

Abstract
The 18-nor (7), 21-nor (8) and 18,21-dinor (9) analogs of (20S)-1α,25-dihydroxy-2-methylene-19-norvitamin D3 (6, 2MD) were prepared by convergent syntheses. The known phosphine oxide 10 was coupled by the Wittig–Horner process with the corresponding C,D-fragments (13–15), obtained by a multi-step procedure from commercial vitamin D2. The goal of our studies was to examine the influence of removal of the methyl groups located at carbons 13 and 20 on the biological potency of 2MD in the hope of finding analogs with improved therapeutic profiles.
Replacement of the 20-methyl with hydrogen in 2-methylene-19-nor-(20S)-1α,25-dihydroxyvitamin D3 (2MD) did not affect binding to the rat vitamin D receptor and had little effect on transcription activity and on HL-60 differentiation. However, the mobilization of calcium from bone was largely eliminated while intestinal calcium transport remained strong. Curiously, removal of both the C-13-methyl and 20-methyl restored slightly the bone calcium mobilizing activity. Thus, the 21-nor analog of 2MD may provide a potent analog with a greater margin of safety than 2MD.
Keywords: Vitamin D analogs, 19-Norvitamin D, Vitamin D receptor, Calcemic activity
1. Introduction
1α,25-Dihydroxyvitamin D3 (1α,25-(OH)2D3, calcitriol, 1; Fig. 1) is the active hormonal form of vitamin D3.1–3 It not only regulates calcium and phosphorus homeostasis but promotes differentiation of various malignant cells,4–7 and suppresses autoimmune diseases.8 Such a broad range of diverse functions of the natural hormone 1 is mediated by its binding to the intracellular vitamin D receptor (VDR), belonging to the nuclear receptor superfamily.9 Therefore, a strong affinity of the vitamin D compound to VDR is of considerable importance to the functions of vitamin D. Formation of a complex between VDR and the ligand changes the conformation of the protein10 and influences its subsequent hetero-dimerization with retinoid X receptor (RXR) and recruitment of several coactivators allowing the transactivation process to begin.10,11 Many analogs of calcitriol have been prepared; most of them are described.12
Figure 1.

Chemical structures of 1α,25-dihydroxyvitamin D3 (calcitriol, 1) and its analogs.
In 1990, we reported the synthesis of the analog 3 of the natural hormone in which the exomethylene group at C-19 was removed. 13 During the subsequent years, similar 19-norvitamins were obtained in our and other laboratories.14 Another very promising structural change of the ring A consisted in a ‘shift’ of the exomethylene group from carbon 10 to carbon 2.15 Thus, the analog 5 exerted a more pronounced ability to elevate serum calcium from bone than 1α,25-(OH)2D3, and such calcemic in vivo activity was significantly enhanced in its epimer 6 with an ‘unnatural’ configuration at C-20. Further biological tests of this analog (2MD) showed its unique ability to induce bone formation.16,17 2MD (6) has proven anabolic for bone in in vivo studies but has a tendency to cause hypercalcemia because of its bone calcium mobilizing activity.15 Because of docking calculations, the idea that removal of the 20-methyl might reduce bone calcium mobilization occurred to us.18 We, therefore, prepared the 21-nor derivative of 2MD (8). As a control, we also removed the 13-methyl (7) or both the 20- and 13-methyl (9). The results are consistent with the idea that the 20-methyl of 2MD is central to its activity on bone calcium mobilization. The synthetic strategy consisted of the Wittig–Horner coupling of the known A-ring fragment, the phosphine oxide 10,14 with the corresponding bicyclic ketones 13–15 which, in turn, were prepared from the hydrindanols 11 and 12 (Fig. 2).
Figure 2.

The building blocks for the synthesis of 7, 8 and 9.
2. Results
2.1. Synthesis
Since the hydroxy compound 11 was previously obtained in our laboratory, by degradation of the commercial vitamin D2 16 followed by reconstruction of the side chain,19 we focused our efforts on the preparation of its 21-nor analog 12 (steroidal numbering). As a starting compound, we have chosen methylketone 17 (Scheme 1) prepared from vitamin D2 16 as described by Posner.20 For the removal of the acetyl substituent, we used the synthetic sequence elaborated by Mourino for the analogous compound.21 Thus, the Bayer–Villiger oxidation of 17 provided the acetate 18 which was hydrolyzed and the resulting alcohol 19 oxidized to the 17-ketone 20. Its subsequent Wittig reaction with the ylide, generated from the phosphonium salt 21,22 yielded a mixture of olefinic compounds. As expected,23 the isomer 22 with Z-geometry of the introduced double bond was a major product. Hydrogenation of the resulted olefinic mixture furnished the protected alcohol 23 which, following removal of the silyl group, gave the desired hydrindanol 12. This compound was a convenient precursor of both Grundmann ketones 14 and 15. Thus, the 8β-alcohol 12 was subjected to the catalytic oxidation with RuO4 which provided the expected hydroxy ketone 24.24 Protection of its tertiary hydroxyl as TES ether was the last step of the hydrindanone 14 synthesis.
Scheme 1.

Synthesis of the Grundmann ketone 14. Reagents: (i) m-CPBA, cyclohexane; (ii) MeONa, MeOH; (iii) PDC, PPTS, CH2Cl2; (iv) tert-BuOK, THF; (v) H2, Pd/C, AcOEt; (vi) CSA, n-BuOH; (vii) RuCl3 × H2O, NaIO4, CCl4, MeCN, H2O; (viii) TESOTf, 2,6-lutidine, CH2Cl2.
For the conversion of 12 to the 18-nor ketone 15, the removal of 13β-methyl group was necessary. The synthetic sequence leading to the 18-nor derivative was initially performed for the homologous compound 11 by applying the method we used satisfactorily in the synthesis of the analog 2.25 Thus, hydrindanol 11 was treated with tert-butyl nitrite and the formed ester 25 (Scheme 2) was irradiated with a UV lamp yielding, as a result of the Barton reaction,26 the E-oxime 27. The unstable primary photolysis product, 18-nitrite, was not isolated. Dehydration of 27 was achieved by its heating in acetic anhydride, resulting in acetylation and the subsequent elimination of the residues of acetic acid. The formed acetoxy nitrile 29 was hydrolyzed and the cyano group in the hydroxy nitrile 31 was removed by reduction with metallic potassium.27 The obtained hydrindanol 33 was subjected to oxidation with RuO4 and the resulted hydroxy ketone 35 was silylated to provide the 18-nor Grundmann ketone 13. By application of the analogous reaction sequence, as indicated in Scheme 2, the 21-nor compound 12 was converted into 18,21-dinor Grundmann ketone 15, thus accomplishing the synthesis of the ‘upper’ building blocks required for the Wittig–Horner coupling with the A-ring fragment. Retention of configuration of the stereogenic center at C-13 in the synthesized hydrindanols and hydrindanones was confirmed by circular dichroism (CD) data. Thus, circular dichroism curve of the hydroxy ketone 36, displaying two negative Cotton effects around 290 and 190 nm, showed close similarity to the CD curves recorded for the Grundmann ketone with a ‘natural’ 20R-configuration and its 18-nor analog.25
Scheme 2.

