

Abstract
The expression of acid ceramidase (AC) – a cysteine amidase that hydrolyses the proapoptotic lipid ceramide – is abnormally high in several human tumors, which is suggestive of a role in chemoresistance. Available AC inhibitors lack, however, the potency and drug-likeness necessary to test this idea. Here we show that the antineoplastic drug carmofur, which is used in the clinic to treat colorectal cancers, is a potent AC inhibitor and that this property is essential to its anti-proliferative effects. Modifications in the chemical scaffold of carmofur yield new AC inhibitors that act synergistically with standard antitumoral drugs to prevent cancer cell proliferation. These findings identify AC as an unexpected target for carmofur, and suggest that this molecule can be used as starting point for the design of novel chemosensitizing agents.
In addition to their roles in cell membrane structure and dynamics, sphingolipids serve important signaling functions in the control of cell growth and differentiation1. Ceramide, a key member of this lipid class, has attracted particular attention for its contributions to the replication and differentiation of neoplastic cells2. In several types of human tumors, ceramide levels are lower than in normal tissues, and are inversely correlated with the degree of malignant progression3,4. Furthermore, various tumor-suppressing signals stimulate the production of ceramide, which has been shown in turn to promote apoptosis of cancer cells3,4. These data suggest that enzyme pathways involved in controlling intracellular ceramide levels might offer potential new targets for antineoplastic therapy5.
Acid ceramidase (AC, also known as N-acylsphingosine amidohydrolase-1, ASAH-1) is a cysteine amidase that catalyzes the hydrolysis of ceramide into sphingosine and fatty acid6. AC is involved in the regulation of ceramide levels in cells and modulates the ability of this lipid messenger to influence the survival, growth and death of tumor cells4,5. Consistent with this possibility, AC is abnormally expressed in various types of human cancer (e.g., prostate, head and neck, and colon) and serum AC levels are elevated in melanoma patients relative to control subjects7. Moreover, AC over-expression renders cells more resistant to pharmacological induction of apoptosis8,9, while inhibition of AC activity sensitizes tumor cells to the effects of antineoplastic agents and radiation9.
Several structural analogs of ceramide have been disclosed, which inhibit AC activity in vitro. These include oleoylethanolamide (also called N-oleylethanolamine)10, (1S,2R)-D-erythro-2-(N-myristoylamino)-1-phenyl-1-propanol (D-e-MAAP)11 and its derivative (1R, 2R)-2-(N-tetradecanoylamino)-1-(4-nitrophenyl)-1,3-propanediol (B-13)12. Though useful experimentally, these compounds suffer from various limitations, including low inhibitory potency and inadequate activity in vivo5. In the present study, we show that the antineoplastic drug, carmofur, which is used in the clinic to treat colorectal cancers13,14,15, is a potent in vivo active inhibitor of intracellular AC activity. We further show that AC inhibition plays an essential role in the anti-proliferative effects of carmofur, and that modifications in the chemical scaffold of this molecule yield new AC inhibitors that act synergistically with standard antitumoral drugs to prevent cancer cell proliferation.
Results
Carmofur is a highly potent AC inhibitor
We screened a commercial chemical library and identified carmofur (1-hexylcarbamoyl-5-fluorouracil) (Table 1) – a 5-fluorouracil (5-FU)-releasing drug used to treat colorectal cancer13,14 – as a highly potent inhibitor of rat recombinant AC. Carmofur inhibited AC activity with a median effective concentration (IC50) of 29 ± 5 nM (mean ± standard error of the mean, s.e.m.; n = 4), whereas 5-FU had no such effect (IC50>1 mM) (Figure 1A). Kinetic studies showed that carmofur decreased the maximal catalytic velocity of AC without influencing its Michaelis-Menten constant (Figure 1B and Table 2). AC inhibition by carmofur developed slowly, with a time to peak of approximately 45 min (Figure 1C) and was significantly, albeit incompletely reversed following 12-h dialysis (Figure 1D). Treating cultures of human colon adenocarcinoma SW403 cells with carmofur caused a concentration- and time-dependent reduction in AC activity (Figure 2A, B). This effect was accompanied by intracellular accumulation of various ceramide species, including cer(d18:1/14:0), cer(d18:1/16:0) and cer(d18:1/18:0), which were identified and quantified by liquid chromatography/mass spectrometry (LC/MS) (Figure 2C and Supplementary Table S1). By contrast, incubation of SW403 cells with 5-FU (3 μM, 3 h) did not cause any detectable changes in AC activity or ceramide content (Figure 2A–C). Similar results were obtained with LNCaP cells, an androgen-sensitive human prostate adenocarcinoma cell line that overexpresses AC8 (Figure 2D–F and Supplementary Table S2). Furthermore, systemic administration of carmofur (10 or 30 mg-kg−1, intraperitoneal, i.p.) to mice produced a dose-dependent inhibition of AC activity in various tissues, including lungs (Figure 3A) and brain cortex (Figure 3B). As seen with cancer cells in cultures, AC inhibition was associated with increases in tissue ceramide content (Figure 3C,D). By contrast, in vivo administration of 5-FU (30 mg-kg−1, i.p.) had no statistically detectable effect on AC activity (Figure 3A). The ceramide levels measured by our LC/MS assay are consistent with those reported in the literature, when analyses are conducted using LC/MS16,17. Higher ceramide levels have been reported, however, when utilizing different analytical methods16,18. The results indicate that carmofur inhibits AC activity and elevates tissue ceramide levels, both in vitro and in vivo, through a mechanism that is independent of 5-FU formation.
Table 1. General structure and inhibitory potencies of N-hexyl-2,4-dioxo-pyrimidine-1-carboxamides against rat AC and human thymidylate synthase (h-TS). IC50 values are expressed as mean ± s.e.m. of 3 determinations.
