndrome. Am J Pathol. 1979;95(2):445–462. [PMC free article] [PubMed] [Google Scholar]
- 19.Kahr WH, Hinckley J, Li L, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. 2011;43(8):738–740. doi: 10.1038/ng.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunay-Aygun M, Falik-Zaccai TC, Vilboux T, et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet alpha-granules. Nat Genet. 2011;43(8):732–734. doi: 10.1038/ng.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albers CA, Cvejic A, Favier R, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. 2011;43(8):735–737. doi: 10.1038/ng.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gissen P, Johnson CA, Morgan NV, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36(4):400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 23.Lo B, Li L, Gissen P, et al. Requirement of VPS33B, a member of the Sec1/Munc18 protein family, in megakaryocyte and platelet α-granule biogenesis. Blood. 2005;106(13):4159–4166. doi: 10.1182/blood-2005-04-1356. [DOI] [PubMed] [Google Scholar]
- 24.Kim SM, Chang HK, Song JW, Koh H, Han SJ. Agranular platelets as a cardinal feature of ARC syndrome. J Pediatr Hematol Oncol. 2010;32(4):253–258. doi: 10.1097/MPH.0b013e3181c3a8d0. [DOI] [PubMed] [Google Scholar]
- 25.Benet B, Lainey E, Fenneteau O, Baudouin V, Hurtaud-Roux MF. Usefulness of gray platelets observation in ARC syndrome. Ann Biol Clin (Paris) 2010;68(4):485–489. doi: 10.1684/abc.2010.0453. [DOI] [PubMed] [Google Scholar]
- 26.Toonen RF, Verhage M. Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol. 2003;13(4):177–186. doi: 10.1016/s0962-8924(03)00031-x. [DOI] [PubMed] [Google Scholar]
- 27.Kim BY, Kramer H, Yamamoto A, Kominami E, Kohsaka S, Akazawa C. Molecular characterization of mammalian homologues of class C Vps proteins that interact with syntaxin-7. J Biol Chem. 2001;276(31):29393–29402. doi: 10.1074/jbc.M101778200. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki T, Oiso N, Gautam R, et al. The mouse organellar biogenesis mutant buff results from a mutation in Vps33a, a homologue of yeast vps33 and Drosophila carnation. Proc Natl Acad Sci U S A. 2003;100(3):1146–1150. doi: 10.1073/pnas.0237292100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nickerson DP, Brett CL, Merz AJ. Vps-C complexes: gatekeepers of endolysosomal traffic. Curr Opin Cell Biol. 2009;21(4):543–551. doi: 10.1016/j.ceb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sevrioukov EA, He JP, Moghrabi N, Sunio A, Kramer H. A role for the deep orange and carnation eye color genes in lysosomal delivery in Drosophila. Mol Cell. 1999;4(4):479–486. doi: 10.1016/s1097-2765(00)80199-9. [DOI] [PubMed] [Google Scholar]
- 31.Lindmo K, Simonsen A, Brech A, Finley K, Rusten TE, Stenmark H. A dual function for Deep orange in programmed autophagy in the Drosophila melanogaster fat body. Exp Cell Res. 2006;312(11):2018–2027. doi: 10.1016/j.yexcr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Pulipparacharuvil S, Akbar MA, Ray S, et al. Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J Cell Sci. 2005;118(Pt 16):3663–3673. doi: 10.1242/jcs.02502. [DOI] [PubMed] [Google Scholar]
- 33.Sriram V, Krishnan KS, Mayor S. deep-orange and carnation define distinct stages in late endosomal biogenesis in Drosophila melanogaster. J Cell Biol. 2003;161(3):593–607. doi: 10.1083/jcb.200210166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akbar MA, Ray S, Kramer H. The SM protein Car/Vps33A regulates SNARE-mediated trafficking to lysosomes and lysosome-related organelles. Mol Biol Cell. 2009;20(6):1705–1714. doi: 10.1091/mbc.E08-03-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akbar MA, Tracy C, Kahr WH, Kramer H. The full-of-bacteria gene is required for phagosome maturation during immune defense in Drosophila. J Cell Biol. 2011;192(3):383–390. doi: 10.1083/jcb.201008119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cullinane AR, Straatman-Iwanowska A, Zaucker A, et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat Genet. 