Synthesis of the Grundmann ketones 13 and 15. Reagents: (i) tert-BuONO, CHCl3; (ii) hm, PhH; IPA; (iii) Ac2O; (iv) MeONa, MeOH; (v) K, HMPA, tert-BuOH, Et2O; (vi) RuCl3 × H2O, NaIO4, CCl4, MeCN, H2O; (vii) TESOTf, 2,6-lutidine, CH2Cl2.
The final reaction between the anion, generated from the phosphine oxide 10 by phenyllithium, and the corresponding C,D-fragments 13–15 provided the protected vitamin D compounds 37–39 (Scheme 3) which, after removal of the silyl groups and purification by HPLC, afforded the target vitamin D analogs 7, 8 and 9, respectively.
Scheme 3.

Synthesis of the vitamin D analogs 7, 8 and 9. Reagents: (i) PhLi, THF; (ii) TBAF, molecular sieves 4 Å, THF for 37→7; CSA, n-BuOH for 38→8, and 39→9.
2.2. Biological evaluation
The synthesized vitamin Danalogs 7, 8 and 9 were tested for their ability to bind the full-length recombinant rat vitamin D receptor (rVDR). In agreement with our expectations, the competitive binding analysis shows (Table 1) that all compounds bind rVDR very effectively.
Table 1.
VDR binding properties,a HL-60 differentiating activities,b and transcriptional activitiesc of the vitamin D compounds 7, 8 and 9
| Compound | Compd no. | VDR binding
|
HL-60 differentiation
|
24-OHase transcription
|
|||
|---|---|---|---|---|---|---|---|
| Ki (M) | Ratio | EC50 (M) | Ratio | EC50 (M) | Ratio | ||
|
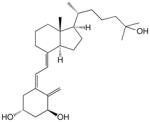
1 | 1 × 10−10 | 1 | 2 × 10−9 | 1 | 3 × 10−10 | 1 |
|
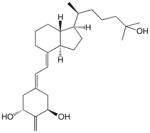
7 | 2 × 10−11 | 0.2 | 4 × 10−10 | 0.2 | 2 × 10−11 | 0.07 |
|
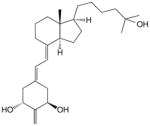
8 | 2 × 10−10 | 2 | 7 × 10−10 | 0.35 | 2 × 10−10 | 0.67 |
|
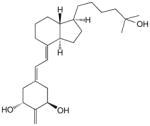
9 | 1 × 10−10 | 1 | 3 × 10−10 | 0.15 | 6 × 10−11 | 0.2 |
Competitive binding of 1α,25-(OH)2D3 (1) and the synthesized vitamin D analogs to the full-length recombinant rat vitamin D receptor. The Ki values are derived from dose–response curves and represent the inhibition constant when radiolabeled 1α,25-(OH)2D3 is present at 1 nM and a Kd of 0.2 nM is used. The binding ratio is the average ratio of the analog Ki to the Ki for 1α,25-(OH)2D3.
Induction of differentiation of HL-60 promyelocytes to monocytes by 1α,25-(OH)2D3 and the synthesized vitamin D analogs. Differentiation state was determined by measuring the percentage of cells reducing nitro blue tetrazolium (NBT). The EC50 values are derived from dose–response curves and represent the analog concentration capable of inducing 50% maturation. The differentiation activity ratio is the average ratio of the analog EC50 to the EC50 for 1α,25-(OH)2D3.
Transcriptional assay in rat osteosarcoma cells stably transfected with a 24-hydroxylase gene reporter plasmid. The EC50 values are derived from dose–response curves and represent the analog concentration capable of increasing the luciferase activity by 50%. The luciferase activity ratio is the average ratio of the EC50 for the analog to the EC50 for 1α,25-(OH)2D3. All the experiments were carried out in duplicate on at least two different occasions.
Studies on the cellular activity of the prepared compounds to induce the differentiation of human promyelocyte HL-60 cells into monocytes show that all vitamins are more potent (3–7 times) than the vitamin D hormone (Table 1).
The results of the transcriptional assay indicate that the analog 7 exhibits a significantly enhanced ability to induce transcription of vitamin D-responsive genes. When the 24-hydroxylase (CY-24) luciferase reporter gene system was used, 18,19-dinorvitamin 7 proved to be 15 times more active than 1α,25-(OH)2D3 (Table 1). The 19,21-dinorvitamin 8 and 18,19,21-trinorvitamin 9 have slightly higher (ca. 1.5 and 5 times, respectively) transcriptional potencies with respect to the native hormone 1.
For in vivo comparison, we have included Figure 3, a summary of the activity of 2MD. While all compounds showed excellent intestinal calcium transport activity, the change in bone calcium mobilization, by removing the 20-methyl group, was dramatic. The 21-nor (8) is essentially devoid of bone calcium mobilization activity (Fig. 4). Removal of C-13 methyl (7) reduced bone calcium mobilizing activity of 2MD (Fig. 5) to a level that is very similar to that of the native hormone. Removal of both the 18- and 21-methyl (9) resulted in a compound with activity on bone in between that observed for the 21-nor compound (8) and the 18-nor analog (7) (Fig. 6).
Figure 3.

Bone calcium mobilization and intestinal calcium transport activity of 2MD (6).
Figure 4.

Bone calcium mobilization and intestinal calcium transport activity of calcitriol (1) and the synthesized analog 8. Letters above the bars indicate statistically significant differences between treatment groups (p <0.05).
Figure 5.

Bone calcium mobilization and intestinal calcium transport activity of calcitriol (1) and the synthesized analog 7.
Figure 6.

Bone calcium mobilization and intestinal calcium transport activity of calcitriol (1) and the synthesized analog 9. Letters above the bars indicate statistically significant differences between treatment groups (p <0.05).
3. Discussion
A few years ago, we reported results of our docking experiments performed with 2-substituted vitamin D analogs and aimed at establishing how the binding pocket of the full-length ligand binding domain (LBD) of rVDR could accommodate these ligands.18 It was found that, in complexes of ligands possessing elevated BCM activity (such as the 2-methylene compounds 5 and 6), short hydrophobic contacts existed between vitamin D methyl group at C-20 and the conserved amino acids L305 and L309. Moreover, the detailed analysis of the VDR binding sites, capable of creating contacts with ligand methyl groups attached to C-13 and C-20, showed the existence of two hydrophobic patches on the receptor surface which could be responsible for specific methyl–methyl interactions. We concluded that the hydrophobic interactions of the VDR with the ligand methyl substituents might modulate its interaction with coactivators and corepressors involved in calcium homeostasis and influence the ability of the analogs to mobilize calcium from bone.