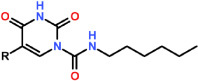 | |||
|---|---|---|---|
| Compound | R | r-AC IC50 (nM) | h-TS IC50 (µM) |
| Carmofur | F | 29 ± 5 | 1212 ± 190 |
| (1) ARN082 | Cl | 67 ± 5 | 496 ± 82 |
| (2) ARN080 | H | 426 ± 104 | 196 ± 27 |
| (3) ARN081 | CH3 | 1500 ± 125 | 762 ± 123 |
| (4) ARN398 | CF3 | 12 ± 2 | 1343 ± 268 |
Figure 1. Carmofur is a potent non-competitive inhibitor of AC activity.

(A) Effects of carmofur ( , n = 3) and 5-fluorouracil (5-FU,
, n = 3) and 5-fluorouracil (5-FU,  , n = 3) on AC activity (rat recombinant).(B) Michaelis-Menten analysis of the AC reaction in the presence of vehicle (
, n = 3) on AC activity (rat recombinant).(B) Michaelis-Menten analysis of the AC reaction in the presence of vehicle ( , DMSO 1%, n = 3) or carmofur (
, DMSO 1%, n = 3) or carmofur ( , 20 nM or
, 20 nM or  , 60 nM; n = 3). (C) Effects of pre-incubation time on AC inhibition by carmofur (60 nM). (D) Overnight dialysis of AC in the presence of vehicle (open bars) or carmofur (200 nM, closed bars). Pre: before dialysis; post: after dialysis. Results are expressed as mean ± s.e.m. of three independent experiments performed in triplicate. *p<0.05, ***p<0.001 vs vehicle; °°°p<0.001 vs carmofur preincubation, two-way ANOVA followed by Bonferroni's test.
, 60 nM; n = 3). (C) Effects of pre-incubation time on AC inhibition by carmofur (60 nM). (D) Overnight dialysis of AC in the presence of vehicle (open bars) or carmofur (200 nM, closed bars). Pre: before dialysis; post: after dialysis. Results are expressed as mean ± s.e.m. of three independent experiments performed in triplicate. *p<0.05, ***p<0.001 vs vehicle; °°°p<0.001 vs carmofur preincubation, two-way ANOVA followed by Bonferroni's test.
Table 2. Michaelis-Menten analysis of rat recombinant AC inhibition in the presence of vehicle (DMSO, 1%) or carmofur (20 or 60 nM). Results are expressed as mean ± s.e.m. (n = 3). **p<0.01, ***p<0.001 vs vehicle, one-way ANOVA followed by Bonferroni's test.
| Carmofur (nM) | 0 | 20 | 60 |
|---|---|---|---|
| Km (μM) | 13.2 ± 1.6 | 18.2 ± 2.4 | 22.3 ± 6.8 |
| Vmax (pmol/min/mg) | 317 ± 15.6 | 246.4 ± 6.8** | 119.4 ± 6.7*** |
Figure 2. Carmofur inhibits AC and increases ceramide levels in human SW403 and LNCaP cells.

Effects of carmofur (closed bars), 5-FU (hatched bars) or vehicle (0.1% DMSO in DMEM, open bars) on AC activity and ceramide levels in SW403 (A–C) and LNCaP (D–F) cells.(A, D) Intact cells were treated with carmofur at different concentrations (0.3–10 µM), 5-FU (3 µM) or vehicle and AC activity was measured 3 h later in cell lysates. (B, E) Intact cells were exposed to carmofur (3 µM, closed bars), 5-FU (3 µM, inset) or vehicle (open bars) and AC activity was measured after various time intervals. (C, F) Effects of a 3-h incubation with carmofur (3 µM), 5-FU (3 µM) or vehicle on ceramide levels. Results are expressed as mean ± s.e.m. (n = 3–6), with each assay performed in duplicate. *p<0.05, **p<0.01, ***p<0.001 vs vehicle, °p<0.05, °°p<0.01, °°°p<0.001 vs 5-FU Student's t test or one-way ANOVA followed by Tukey's test.
Figure 3. Carmofur inhibits AC and increases ceramide levels in mice.
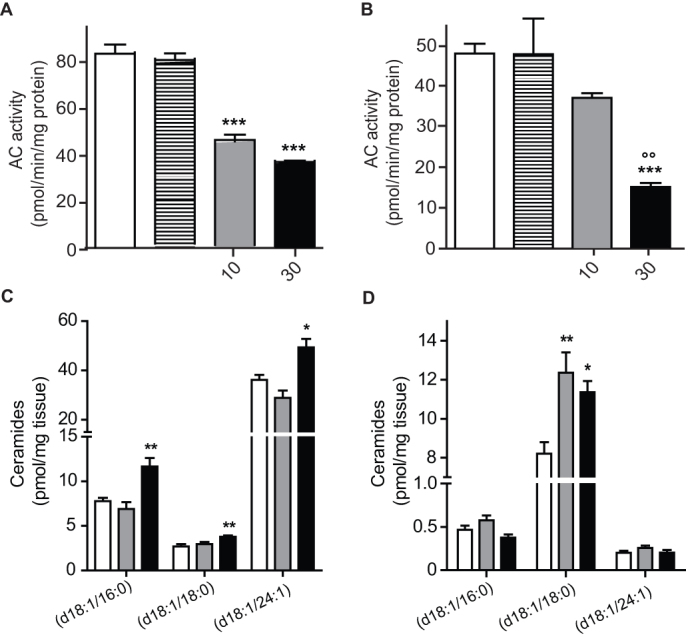
Effects of carmofur (closed bars), 5-FU (hatched bars) or vehicle (15% polyethylene glycol, 15% Tween80, 70% saline, open bars) on AC activity and ceramide levels in mouse tissues (lungs and cerebral cortex).(A–B) AC activity measured ex vivo 2 h after intraperitoneal injection of carmofur (10 mg-kg−1, shaded bars; 30 mg-kg−1, closed bars), 5-FU (30 mg-kg−1, hatched bars) or vehicle in lungs (A) and brain cortex (B). (C−D) Ceramide levels in (C) lungs and (D) brain cortex. Results are expressed as mean ± s.e.m. (n = 6). *p<0.05, **p<0.01, ***p<0.001 vs vehicle, one-way ANOVA followed by Tukey's test.