2010;42(4):303–312. doi: 10.1038/ng.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu GD, Salazar G, Zlatic SA, et al. SPE-39 family proteins interact with the HOPS complex and function in lysosomal delivery. Mol Biol Cell. 2009;20(4):1223–1240. doi: 10.1091/mbc.E08-07-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Licht C, Pluthero FG, Li L, et al. Platelet-associated complement factor H in healthy persons and patients with atypical HUS. Blood. 2009;114(20):4538–4545. doi: 10.1182/blood-2009-03-205096. [DOI] [PubMed] [Google Scholar]
- 39.Nishibori M, Cham B, McNicol A, Shalev A, Jain N, Gerrard JM. The protein CD63 is in platelet dense granules, is deficient in a patient with Hermansky-Pudlak syndrome, and appears identical to granulophysin. J Clin Invest. 1993;91(4):1775–1782. doi: 10.1172/JCI116388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwertz H, Koster S, Kahr WH, et al. Anucleate platelets generate progeny. Blood. 2010;115(18):3801–3809. doi: 10.1182/blood-2009-08-239558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNicol A, Israels SJ. Platelet dense granules: structure, function and implications for haemostasis. Thromb Res. 1999;95(1):1–18. doi: 10.1016/s0049-3848(99)00015-8. [DOI] [PubMed] [Google Scholar]
- 42.Weyrich AS, Schwertz H, Kraiss LW, Zimmerman GA. Protein synthesis by platelets: historical and new perspectives. J Thromb Haemost. 2009;7(2):241–246. doi: 10.1111/j.1538-7836.2008.03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Italiano JE, Jr, Richardson JL, Patel-Hett S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111(3):1227–1233. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sehgal S, Storrie B. Evidence that differential packaging of the major platelet granule proteins von Willebrand factor and fibrinogen can support their differential release. J Thromb Haemost. 2007;5(10):2009–2016. doi: 10.1111/j.1538-7836.2007.02698.x. [DOI] [PubMed] [Google Scholar]
- 45.Kamykowski J, Carlton P, Sehgal S, Storrie B. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet alpha-granules. Blood. 2011;118(5):1370–1373. doi: 10.1182/blood-2011-01-330910. [DOI] [PubMed] [Google Scholar]
- 46.van Nispen tot Pannerden H, de Haas F, Geerts W, Posthuma G, van Dijk S, Heijnen HF. The platelet interior revisited: electron tomography reveals tubular alpha-granule subtypes. Blood. 2010;116(7):1147–1156. doi: 10.1182/blood-2010-02-268680. [DOI] [PubMed] [Google Scholar]
- 47.Nightingale TD, White IJ, Doyle EL, et al. Actomyosin II contractility expels von Willebrand factor from Weibel-Palade bodies during exocytosis. J Cell Biol. 2011;194(4):613–629. doi: 10.1083/jcb.201011119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furie B, Furie BC, Flaumenhaft R. A journey with platelet P-selectin: the molecular basis of granule secretion, signalling and cell adhesion. Thromb Haemost. 2001;86(1):214–221. [PubMed] [Google Scholar]
- 49.Rosa JP, George JN, Bainton DF, Nurden AT, Caen JP, McEver RP. Gray platelet syndrome. Demonstration of alpha granule membranes that can fuse with the cell surface. J Clin Invest. 1987;80(4):1138–1146. doi: 10.1172/JCI113171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith MP, Cramer EM, Savidge GF. Megakaryocytes and platelets in alpha-granule disorders. Baillieres Clin Haematol. 1997;10(1):125–148. doi: 10.1016/s0950-3536(97)80054-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.