An experimental confirmation of these predictions, based on the docking simulations, was obtained soon afterwards when we determined the X-ray crystallographic data of a ternary complex of 2MD with the ligand binding domain (LBD) of rVDR and a LXXLL-containing coactivator peptide.28 Inspection of this complex reveals that the angular methyl group, located at C-13 in the 2MD molecule, as well as its 20-methyl substituent, are surrounded in the LBD by the most hydrophobic amino acids. Thus, strong hydrophobic contacts (distances between hydrogens shorter than 3.5 Å) were found29 between the angular methyl at C-13 and two amino acids (V230 and I267) whereas, for the methyl attached to C-20, four residues (I264, M268, L305 and L309) were involved in such interactions. That all these amino acids influence the transactivation activity of 2MD (6) was nicely demonstrated by the results of alanine scanning mutational analysis (ASMA). Yamada and Yamamoto established that Ala mutations of I267 and L305 (rVDR numbering) in the ligand binding pocket of the human vitamin D receptor (hVDR) nearly abolished the mutant transactivation potency (less than 20% of the activity of the wild-type VDR) induced by 2MD, mutation of V230 and I264 had also significant effect, whereas the remaining residues M268 and L309 seemed to play less important role (more than 90% of the wild-type receptor activity).30
Thus, docking calculations suggest that removal of these methyl constituents, especially C-21, from 2MD may result in a reduction of bone calcium mobilizing activity. Clearly, removal of the 20-methyl abolishes bone calcium mobilizing activity. Removal of the 13-methyl reduces but does not eliminate bone calcium mobilizing activity. When both are removed, some restoration of bone calcium mobilizing activity occurs.
4. Conclusions
Removal of the 20-methyl from 2MD essentially eliminates its bone calcium mobilizing activity specifically while not affecting all other measured activities including intestinal calcium transport. Removal of 13-methyl also reduces somewhat bone calcium mobilizing activity while removal of both methyls is not additive in lowering bone calcium activity. These results suggest that the 20-methyl is an essential group for 2MD’s strong activity on bone.
5. Experimental
5.1. Chemistry
Melting points (uncorrected) were determined on a Thomas–Hoover capillary melting-point apparatus. Infrared (IR) spectra were recorded with a Perkin–Elmer FT-IR Spectrum 2000. Ultraviolet (UV) absorption spectra were recorded with a Perkin–Elmer Lambda 3B UV–vis spectrophotometer in ethanol. Circular dichroism (CD) spectra were recorded between 185 and 300 nm at room temperature with a JASCO J-715 spectropolarimeter in hexane solutions. 1H nuclear magnetic resonance (NMR) spectra were recorded at 400 and 500 MHz with Bruker Instruments DMX-400 and DMX-500 Avance console spectrometers in deuteriochloroform. 13C nuclear magnetic resonance (NMR) spectra were recorded at 100 and 125 MHz with the same Bruker Instruments in deuteriochloroform. Chemical shifts (δ) are reported downfield from internal Me4Si (δ 0.00). Electron impact (EI) mass spectra were obtained with a Micromass AutoSpec (Beverly, MA) instrument. High-performance liquid chromatography (HPLC) was performed on a Waters Associates liquid chromatograph equipped with a Waters 600 Controller, a Rheodyne 7725i injector and Waters 2487 Dual λ Absorbance Detector. THF was freshly distilled before use from sodium benzophenone ketyl under argon.
The starting compounds 11 and 17 were obtained from commercial vitamin D2 (16) according to the published procedures.19,20
5.2. des-A,B-8β-[(Triethylsilyl)oxy]-testosterone acetate (18)
To a stirred solution of the methylketone 17 (1.13 g; 3.65 mmol) in cyclohexane (50 mL), meta-chloroperbenzoic acid (77%, 1.50 g) was added at 0 °C. Then the reaction mixture was allowed to warm up to room temperature and left stirred for five days. Next portions of meta-chloroperbenzoic acid (1.0 g, 0.8 g, and 0.6 g) were added after one day, two days, and four days, respectively. The suspension was filtered off and the filtrate was washed with a saturated aqueous solution of NaHCO3 (20 mL). The organic phase was dried over anhydrous MgSO4, concentrated under reduced pressure and the residue was purified by column chromatography on silica (1→3% ethyl acetate/hexane) to give the acetate 18 (0.89 g, 58%); +18.7 (c 0.90, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.56 (6H, q, J = 7.9 Hz), 0.95 (9H, t, J = 7.9 Hz), 1.11 (3H, s), 2.03 (3H, s), 4.05 (1H, narrow m); 13C NMR (100 MHz, CDCl3) δ 4.9, 6.9, 13.6, 17.2, 21.2, 22.2, 26.7, 34.5, 37.8, 42.0, 47.8, 69.0, 82.9, 171.3; MS (EI) m/z (relative intensity) 326 (M+, 3), 297 (18), 283 (8), 145 (70), 135 (100); exact mass calculated for C18H34O3Si 326.2277, measured 326.2269.
5.3. des-A,B-8β-[(Triethylsilyl)oxy]-testosterone (19)
The acetate 18 (972 mg, 2.98 mmol) was dissolved in methanol (25 mL) and treated with 10% solution of sodium methoxide in methanol (5 mL) for 2.5 h. A saturated aqueous solutions of NH4Cl (10 mL) and water (15 mL) were added and the product was extracted with methylene chloride (5 × 75 mL). The organic phase was dried over anhydrous MgSO4, concentrated under reduced pressure and the residue was purified by column chromatography on silica (5→15% ethyl acetate/hexane) to give the semi-crystalline alcohol 19 (764 mg, 90%); +39.6 (c 0.95, CHCl3); mp 93–95 °C; 1H NMR (400 MHz, CDCl3) δ 0.56 (6H, q, J = 7.9 Hz), 0.95 (9H, t, J = 7.9 Hz), 0.96 (3H, s), 3.56 (1H, m), 4.02 (1H, narrow m); 13C NMR (100 MHz, CDCl3) δ 5.0, 6.9, 12.4, 17.3, 22.2, 29.9, 34.6, 37.5, 42.2, 48.1, 69.1, 82.2; MS (EI) m/z (relative intensity) 284 (M+, 9), 255 (100), 237 (40), 135 (42), 103 (66); exact mass calculated for C14H27O2Si (M+−C2H5) 255.1780, measured 255.1770.