AC inhibition contributes to the anti-proliferative effects of carmofur
To test whether AC inhibition contributes to the anti-proliferative properties of carmofur, we examined the effects of this compound on human embryonic kidney 293 (HEK 293) cells that were genetically modified to stably overexpress rat AC (Figure 4A). In the absence of pharmacological treatment, AC-overexpressing HEK 293 cells contained substantially reduced levels of cer(d18:1/16:0) (Figure 4B) and proliferated more rapidly than did native control cells (Figure 4C). Importantly, when exposed for 24 h to carmofur (1–100 µM), AC-overexpressing HEK 293 cells showed a markedly reduced ceramide accumulation (Figure 4D) and cell death (Figure 4E) relative to control cells subjected to the same treatment: the median effective concentration (EC50) of carmofur was 50-fold higher in AC-overexpressing cells than native cells (66 µM vs 1.4 µM, n = 4, Figure 4E). A plausible interpretation of these experiments is that AC inhibition is an important determinant of carmofur-induced cytotoxicity. We next examined whether inhibition of ceramide synthesis via the de novo pathway might prevent the cytotoxic effect of carmofur. To this aim we measured the viability of HEK 923 cells exposed to carmofur, fumonisin B1 (FB1, a ceramide synthase inhibitor) and a combination of carmofur plus FB1. The results show that FB1 does not decrease carmofur induced cell death (Supplementary Figure S1). The lack of effect of FB1 is likely due to the existence of multiple pathways of ceramide biosynthesis (e.g. sphingomyelinase activation)19.
Figure 4. AC overexpression in HEK 293 cells confers resistance to carmofur.
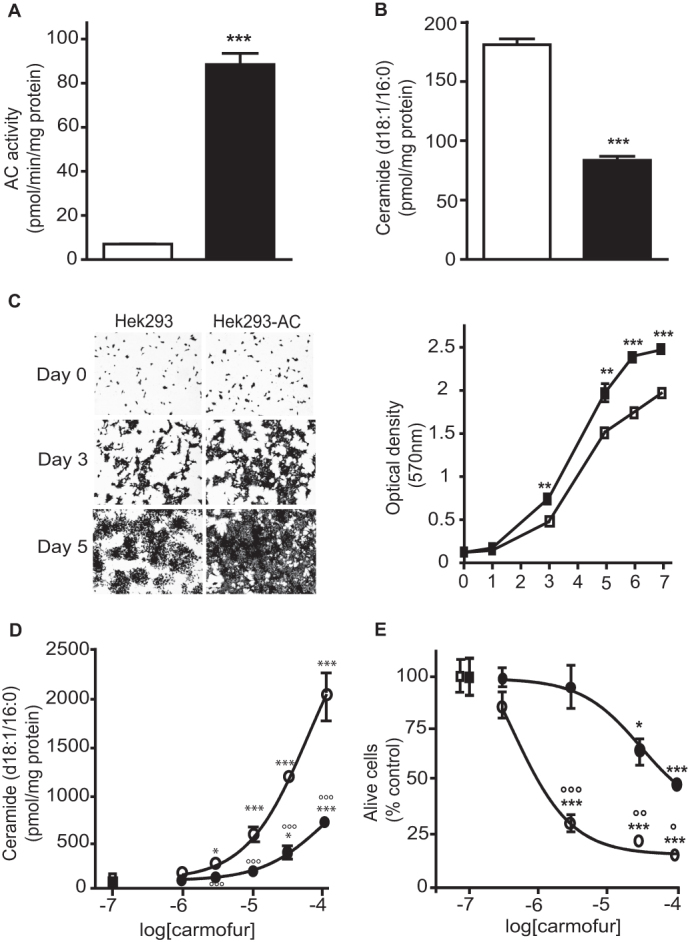
(A) AC activity and (B) ceramide (d18:1/16:0) levels in native HEK 293 cells (open bars) or AC-over-expressing HEK 293 cells (closed bars).(C) Representative images and quantification of cell proliferation assessed by Crystal violet staining. Concentration-dependent effects of carmofur on (D) ceramide (d18:1/16:0) levels and (E) cell viability evaluated by Trypan blue exclusion [ untreated HEK 293,
untreated HEK 293,  HEK 293 treated with carmofur (3 µM),
HEK 293 treated with carmofur (3 µM),  untreated HEK 293-AC,
untreated HEK 293-AC,  HEK 293-AC treated with carmofur (3 µM)]. Results are expressed as mean ± s.e.m. (n = 4). *p<0.05, **p<0.01, ***p<0.001 vs vehicle, °p<0.05, °°p<0.001, °°°p<0.001 vs carmofur on HEK 293. Student's t test or two-way ANOVA followed by Tukey's test.
HEK 293-AC treated with carmofur (3 µM)]. Results are expressed as mean ± s.e.m. (n = 4). *p<0.05, **p<0.01, ***p<0.001 vs vehicle, °p<0.05, °°p<0.001, °°°p<0.001 vs carmofur on HEK 293. Student's t test or two-way ANOVA followed by Tukey's test.
Identification of novel AC inhibitors
Carmofur releases 5-FU, which blocks tumor cell proliferation by inhibiting the DNA-synthesizing enzyme thymidylate synthetase13. Therefore, to further evaluate the contribution of AC inhibition to the anti-proliferative effects of carmofur, we synthesized a small set of carmofur derivatives that were rendered unable to release 5-FU through replacement of the fluorine atom at the 5 position of the pyrimidine ring with one of several substituent groups (Table 1). The new compounds inhibited AC activity with potencies that were markedly influenced by the stereo-electronic properties of the 5-substituent (Table 1, Figure 5A). Replacing fluorine with chlorine (compound 1, ARN082) or hydrogen (2, ARN080) caused a decrease in potency, while substitution with an electron-donating methyl group (3, ARN081) resulted in an almost complete loss of inhibitory activity (Table 1). On the other hand, replacement of fluorine with a strongly electron-withdrawing trifluoromethyl group yielded the highly potent AC inhibitor 4 (ARN398) (Table 1, Figure 5A). The new compounds did not affect human thymidylate synthetase activity (Table 1). LC/MS analyses showed that both ARN080 and ARN398 were subject to rapid degradation when incubated in mouse plasma at 37°C. ARN080 displayed an in vitro plasma half-life time (t1/2) of 3.5 min (Supplementary Figure S2); nevertheless, when administered systemically in mice at the doses of 10 and 30 mg-kg−1 (i.p.), ARN080 substantially reduced AC activity in lungs and brain cortex (Supplementary Figure S3), indicating that it was able to engage AC in vivo. ARN398 was degraded in plasma even more rapidly than ARN080 (t1/2 less than 1 min) and was not further investigated. The results identify the 5-substituted pyrimidine, ARN080, as a prototype for a new class of inhibitors of intracellular AC activity.
Figure 5. Pharmacological profile of novel AC inhibitors.
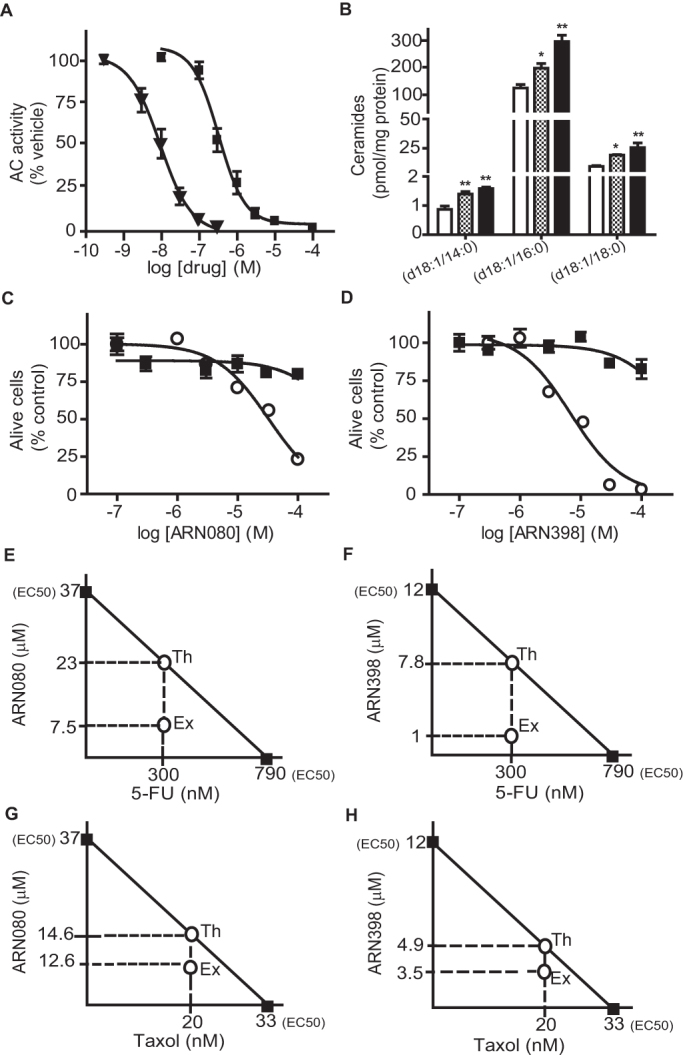
(A) Effects of ARN080 ( , n = 3) and ARN398 (
, n = 3) and ARN398 ( , n = 3) on rat recombinant AC activity.(B) Effects of a 3-h incubation with ARN080 (3 µM, dotted bars), ARN398 (3 µM, closed bars) or vehicle (open bars) on ceramide levels in SW403 cells. (C−D) Effects of single (
, n = 3) on rat recombinant AC activity.(B) Effects of a 3-h incubation with ARN080 (3 µM, dotted bars), ARN398 (3 µM, closed bars) or vehicle (open bars) on ceramide levels in SW403 cells. (C−D) Effects of single ( ) or multiple (
) or multiple ( ) exposure to ARN080 or ARN398 on SW403 cell viability. Isobolographic analyses of data obtained after multiple exposure of SW403 cells to (E) ARN080, 5-FU, or a combination of the two; (F) ARN398, 5-FU, or a combination of the two; (G) ARN080, taxol, or a combination of the two; (H) ARN398, taxol, or a combination of the two. Th, IC50 values expected if the effects were additive; Ex, experimental values actually obtained. Results are expressed as mean ± s.e.m. (n = 3), with each experiment performed twice. *p<0.05, **p<0.01, vs vehicle, one-way ANOVA followed by Tukey's test.
) exposure to ARN080 or ARN398 on SW403 cell viability. Isobolographic analyses of data obtained after multiple exposure of SW403 cells to (E) ARN080, 5-FU, or a combination of the two; (F) ARN398, 5-FU, or a combination of the two; (G) ARN080, taxol, or a combination of the two; (H) ARN398, taxol, or a combination of the two. Th, IC50 values expected if the effects were additive; Ex, experimental values actually obtained. Results are expressed as mean ± s.e.m. (n = 3), with each experiment performed twice. *p<0.05, **p<0.01, vs vehicle, one-way ANOVA followed by Tukey's test.