5.4. des-A,B-8β-[(Triethylsilyl)oxy]-androstane-17-one (20)
To a stirred solution of the alcohol 19 (760 mg, 2.68 mmol) and pyridinium p-toluenesulfonate (30 mg, 0.12 mmol) in methylene dichloride (90 mL), pyridinium dichromate (2.25 g, 5.98 mmol) was added at 0 °C. The cooling bath was removed and the reaction mixture was stirred for 9 h. Then the solvent was removed under reduced pressure and the residue was purified by column chromatography on silica (5→10% ethyl acetate/hexane) to give the ketone 20 (642 mg, 85%); +82.9 (c 0.90, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.60 (6H, q, J = 7.9 Hz), 0.97 (9H, t, J = 7.9 Hz), 1.11 (3H, s), 2.43–2.47 (1H, m), 4.19 (1H, narrow m); 13C NMR (125 MHz, CDCl3) δ 4.9, 6.9, 16.3, 17.0, 21.3, 32.3, 34.5, 35.3, 47.5, 48.8, 69.9, 221.2; MS (EI) m/z (relative intensity) 282 (M+, 10), 252 (100), 133 (17), 103 (39); exact mass calculated for C16H30O2Si 282.2015, measured 282.2013.
5.5. des-A,B-8β-[(Triethylsilyl)oxy]-21-norcholest-17-ene (22, mixture of Z and E isomers)
To a stirred suspension of 5-methylhexyltriphenylphosphonium bromide23 (21; 4.10 g, 9.35 mmol) in anhydrous THF (13.5 mL), a solution of potassium tert-butoxide (1 M in THF, 8.9 mL, 8.9 mmol) was added dropwise at −10 °C. The suspension was stirred and allowed to warm up to 0 °C over 30 min. Then a solution of the ketone 20 (716 mg, 2.53 mmol) in anhydrous THF (3.0 mL) was added via cannula and the resulting mixture was stirred at 45 °C for four days. The saturated aqueous solution of NH4Cl (20 mL) and water (30 mL) were added and the mixture was extracted with diethyl ether (3 × 100 mL). The organic phase was dried over anhydrous MgSO4, concentrated under reduced pressure, and the residue was purified by column chromatography on silica (0→5% ethyl acetate/hexane) to give a mixture of isomeric olefins 22 (572 mg, 63%; Z/E ratio of 5:1); 1H NMR (400 MHz, CDCl3) δ 0.56 (6H, q, J = 7.9 Hz), 0.86 (6H, d, J = 6.7 Hz), 0.95 (9H, t, J = 7.9 Hz), 1.10 (3H, s), 4.11 (1H, narrow m), 4.94 (0.83H, t, J = 7.3 Hz), 5.36 (0.17H, t, J = 4.7 Hz); 13C NMR (100 MHz, CDCl3) δ 5.0, 7.0, 18.0, 19.9, 22.6, 22.7, 23.8, 27.5, 27.6, 27.7, 28.0, 28.7, 30.6, 34.6, 38.3, 38.7, 38.9, 44.2, 52.8, 69.7, 119.3, 130.0, 149.9; MS (EI) m/z (relative intensity) 364 (M+, 6), 335, (23), 321 (10), 279 (37), 232 (54), 205 (43), 171 (51), 147 (100); exact mass calculated for C23H44OSi 364.3161, measured 364.3175.
5.6. des-A,B-8β-[(Triethylsilyl)oxy]-21-norcholestane (23)
To a solution of the olefins 22 (552 mg, 1.52 mmol) in ethyl acetate (20 mL), 5% palladium on activated carbon (160 mg) was added and the mixture was hydrogenated overnight. The catalyst was filtered off and the filtrate was concentrated under reduced pressure. The residue was purified on a Waters silica Sep-Pak cartridge (washed with hexane) to give the saturated silyl ether 23 (515 mg, 93%); +41.8 (c 1.15, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.55 (6H, q, J = 7.9 Hz), 0.81 (3H, s), 0.86 (6H, d, J = 6.6 Hz), 0.95 (9H, t, J = 7.9 Hz), 4.05 (1H, narrow m); 13C NMR (100 MHz, CDCl3) δ 5.0, 7.0, 14.1, 17.6, 2 × 22.7, 23.4, 27.8, 28.0, 29.1, 30.0, 35.0, 38.9, 39.1, 41.5, 51.7, 52.8, 69.2; MS (EI) m/z (relative intensity) 337 (100), 323 (59), 271 (67), 233 (77); exact mass calculated for C21H41OSi (M+−C2H5) 337.2927, measured 337.2911.
5.7. des-A,B-21-Norcholestane-8β-ol (12)
To a stirred solution of the silyl ether 23 (485 mg, 1.33 mmol) in n-butanol (25 mL) was added (+)-10-camphorsulfonic acid (330 mg, 1.42 mmol). After one day, the solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica (5→15% ethyl acetate/hexane) to give the 8β-alcohol 12 (328 mg, 98%); +34.1 (c 1.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.84 (3H, s), 0.86 (6H, d, J = 6.6 Hz), 4.10 (1H, narrow m); 13C NMR (100 MHz, CDCl3) δ 14.0, 17.4, 2 × 22.7, 22.9, 27.6, 2 × 28.0, 29.0, 29.9, 33.9, 38.5, 39.1, 41.3, 51.5, 52.3, 69.3; MS (EI) m/z (relative intensity) 252 (M+, 43), 237 (44), 219 (22), 209 (19), 125 (49), 111 (100); exact mass calculated for C17H32O 252.2453, measured 252.2446.
5.8. des-A,B-25-Hydroxy-21-norcholestane-8-one (24)
A solution of hydrindanol 12 (74 mg, 0.29 mmol) in acetonitrile/carbon tetrachloride (1:1; 1.5 mL) was added to a vigorously stirred solution of RuCl3 × H2O (10 mg, 0.05 mmol) and NaIO4 (227 mg, 1.06 mmol) in water (1 mL). After 2 h, the next portion of RuCl3 × H2O (8 mg, 0.04 mmol) was added and the reaction mixture was stirred for three days. Then a few drops of 2-propanol were added, followed by water (5 mL) and the mixture was extracted with diethyl ether (3 × 15 mL). The organic phase was dried over anhydrous MgSO4, concentrated under reduced pressure and the residue was purified on a Waters silica Sep-Pak cartridge (3→25% ethyl acetate/hexane) to give the hydroxy ketone 24 (24 mg, 31%); −14.0 (c 1.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.55 (3H, s), 1.21 (6H, s), 2.42 (1H, dd, J = 11.2, 7.7 Hz); 13C NMR (100 MHz, CDCl3) δ 12.9, 19.6, 23.9, 24.8, 27.8, 29.2, 2 × 29.3, 29.9, 30.2, 37.1, 41.0, 44.0, 49.7, 51.5, 61.4, 71.0, 211.9; MS (EI) m/z (relative intensity) 266 (M+, 18), 248 (87), 233 (84), 151 (97), 111 (94), 97 (95), 81 (100); exact mass calculated for C17H28O (M+−H2O) 248.2140, measured 248.2131.