Chemosensitizing effects of AC inhibitors
A single treatment of SW403 cells with ARN080 or ARN398 (3 μM, 3 h) increased ceramide levels (Figure 5B), but did not result in any statistically detectable change in cell viability (Figure 5C, D). Cell viability was reduced, however, when the compounds were applied three times over a period of 72 h (once every 24 h) (Figure 5C, D). Under the latter conditions, ARN080 and ARN398 decreased SW403 cell viability with EC50 values of 12.6 ± 3 µM and 37 ± 2.6 µM, respectively (n = 3). The cytotoxic potency of 5-FU was also slightly enhanced when using this prolonged exposure protocol [EC50 = 0.79 ± 0.18 µM (n = 2); compare to EC50 = 1.36 ± 0.06 µM (n = 3) in the single-exposure protocol] (Supplementary Figure S4). The weak and gradual effects of ARN080 and ARN398 in SW403 cells (Figure 5C,D) were clearly distinguishable from the rapid and potent anti-proliferative response elicited by carmofur (Supplementary Figure S4), implying that AC blockade might be most effective when combined with other cytotoxic mechanisms (in the case of carmofur, 5-FU release). To test whether AC inhibition potentiates the effects of 5-FU in a synergistic manner, we incubated SW403 cells with a sub-effective concentration of 5-FU (0.3 µM) along with varying concentrations of ARN080 or ARN398 (0.1–100 µM). The compounds were added to the cultures once every 24 h over a period of 72 h and the effects of the combined treatments were evaluated using isobolographic analyses20. The analyses showed that ARN080 and ARN398 acted synergistically with 5-FU to reduce SW403 cell viability (Figure 5E,F). Indeed, experimental EC50 values were 1.0 µM for the combination ARN398 plus 5-FU and 7.5 µM for the combination ARN080 plus 5-FU; these values were markedly lower than the theoretical EC50s calculated using the isobole equation (7.8 µM and 23.0 µM, respectively; Figure 5E,F). Super-additive effects of ARN080 and ARN398 similar to those seen with 5-FU were also observed with taxol, a chemotherapeutic agent that acts by interfering with microtubule stability (Figure 5G,H). These findings are consistent with available data suggesting that AC blockade sensitizes cancer cells to the actions of cytotoxic agents and radiation therapy9,21,22,23.
Discussion
In the present report, we identify carmofur as a highly potent inhibitor of AC activity, and demonstrate that AC blockade plays an important role in the anti-proliferative effects of this chemotherapeutic agent. We further show that the chemical scaffold of carmofur provides a viable starting point for the discovery of novel AC inhibitors that act synergistically with other antineoplastic drugs, such as 5-FU and taxol, to stop the proliferation of cancer cells in vitro. By making cells in AC-over-expressing tumors more sensitive to the actions of chemotherapy or radiotherapy, future AC inhibitors designed on the 5-substituted pyrimidine scaffold might increase the efficacy and decrease the potential side effects of those treatments.
The anticancer properties of carmofur have been attributed to the ability of this agent to generate 5–FU13,14,24,25,26, a pyrimidine analog that inhibits DNA synthesis in tumor cells by blocking thymidylate synthase27. Nevertheless, the lack of positive correlation between carmofur-induced inhibition of thymidylate synthetase activity and cell proliferation is suggestive of a more complex mechanism of action14. This idea finds additional support in data indicating that carmofur is toxic for human cancer cells that have developed resistance to 5-FU28. In the present study, we show that AC overexpression (i) accelerates proliferation in HEK 293 cells, and (ii) renders these cells markedly resistant to the cytotoxic effects of carmofur. These results suggest that the antitumoral properties of carmofur might result from a concomitant action of this compound on DNA synthesis (caused by 5-FU release) and AC inhibition.
To further probe the role of AC inhibition in the anti-tumoral effects of carmofur, we synthesized a set of carmofur derivatives that lack the ability to block thymidylate synthetase, either directly or through 5-FU release. One of the new molecules, ARN080, inhibited AC activity both in vitro (IC50 = 426 ± 104 nM) and in vivo (median inhibitory dose, ID50 ~ 10 mg-kg−1, i.p.), and acted synergistically with two different antineoplastic drugs, 5-FU and taxol, to reduce proliferation of SW403 human colon carcinoma cells. Consistent with these findings, previous work has shown that blocking ceramide metabolism potentiates the cytotoxic effects of antineoplastic agents21,23,29. For example, the ceramide analog B13, which inhibits AC in vitro with an IC50 of approximately 10 μM in keratinocytes30, induces apoptosis in metastatic human colon cancer cell lines31 and sensitizes tumors to the effects of radiation in a model of xenografted androgen-insensitive prostate cancer32. Similarly, the AC inhibitor DM102 [(2R,3Z)-N-(1-hydroxyoctadec-3-en-2-yl)pivaloylamide] acts in synergy with fenretinide [N-(4-hydroxyphenyl) retinamide] to decrease prostate cancer cell proliferation23.
In conclusion, our results identify carmofur, an antineoplastic drug currently used in the clinic to treat colorectal cancers, as the first highly potent inhibitor of intracellular AC. Our findings further indicate that the ability to interfere with AC activity is an essential component of the anti-proliferative effects of carmorfur, and that modifications in the chemical scaffold of this molecule yield AC inhibitors that might enhance the therapeutic efficacy of standard antitumoral drugs.
Methods
Animals and treatments
Male Swiss Webster mice (20–35 g, Charles River) were group-housed at room temperature on a 12 h light/dark cycle. Water and standard chow pellets were freely available. Drugs were dissolved in 15% polyethylene glycol, 15% Tween-80 and 70% saline (injection volume, 1 ml-kg−1; i.p.). Animals were killed by cervical dislocation 2 h after drug administration, tissues were collected, frozen in liquid nitrogen and stored at −80°C. Experiments were performed in an AAALAC-accredited facility at the University of California, Irvine (USA). They met the National Institutes of Health guidelines for the care and use of laboratory animals, and were approved by the University's Institutional Animal Care and Use Committee.