5.9. des-A,B-25-[(Triethylsilyl)oxy]-21-norcholestane-8-one (14)
To a stirred solution of the hydroxy ketone 24 (21 mg, 79 μmol) and 2,6-lutidine (20 μL, 18 mg, 152 μmol) in anhydrous methylene chloride (500 μL), dropwise triethylsilyl trifluoromethanesulfonate (32 μL, 37 mg, 132 μmol) was added at −50 °C. After 20 min, a few drops of wet methylene chloride were added, followed by water (5 mL) and the mixture was extracted with methylene chloride (3 × 15 mL). The combined organic phases were washed with a saturated aqueous solution of CuSO4 (5 mL), dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified on a Waters silica Sep-Pak cartridge (0→5% ethyl acetate/hexane) to give the protected hydroxy ketone 14 (20 mg, 67%); −7.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.55 (3H, s), 0.56 (6H, q, J = 7.9 Hz), 0.95 (9H, t, J = 7.9 Hz), 1.19 (6H, s), 2.42 (1H, dd, J = 11.1, 7.7 Hz); 13C NMR (100 MHz, CDCl3) δ 6.8, 7.1, 24.0, 24.8, 27.7, 29.2, 29.9, 30.2, 37.1, 41.1, 45.1, 49.7, 51.5, 61.4, 73.4, 212.1; MS (EI) m/z (relative intensity) 365 (32), 351 (54), 322 (45), 231 (66), 173 (92), 135 (86), 103 (100).
5.10. (18E)-(20S)-18-(Hydroxyimino)-des-A,B-cholestan-8β-ol (27)
A solution of the hydrindanol 11 (185 mg, 0.69 mmol) in chloroform (5 mL) was treated with tert-butyl nitrite (1 mL) for 1 h in darkness. Then anhydrous benzene (10 mL) was added and the solvents were removed under reduced pressure. The crude oily (20S)-des-A,B-cholestan-8β-yl nitrite (25) was sufficiently pure to be used in the next synthetic step without purification.
Compound 25: 1H NMR (500 MHz, CDCl3) δ 0.76 (3H, s), 0.81 (3H, d, J = 6.5 Hz), 0.87 (6H, d, J = 6.6 Hz), 5.78 (1H, narrow m); 13C NMR (125 MHz, CDCl3) δ 13.1, 17.9, 18.5, 22.2, 22.6, 22.7, 23.9, 27.1, 28.0, 31.5, 34.9, 35.3, 39.3, 39.7, 41.9, 51.9, 56.0.
A solution of the crude nitrite 25 in anhydrous benzene (150 mL) was irradiated in an apparatus consisting of a Pyrex vessel with a water-cooled immersion well and a Hanovia high-pressure mercury arc lamp equipped with a Pyrex filter. A slow stream of argon was passed through the solution and the temperature was maintained at about 10 °C. Monitoring of the reaction progress by TLC indicated that the reaction was completed within 30 min. Benzene was removed under reduced pressure, the residue was dissolved in 2-propanol (5 mL) and refluxed for 2 h. Then the mixture was allowed to cool and it was left overnight to accomplish the isomerization of the initially formed nitroso compound to the desired oxime form. The solvent was evaporated and the residue was purified on a Waters silica Sep-Pak cartridge (25% ethyl acetate/hexane) to give the oxime 27 (102 mg, 51% yield with respect to 11); +8.2 (c 0.80, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.84 (3H, d, J = 6.3 Hz), 0.87 (6H, d, J = 6.6 Hz), 2.20 (1H, br d, J = 13.1 Hz), 4.04 (1H, narrow m), 7.33 (1H, s), 10.8 (1H, br s); 13C NMR (100 MHz, CDCl3) δ 17.5, 18.6, 21.8, 22.6, 22.7, 24.1, 27.2, 28.0, 34.3, 35.0, 35.6, 39.3, 49.5, 52.6, 56.7, 67.6, 152.2; MS (EI) m/z (relative intensity) 295 (M+, 2), 278 (28), 260 (20), 245 (8), 206 (19), 183 (38), 165 (13), 148 (15), 121 (100); exact mass calculated for C18H33NO2Na (M++Na) 318.2409, measured 318.2412.
5.11. (18E)-18-(Hydroxyimino)-des-A,B-21-norcholestane-8β-ol (28)
The des-A,B-21-norcholestane-8β-yl nitrite (26) was obtained from the alcohol 12 by the trans-esterification with tert-butyl nitrite, performed analogously to the process described above for 11. Photolysis of the crude nitrite with the mercury arc lamp followed by rearrangement of the intermediate nitroso compound was carried out as described above for 25. After evaporation of the solvents, the residue was purified on a Waters silica Sep-Pak cartridge (15→25% ethyl acetate/hexane) to give the oxime 28 (40% yield with respect to 12); +35.5 (c 0.90, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.86 (6H, d, J = 6.6 Hz), 4.06 (1H, narrow m), 6.81 (1H, br s), 7.23 (1H, s), 10.91 (1H, s); 13C NMR (100 MHz, CDCl3) δ 17.3, 22.0, 2 × 22.6, 27.7, 27.9, 28.1, 29.1, 34.4, 34.6, 38.9, 49.1, 51.8, 52.0, 67.4, 152.4; MS (EI) m/z (relative intensity) 281 (M+, 11), 264 (72), 246 (57), 205 (49), 183 (100); exact mass calculated for C17H30NO (M+−OH) 264.2327, measured 264.2326.
5.12. (20S)-8β-(Acetoxy)-des-A,B-cholestan-18-nitrile (29)
A solution of 27 (100 mg, 0.34 mmol) in acetic anhydride (5 mL) was refluxed for 1.5 h. The reaction mixture was cooled, poured carefully into ice water (ca. 25 mL) and extracted with benzene (3 × 40 mL). The combined organic phases were washed with a saturated aqueous solution of NaHCO3 (2 × 40 mL), water (30 mL), dried over anhydrous Na2SO4 and evaporated. The residue was purified on a Waters silica Sep-Pak cartridge (5% ethyl acetate/hexane) to give the nitrile 29 (91 mg, 84%); −26.4 (c 0.75, CHCl3); IR (CHCl3) νmax 2228, 1741, 1241 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.87 (6H, d, J = 6.6 Hz), 0.91 (3H, d, J = 6.6 Hz), 2.15 (3H, s), 2.46 (1H, br d, J = 3.2 Hz), 5.20 (1H, narrow m); 13C NMR (125 MHz, CDCl3) δ 17.9, 18.8, 22.6, 22.7, 23.3, 23.8, 27.1, 28.0, 29.9, 35.6, 36.2, 36.3, 39.1, 45.6, 51.9, 54.1, 68.7, 121.2, 171.0; MS (EI) m/z (relative intensity) 319 (M+, 18), 304 (10), 290 (3), 277 (84), 259 (100), 244 (54), 234 (27), 216 (40), 202 (33), 188 (60), 174 (47), 147 (39), 134 (34), 121 (95); exact mass (ESI) calculated for C20H33NO2Na (M++Na) 342.2409, measured 342.2413.