Chemical syntheses
Compounds were synthesized by reaction of an appropriate 2,4-dioxo-pyrimidine with hexyl isocyanate in a suitable solvent, as depicted in Supplementary Figure S5, according to either method A or B. Column chromatography was performed on pre-packed silica cartridges (2 g or 5 g) from Biotage or on glass columns using Merck silica gel 60 (230–400 mesh) as stationary phase. NMR spectra were recorded on a Bruker Avance III 400 system (400.13 MHz for 1H, and 100.62 MHz for 13C), equipped with a BBI inverse probe and Z-gradients, using deuterated solvents. Splitting parameters are designated as singlet (s), broad singlet (bs), doublet (d), triplet (t), and multiplet (m). NMR coupling constants (J) are in Hertz.
UPLC-MS analyses were run on a Waters ACQUITY UPLC-MS system consisting of a SQD (Single Quadrupole Detector) Mass Spectrometer equipped with an Electrospray Ionization interface and a Photodiode Array Detector. UV spectra acquisition range was 210–400 nm. Analyses were performed on an ACQUITY UPLC HSS T3 C18 column (50×2.1mmID, particle size 1.8 µm) with a VanGuard HSS T3 C18 pre-column (5×2.1mmID, particle size 1.8 µm). Mobile phase was either 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN–H2O (95:5) at pH 5. Electrospray ionization in positive and negative mode was applied. All final compounds showed ≥ 95% purity by NMR and UPLC-MS analysis.
Method A: The compounds were prepared based on literature procedures33. A properly substituted 2,4-dioxo-pyrimidine (1.0 g, 1.0 equiv) and hexyl isocyanate (1.5 equiv) were heated in dry DMSO (15 mL) at 50°C for 18 h. The reaction mixture was cooled to room temperature and quenched by addition of water. The precipitate was filtered under reduced pressure, and crystallised from refluxing ethanol (20 mL) to yield a white solid. Method B: A properly substituted 2,4-dioxo-pyrimidine (0.1 g, 1.0 equiv) and 4-dimethylaminopyridine (1.1 equiv) were dissolved in dry pyridine (5 mL). After 30 min, hexyl isocyanate (1.2 equiv) was added and the resulting solution was stirred under nitrogen at room temperature for 20 h. The solvent was evaporated under reduced pressure, and the crude residue was purified by silica gel column chromatography. Evaporation of the fractions containing the compound yielded the product as white solid.
5-chloro-N-hexyl-2,4-dioxo-pyrimidine-1-carboxamide (Compound 1, ARN082)
The compound was prepared according to method B. The final compound was obtained by column chromatography purification (petroleum ether/ethyl acetate 7:3 as eluent) in 31% yield. 1H NMR (DMSO-d6, 400 MHz): δ 0.86 (t, J = 6.8 Hz, 3H), 1.21–1.35 (m, 6H), 1.46–1.54 (m, 2H), 3.24–3.29 (m, 2H), 8.41 (s, 1H), 9.07 (t, J = 5.3 Hz, 1H), 12.24 (s, 1H). 13C NMR (101 MHz, DMSO) δ 13.85, 21.97, 25.81, 28.57, 30.82, 40.55, 109.55, 135.56, 149.21, 150.61, 158.78; MS (ES−): C11H16ClN3O3 requires 273, found 272.
N-hexyl-2,4-dioxo-pyrimidine-1-carboxamide (Compound 2, ARN080)
The compound was prepared according to method A in 28% yield. 1H NMR (DMSO-d6, 400 MHz): δ 0.89 (t, J = 7.2 Hz, 3H), 1.18–1.42 (m, 6H), 1.47–1.63 (m, 2H), 3.26–3.34 (m, 2H), 5.83 (d, J = 8.4 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 9.15 (t, J = 5.3 Hz, 1H), 11.75 (bs, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 13.8, 22.0, 25.8, 28.6, 30.8, 40.3, 103.4, 138.7, 149.9, 151.5, 162.8; MS (ES−): C11H17N3O3 requires 239, found 238.
N-hexyl-5-methyl-2,4-dioxo-pyrimidine-1-carboxamide (Compound 3, ARN081)
The compound was prepared according to method A in 35% yield. 1H NMR (DMSO-d6, 400 MHz): δ 0.86 (t, J = 6.8 Hz, 3H), 1.17–1.36 (m, 6H), 1.41–1.60 (m, 2H); 1.83 (d, J = 1.3 Hz, 3H), 3.23–3.28 (m, 2H), 7.88–8.31 (m, 1H), 9.15 (t, J = 5.7 Hz, 1H), 11.72 (bs, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 12.1, 13.8, 22.0, 25.8, 28.6, 30.8, 40.3, 111.1, 134.0, 150.1, 151.5, 163.5; MS (ES−): C12H19N3O3 requires 253, found 252.
5-trifluoromethyl-N-hexyl-2,4-dioxo-pyrimidine-1-carboxamide (Compound 4, ARN398)
The compound was prepared according to method B. The final compound was obtained by column chromatography purification (cyclohexane/ethyl acetate 10:1 as eluent) in 34% yield. 1H NMR (CDCl3, 400 MHz): δ 0.90 (t, J = 6.7 Hz, 3H), 1.16–1.49 (m, 6H), 1.49–1.75 (m, 2H), 3.33–3.58 (m, 2H), 8.10 (bs, 1H), 8.85–8.95 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 14.14, 22.56, 26.56, 31.33, 31.41, 41.71, 108.21, 121.27 (q, J = 270.7 Hz), 140.13, 148.48, 150.25, 157.41. MS (ES−): C12H16F3N3O3 requires 307, found 306.
Other chemicals
Carmofur and 5-FU were purchased from LKT Laboratories (St. Paul, MN, USA). All solvents and reagents were obtained from commercial suppliers and were used without further purification.