5.13. 8β-(Acetoxy)-des-A,B-21-norcholestane-18-nitrile (30)
Transformation of the oxime 28 to the corresponding nitrile by heating in acetic anhydride was accomplished as described above for 27. The product was purified on a Waters silica Sep-Pak cartridge (3→5% ethyl acetate/hexane) to give the nitrile 30 in 92% yield; +5.2 (c 1.30, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.86 (6H, d, J = 6.6 Hz), 2.13 (3H, s), 2.25–2.28 (1H, m), 5.21 (1H, narrow m); 13C NMR (100 MHz, CDCl3) δ 18.5, 20.9, 22.6, 23.3, 27.2, 27.6, 27.9, 28.2, 30.1, 30.8, 34.0, 38.9, 46.5, 49.1, 51.2, 68.4, 121.4, 170.9; MS (EI) m/z (relative intensity) 305 (M+, 7), 288 (3), 263 (83), 245 (44), 220 (79), 205 (100); exact mass calculated for C19H31NO2Na 328.2252, measured 328.2249.
5.14. (20S)-des-A,B-Cholestan-18-nitrile-8β-ol (31)
A solution of the acetoxy nitrile 29 (90 mg, 0.28 mmol) in methanol (3 mL) was treated with 5% solution of sodium methoxide in methanol (3 mL) for 2 h. A saturated aqueous solution of NH4Cl (5 mL) was added, followed by water (10 mL), and the mixture was extracted with methylene chloride (5 × 40 mL), dried over anhydrous Na2SO4 and evaporated. The residue was purified on a Waters silica Sep-Pak cartridge (20% ethyl acetate/hexane) to give the hydroxy nitrile 31 (73 mg, 94%); −6.1 (c 0.75, CHCl3); IR (CHCl3) 3486, 2228; 1H NMR (500 MHz, CDCl3) δ 0.87 (6H, d, J = 6.6 Hz), 0.92 (3H, d, J = 6.7 Hz), 2.43 (1H, br d, J = 3.1 Hz), 4.10 (1H, narrow m); 13C NMR (125 MHz, CDCl3) δ 17.9, 22.6, 22.7, 22.9, 23.9, 27.1, 28.0, 32.8, 35.7, 36.2, 36.3, 44.7, 53.4, 54.2, 122.5; MS (EI) m/z (relative intensity) 277 (M+, 28), 262 (34), 259 (18), 248 (16), 244 (24), 220 (30), 216 (18), 206 (100); exact mass calculated for C18H31NO 277.2496, measured 277.2395.
5.15. des-A,B-21-Norcholestane-18-nitrile-8β-ol (32)
Hydrolysis of the acetoxy nitrile 30 with sodium methoxide in methanol was performed analogously as it was described above for 29. The crude product was purified on a Waters silica gel Sep-Pack cartridge (15→25% ethyl acetate/hexane) to give the hydroxy nitrile 32 in 89% yield; +23.0 (c 1.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.86 (6H, d, J = 6.6 Hz), 2.24 (1H, d, J = 13.0 Hz), 4.12 (1H, narrow m); 13C NMR (100 MHz, CDCl3) δ 17.8, 22.6, 22.9, 27.3, 27.6, 27.9, 28.3, 30.7, 33.0, 34.2, 38.9, 45.7, 49.0, 52.6, 67.2, 122.5; MS (EI) m/z (relative intensity) 263 (M+, 64), 246 (39), 236 (77), 220 (92), 206 (84), 193 (95), 134 (100); exact mass calculated for C17H29NO 263.2249, measured 263.2256.
5.16. (20S)-des-A,B-18-Norcholestan-8β-ol (33)
To a stirred mixture of potassium (110 mg, 2.82 mmol) in hexamethylphosphoramide (280 μL, 288 mg, 1.62 mmol) and anhydrous diethyl ether (700 μL), a solution of the hydroxy nitrile 31 (70 mg, 0.25 mmol) in anhydrous tert-butyl alcohol (65 μL) and anhydrous diethyl ether (250 μL) was added dropwise at 0 °C under argon. The mixture was allowed to warm to the room temperature and then stirred for 5 h. The remaining potassium was removed, and a few drops of 2-propanol were added followed by benzene (20 mL). The organic phase was washed with water (10 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified on a Waters silica Sep-Pak cartridge (10% ethyl acetate/hexane) to give the alcohol 33 (54 mg, 85%); +32.6 (c 0.90, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.78 (3H, d, J = 6.8 Hz), 0.87 (6H, d, J = 6.6 Hz), 4.06 (1H, narrow m); 13C NMR (125 MHz, CDCl3) δ 14.7, 20.2, 22.7, 22.9, 24.7, 25.3, 28.0, 30.8, 33.1, 33.5, 36.3, 39.3, 39.7, 48.6, 50.3, 67.9; MS (EI) m/z (relative intensity) 252 (M+, 6), 234 (21), 219 (23), 209 (26), 191 (8), 179 (4), 167 (13), 149 (89), 139 (47), 122 (90), 107 (35), 95 (80), 79 (87), 67 (88), 58 (100); exact mass calculated for C17H32O 252.2453, measured 252.2448.
5.17. des-A,B-18,21-Dinorcholestane-8β-ol (34)
Decyanation of the hydroxy nitrile 32 to the hydrindanol 34 was performed analogously to the process described above for 31. The crude product was purified on a Waters silica Sep-Pak cartridge (5→15% ethyl acetate/hexane) to give the alcohol 34 in 80% yield; +66.8 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.86 (6H, d, J = 6.6 Hz), 4.04 (1H, narrow m); 13C NMR (100 MHz, CDCl3) δ 20.2, 2 × 22.6, 24.4, 27.7, 27.9, 28.6, 29.2, 30.6, 33.5, 34.4, 39.0, 43.6, 44.6, 50.3, 67.8; MS (EI) m/z (relative intensity) 238 (M+, 50), 220 (76), 205 (41), 195 (79), 135 (82), 122 (86), 93 (100); exact mass calculated for C16H30O 238.2297, measured 238.2323.
5.18. (20S)-des-A,B-25-Hydroxy-18-norcholestane-8-one (35)
To a stirred solution of RuCl3 × H2O (10 mg, 0.05 mmol) and NaIO4 (227 mg, 1.06 mmol) in water (1 mL), a solution of the 8β-alcohol 33 (74 mg, 0.29 mmol) in tetrachloromethane (0.75 mL) and acetonitrile (0.75 mL) was added. The reaction mixture was stirred vigorously for three days. Then a few drops of 2-propanol were added followed by water (10 mL). The reaction products were extracted with methylene chloride (3 × 20 mL). The combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The mixture of products was separated on a Waters silica Sep-Pak cartridge (10→60% ethyl acetate/hexane) to give the desired hydroxy ketone 35 (13 mg, 17%) and (20S)-des-A,B-18-norcholestane-8-one (27 mg, 37%).
Compound 35: 1H NMR (400 MHz, CDCl3) δ 0.78 (3H, d, J = 6.7 Hz), 1.22 (6H, s), 2.01 (1H, br d, J = 12.3 Hz); 13C NMR (100 MHz, CDCl3) δ 14.3, 21.3, 22.2, 22.6, 27.8, 29.3, 29.7, 33.0, 36.5, 41.6, 44.1, 49.6, 51.0, 58.0, 71.0, 212.0; MS (EI) m/z (relative intensity) 266 (M+, <1), 248 (57), 233 (19), 215 (4), 208 (15), 163 (29), 137 (100); exact mass (ESI) calculated for C17H30O2Na (M++Na) 289.2144, measured 289.2136.
5.19. des-A,B-25-Hydroxy-18,21-dinorcholestane-8-one (36)
The ruthenium tetroxide oxidation of the alcohol 34 was performed analogously to the process described above for 33. The products were separated on a Waters silica Sep-Pak cartridge (5→30% ethyl acetate/hexane) to give the hydroxy ketone 36 in 31% yield; +9.0 (c 0.95, CHCl3); CD Δε (λmax) −1.70 (312), −2.86 (300), −2.93 (293), −3.10 (191); 1H NMR (400 MHz, CDCl3) δ 1.21 (6H, s), 13C NMR (100 MHz, CDCl3) δ 21.1, 24.9, 27.7, 28.7, 29.1, 29.2, 29.6, 34.1, 41.5, 43.9, 45.7, 54.8, 56.8, 58.1, 211.79; MS (EI) m/z (relative intensity) 252 (M+, 47), 234 (68), 219 (63), 194 (75), 67 (100); exact mass calculated for C16H28O2 252.2089, measured 252.2080.
5.20. (20S)-25-[(Triethylsilyl)oxy]-des-A,B-18-norcholestane-8-one (13)
To a stirred solution of 35 (12 mg, 45 μmol) and 2,6-lutidine (13 μL, 12 mg; 111 μmol) in anhydrous methylene chloride (250 μL), triethylsilyl trifluoromethanesulfonate was added dropwise at −50 °C under argon. After 20 min, a few drops of wet methylene chloride were added, followed by water (7 mL). The reaction mixture was extracted with methylene chloride (3 × 7 mL), the combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified on a Waters silica Sep-Pak cartridge (3% ethyl acetate/hexane). Final purification was performed by HPLC (5% ethyl acetate/hexane, 4 mL/min, 10 mm × 25 cm Zorbax-Sil column; Rt = 5.42 min) to give the TES-protected hydroxy ketone 13 (13 mg, 76%); H NMR (500 MHz, CDCl3) δ 0.56 (6H, q, J = 7.9 Hz), 0.77 (3H, d, J = 6.8 Hz), 0.94 (9H, t, J = 7.9 Hz), 1.19 (6H, s), 13C NMR (125 MHz, CDCl3) δ 6.8, 7.1, 14.3, 21.4, 22.2, 22.7, 27.8, 29.7, 29.8, 29.9, 32.9, 36.4, 41.6, 45.2, 49.6, 51.1, 58.0, 73.4, 212.1; MS (EI) m/z (relative intensity) 365 (M+−Me, 8), 351 (100), 322 (6), 239 (2), 231 (25), 220 (4), 205 (15), 189 (4), 173 (92); exact mass (ESI) calculated for C23H44O2SiNa (M++Na) 403.3008, measured 403.2995.
5.21. des-A,B-25-[(Triethylsilyl)oxy]-18,21-dinorcholestane-8-one (15)
Silylation of the hydroxy ketone 36 was performed analogously to the process described above for 35. The crude product was purified on a Waters silica Sep-Pak cartridge (2→5% ethyl acetate/hexane) to give the silylated compound 15 (15 mg, 58%); 1H NMR (400 MHz, CDCl3) δ 0.56 (6H, q, J = 7.9 Hz), 0.94 (9H, t, J = 7.9 Hz), 1.18 (6H, s), 13C NMR (100 MHz, CDCl3) δ 6.8, 7.1, 21.4, 24.7, 27.8, 28.7, 29.1, 29.6, 29.9, 34.2, 41.5, 45.1, 45.8, 54.8, 58.1, 73.4, 211.7; MS (EI) m/z (relative intensity) 366 (M+, 1), 351 (33), 337 (76), 308 (13), 217 (64), 173 (100); exact mass (ESI) calculated for C22H42O2SiNa 389.2852, measured 389.2840.
5.22. (20S)-1α,25-Dihydroxy-2-methylene-18,19-dinorvitamin D3 (7)
To a stirred solution of the phosphine oxide 10 (46 mg; 79 μmol) in anhydrous THF (600 μL), a solution of phenyllithium (1.5 M in THF, 63 μL, 95 μmol) was added at −20 °C under argon. The mixture was stirred for 20 min and then cooled to −78 °C. A pre-cooled solution of the Grundmann ketone 13 (13 mg; 34 μmol) in anhydrous THF (300 μL) was added via cannula and the reaction mixture was stirred for 3 h at −78 °C. Then the reaction mixture was stirred at 4 °C overnight, ethyl acetate (20 mL) was added and the organic phase was washed with brine (3 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified on a Waters silica Sep-Pak cartridge (0→2% ethyl acetate/hexane). Final purification was performed by HPLC (0.5% 2-propanol/hexane; 4 mL/min, 10 mm × 25 cm Zorbax-Sil column; Rt = 6.59 min) to give the protected vitamin D compound 37 (13.5 mg, 53%); UV (hexane) λmax = 242, 251, 261 nm; 1H NMR (500 MHz, CDCl3) δ 0.06 (3H, s), 0.11 (3H, s), 0.17 (3H, s), 0.19 (3H, s), 0.56 (6H, q, J = 8.0 Hz), 0.76 (3H, d, J = 6.7 Hz), 0.86 (9H, s), 0.89 (9H, s), 0.94 (9H, t, J = 8.0 Hz), 1.19 (6H, s), 2.18 (1H, dd, J = 12.5, 8.1 Hz), 2.86 (1H, br d, J = 13.8 Hz), 4.93 (1H, s), 4.96 (1H, s), 5.92 (1H, d, J = 11.1 Hz), 6.19 (1H, d, J = 11.1 Hz); 13C NMR (125 MHz, CDCl3) δ −5.1, −4.9, −4.9, −4.8, 6.8, 7.1, 18.2, 18.2, 22.3, 23.1, 25.8, 25.8, 27.8, 29.0, 29.7, 29.8, 29.9, 31.3, 33.6, 36.5, 38.7, 45.3, 47.5, 49.0, 50.2, 52.3, 71.9, 72.3, 73.4, 106.3, 113.7, 122.4, 132.9, 143.8, 152.9; MS (EI) m/z (relative intensity) 687 (M+−t-Bu, 6), 628 (2), 612 (100), 583 (6), 555 (4), 480 (29), 366 (44); exact mass calculated for C40H75O3Si3 (M+−t-Bu) 687.5024, measured 687.5028.