Plasma stability
Compounds were dissolved in mouse plasma pre-heated at 37°C (Tebu-bio, Le-Perray-en-Yvelines, France) to a final concentration of 0.1 mM, and incubated at 37°C for 1h with mild agitation. Samples (50 μl) were removed at various time points, diluted with acetonitrile (0.15 ml), spiked with warfarin (500 nM) as internal standard, stirred for 30 s and centrifuged at 18,000xg at 4°C for 5 min. Samples (2 µl) of the supernatants were injected into a Waters UPLC/MS system equipped with a TQD detector. The following conditions were used: Mobile Phase (A) 0.1% formic acid in water; (B) 0.1% formic acid in acetonitrile. Gradient: 0–0.5 min: 5% B, 0.5–2.5 min: 5–100% B, 2.5–3.0 min: 100% B, 3.1–3.5 min: 5% B. Flow rate 0.5 mL-min−1. The column was an Acquity UPLC BEH C18 1.7 µm 2.1×50mm equipped with a VanGuard BEH C18 1.7 µm pre-column. Temperature was set at 40°C. Both UV (260 nm) and ESI/MS traces were used for quantification. Data were fitted with a one-phase decay curve, using PRISM software suite (GraphPad Software Inc, USA).
Cells and treatments
SW403 cells were purchased from American Type Culture Collection (Manassas, VA) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum at 37°C and 5% CO2. Drugs were added in dimethylsulphoxide (DMSO, final concentration, 0.1%).
Recombinant acid ceramidase expression
Rat AC was cloned from a brain cDNA library using primers based on the sequence obtained from the National Center for Biotechnology Information (NCBI) database: 5′rAC (5′-GACCATGCTGGGCCGTAGT-3′) and 3′rAC (5′-CCAGCCTATACAAGGGTCT-3′). The PCR (High Fidelity PCR Master, Roche) product was subcloned into a pEF6-V5/His vector (Invitrogen) to construct a mammalian expression vector encoding V5/His-tagged rat AC. HEK293 cells were transfected with pEF6-rAC-V5/His using Super-Fect reagent (Qiagen) and screened with G418 (0.3 mg/mL).
Cell viability
Trypan blue assay: SW403 cells (2×105) were seeded onto 12-well plates the day before the experiments and incubated with appropriate concentrations of drugs in serum-free DMEM. Following treatment, cells were harvested, centrifuged at 800xg for 10 min and the pellets were suspended in PBS. Cell suspensions were diluted 1:1 with 0.4% Trypan Blue (Sigma), incubated for 1 min and viable cells were counted with a hemocytometer. Crystal violet assay: cells were stained with 0.4% Crystal violet in 50% MeOH for 20 min and washed with water. DMSO (0.1 ml per well) was added to dissolve cells and absorbance was measured at 570 nm.
Acid ceramidase activity
AC activity was measured as described in Supplementary Material and Methods34.
Tissue lipid analyses
Lipid analyses were conducted as described in Supplementary Material and Methods35.
Statistics
We assessed the interaction between drugs using a standard isobolographic analyses36,37. GraphPad Prism software (GraphPad Software, Inc., USA) was used for all other statistical analysis. Statistical significance of differences was assessed using Student's t test or one-way ANOVA followed by a Bonferroni's post hoc test for multiple comparisons. Two-way ANOVA was used to compare the means of data with two independent variables. Differences between groups were considered statistically significant at values of p < 0.05. Results are expressed as mean ± s.e.m.
Author Contributions
DP (Piomelli) and TB overviewed the project and supervised the work; NR and CS designed and performed experiments; CP and DP (Pizzirani) performed chemical synthesis; AA performed stability experiments; RL and MPC performed thymidylate synthase assays; DP (Piomelli), NR, TB wrote the manuscript with contributions from all coauthors.
Supplementary Material
Supplementary dataset
Acknowledgments
We thank Agnesa Avanesian, Dr. Ana Guijarro and Dr. Guillermo Moreno-Sanz for discussion and help with initial experiments. This work was partially supported by the National Institute on Drug Abuse (RC2 DA028902-01, to D.P.).
Footnotes
The following authors declare a financial interest: NR, CP, D Pizzirani, TB, and D Piomelli are inventors in patent applications, filed by the University of California Irvine and the Istituto Italiano di Tecnologia, which disclose AC inhibitors as anticancer agents.
References
- Zeidan Y. H. & Hannun Y. A. The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr. Mol. Med. 10, 454–466 (2010). [DOI] [PubMed] [Google Scholar]
- Furuya H., Shimizu Y. & Kawamori T. Sphingolipids in cancer. Cancer Metastasis Rev 30, 567–576 (2011). [DOI] [PubMed] [Google Scholar]
- Riboni L. et al. Ceramide levels are inversely associated with malignant progression of human glial tumors. Glia 39, 105–113 (2002). [DOI] [PubMed] [Google Scholar]
- Ogretmen B. & Hannun Y. A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 4, 604–616 (2004). [DOI] [PubMed] [Google Scholar]
- Canals D., Perry D. M., Jenkins R. W. & Hannun Y. A. Drug targeting of sphingolipid metabolism: sphingomyelinases and ceramidases. Br. J. Pharmacol. 163, 694–712 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault C. R., Obeid L. M. & Hannun Y. A. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv. Exp. Med. Biol. 688, 1–23 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. et al. Serum autoantibody profiling using a natural glycoprotein microarray for the prognosis of early melanoma. J Proteome Res 9, 6044–6051 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelan R. S. et al. Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes Chromosomes Cancer 29, 137–146 (2000). [DOI] [PubMed] [Google Scholar]
- Mahdy A. E. et al. Acid ceramidase upregulation in prostate cancer cells confers resistance to radiation: AC inhibition, a potential radiosensitizer. Mol. Ther. 17, 430–438 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita M., Willians M., Dulaney J. T. & Moser H. W. Ceramidase and ceramide synthesis in human kidney and cerebellum. Description of a new alkaline ceramidase. Biochim. Biophys. Acta 398, 125–131 (1975). [DOI] [PubMed] [Google Scholar]
- Bielawska A. et al. (1S,2R)-D-erythro-2-(N-myristoylamino)-1-phenyl-1-propanol as an inhibitor of ceramidase. J. Biol. Chem. 271, 12646–12654 (1996). [DOI] [PubMed] [Google Scholar]
- Bielawska A. et al. Novel analogs of D-e-MAPP and B13. Part 2: signature effects on bioactive sphingolipids. Bioorg. Med. Chem. 16, 1032–1045 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T. et al. Antitumor activity of fluoropyrimidines and thymidylate synthetase inhibition. Jpn. J. Cancer Res. 82, 476–482 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M. et al. [Inhibition of thymidylate synthetase and antiproliferative effect by 1-hexylcarbamoyl-5-fluorouracil]. Gan. To Kagaku Ryoho 15, 3109–3113 (1988). [PubMed] [Google Scholar]
- Watanabe M. et al. Randomized trial of the efficacy of adjuvant chemotherapy for colon cancer with combination therapy incorporating the oral pyrimidine 1-hexylcarbamoyl-5-fluorouracil. Langenbecks Arch. Surg. 391, 330–337 (2006). [DOI] [PubMed] [Google Scholar]
- Nikolova-Karakashian M. N. & Rozenova K. A. Ceramide in stress response. Adv. in Exp. Med. Biol. 688, 88–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland W. L. et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 17, 55–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichgraber V. et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 14, 382–391 (2008). [DOI] [PubMed] [Google Scholar]
- Hannun Y. A. & Obeid L. M. Many ceramides. J. Biol. Chem. 286, 27855–27862 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabovsky Y. & Tallarida R. J. Isobolographic analysis for combinations of a full and partial agonist: curved isoboles. J. Pharmacol. Exp. Ther. 310, 981–986 (2004). [DOI] [PubMed] [Google Scholar]
- Maurer B. J., Melton L., Billups C., Cabot M. C. & Reynolds C. P. Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl)retinamide and modulators of ceramide metabolism. J. Natl. Cancer Inst. 92, 1897–1909 (2000). [DOI] [PubMed] [Google Scholar]
- Roberts C. G., Gurisik E., Biden T. J., Sutherland R. L. & Butt A. J. Synergistic cytotoxicity between tamoxifen and the plant toxin persin in human breast cancer cells is dependent on Bim expression and mediated by modulation of ceramide metabolism. Mol. Cancer Ther. 6, 2777–2785 (2007). [DOI] [PubMed] [Google Scholar]
- Gouaze-Andersson V. et al. Inhibition of acid ceramidase by a 2-substituted aminoethanol amide synergistically sensitizes prostate cancer cells to N-(4-hydroxyphenyl) retinamide. Prostate 71, 1064–1073 (2011). [DOI] [PubMed] [Google Scholar]
- Hoshi A., Iigo M., Nakamura A., Yoshida M. & Kuretani K. Antitumor activity of 1-hexylcarbamoyl-5-fluorouracil in a variety of experimental tumors. Gann 67, 725–731 (1976). [PubMed] [Google Scholar]
- Kobari T. et al. Metabolic fate of 1-hexylcarbamoyl-5-fluorouracil in rats. Xenobiotica 8, 547–556 (1978). [DOI] [PubMed] [Google Scholar]
- Iigo M., Nakamura A., Kuretani K. & Hoshi A. Metabolic fate of 1-hexylcarbamoyl-5-fluorouracil after oral administration in mice. Xenobiotica 10, 847–854 (1980). [DOI] [PubMed] [Google Scholar]
- Longley D. B., Harkin D. P. & Johnston P. G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 (2003). [DOI] [PubMed] [Google Scholar]
- Sato S., Ueyama T., Fukui H., Miyazaki K. & Kuwano M. [Anti-tumor effects of carmofur on human 5-FU resistant cells]. Gan. To Kagaku Ryoho 26, 1613–1616 (1999). [PubMed] [Google Scholar]
- Awad A. B., Barta S. L., Fink C. S. & Bradford P. G. beta-Sitosterol enhances tamoxifen effectiveness on breast cancer cells by affecting ceramide metabolism. Mol. Nutr. Food Res. 52, 419–426 (2008). [DOI] [PubMed] [Google Scholar]
- Raisova M. et al. Bcl-2 overexpression prevents apoptosis induced by ceramidase inhibitors in malignant melanoma and HaCaT keratinocytes. FEBS Lett. 516, 47–52 (2002). [DOI] [PubMed] [Google Scholar]
- Selzner M. et al. Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 61, 1233–1240 (2001). [PubMed] [Google Scholar]
- Samsel L. et al. The ceramide analog, B13, induces apoptosis in prostate cancer cell lines and inhibits tumor growth in prostate cancer xenografts. Prostate 58, 382–393 (2004). [DOI] [PubMed] [Google Scholar]
- Arnal-Herault C. et al. Constitutional self-organization of adenine-uracil-derived hybrid materials. Chemistry 13, 6792–6800 (2007). [DOI] [PubMed] [Google Scholar]
- Solorzano C. et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci U S A 106, 20966–20971 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snigdha S. et al. Dietary and behavioral interventions protect against age related activation of caspase cascades in the canine brain. PLoS One 6, e24652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida R. J. & Raffa R. B. The application of drug dose equivalence in the quantitative analysis of receptor occupation and drug combinations. Pharmacol. Ther. 127, 165–174 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida R. J. Interactions between drugs and occupied receptors. Pharmacol Ther 113, 197–209 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary dataset