To a solution of the protected vitamin 37 (13 mg, 17 μmol) in anhydrous THF (5 mL) tetra-n-butylammonium fluoride (1 M in THF, 260 μL, 260 μmol) was added dropwise, followed by addition of activated molecular sieves 4 Å (200 mg). The reaction mixture was stirred under argon for 2 h. Then the solvent was removed under reduced pressure, and the residue was purified on a Waters silica Sep-Pak cartridge (40→50% ethyl acetate/hexane). Final purification of the vitamin D compound 7 was performed by HPLC (20% 2-propanol/hexane; 4 mL/min; 10 mm × 25 cm Zorbax-Sil column; Rt = 5.58 min) to give the analytically pure 18,19-dinorvitamin D analog 7 (3.8 mg, 56%); UV (EtOH) λmax = 242, 250, 260 nm; 1H NMR (500 MHz, CDCl3) δ 0.77 (3H, d, J = 6.6 Hz), 1.21 (6H, s), 2.58 (1H, dd, J = 13.2 Hz, 3.9 Hz), 2.81 (1H, dd, J = 13.3, 4.4 Hz), 2.87 (1H, br d, J =13.9 Hz), 5.10 (1H, s), 5.11 (1H, s), 5.97 (1H, d, J = 11.3 Hz), 6.35 (1H, d, J = 11.3 Hz); MS (EI) m/z (relative intensity) 402 (M+, 39), 384 (41), 366 (14), 351 (11), 299 (58), 231 (36), 142 (58), 69 (100); exact mass calculated for C26H42O3 402.3134, measured 402.3121.
5.23. 1α,25-Dihydroxy-2-methylene-19,21-dinorvitamin D3 (8)
The protected vitamin D compound 38 was prepared in the same way as 37, except starting from the corresponding Grundmann ketone 14. The crude product was purified on a Waters silica Sep-Pak cartridge (0→2% ethyl acetate/hexane) to give the vitamin 38 in 90% yield; +20.4 (c 1.05, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.02 (3H, s), 0.05 (3H, s), 0.07 (3H, s), 0.08 (3H, s), 0.45 (3H, s), 0.56 (6H, q, J = 7.9 Hz), 0.86 (9H, s), 0.90 (9H, s), 0.94 (9H, q, J = 7.9 Hz), 1.18 (6H, s), 2.18 (1H, dd, J = 12.2, 8.3 Hz), 2.33 (1H, m), 2.50 (2H, m), 2.86 (1H, m), 4.42 (2H, m), 4.92 (1H, s), 4.97 (1H, s), 5.84 (1H, d, J = 11.0 Hz), 6.22 (1H, d, J = 11.0 Hz); 13C NMR (125 MHz, CDCl3) δ −5.1, −4.9, 6.8, 7.1, 12.4, 18.2, 18.3, 22.6, 23.2, 24.9, 25.8, 25.9, 27.9, 28.9, 29.4, 29.9, 30.6, 38.6, 45.2, 45.3, 47.6, 51.3, 55.7, 71.7, 72.5, 73.5, 106.3, 115.9, 122.4, 132.7, 141.4, 153.0; MS (EI) m/z (relative intensity) 687 (M+−t-Bu, 34), 628 (12), 612 (100), 583 (38), 480 (49), 366 (86); exact mass calculated for C40H75O3Si3 (M+−t-Bu) 687.5024, measured 687.5052.
To a stirred solution of the protected vitamin 38 (30 mg, 40 μmol) in n-butanol (3 mL), (+)-10-camphorsulfonic acid (21 mg, 90 μmol) was added at 0 °C. Then the cooling bath was removed and the reaction mixture was stirred for 1 week. A few drops of a saturated aqueous solution of NaHCO3 were added, followed by water (5 mL), and the mixture was extracted with ethyl acetate (5 × 10 mL). The combined organic phases were dried over anhydrous MgSO4, concentrated under reduced pressure, and the residue was purified on a Waters silica Sep-Pak cartridge (20→50% ethyl acetate/hexane) to give 17 mg of the crude compound 8. The vitamin was further purified by HPLC (13% 2-propanol/hexane, 4 mL/min, 10 mm × 25 cm Zorbax-Sil column; Rt = 7.66 min) to give the analytically pure 19,21-dinorvitamin D analog 8 (11.9 mg, 74%); +8.0 (c 0.80, CHCl3); UV (EtOH) λmax = 243, 251, 261 nm; 1H NMR (400 MHz, CDCl3) δ 0.45 (3H, s), 1.21 (6H, s), 2.26–2.35 (2H, m), 2.57 (1H, dd, J = 13.4, 3.5 Hz), 2.84 (2H, m), 4.48 (2H, m), 5.09 (1H, s), 5.11 (1H, s), 5.89 (1H, d, J = 11.2 Hz), 6.38 (1H, d, J = 11.2 Hz); 13C NMR (100 MHz, CDCl3) δ 12.6, 22.6, 23.3, 27.8, 29.1, 29.2, 2 × 29.3, 30.5, 38.2, 38.5, 44.0, 45.4, 45.8, 51.3, 55.8, 70.7, 71.1, 71.8, 107.7, 115.2, 124.3, 130.4, 143.5, 152.0; MS (EI) m/z (relative intensity) 402 (M+, 36), 299 (15), 231 (17), 107 (50), 91 (64), 49 (100); exact mass calculated for C26H42O3 402.3134, measured 402.3145.
5.24. 1α,25-Dihydroxy-2-methylene-18,19,21-trinorvitamin D3 (9)
The protected vitamin D compound 39 was prepared in the same way as 37, except starting from the corresponding Grundmann ketone 15 and using 1.8 M solution of phenyl lithium in di-n-butyl ether. The crude product was purified on a Waters silica Sep-Pak cartridge (0→2% ethyl acetate/hexane) to give the vitamin 39 in 72% yield; +4.5 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.03 (3H, s), 0.04 (3H, s), 0.07 (3H, s), 0.08 (3H, s), 0.56 (6H, q, J = 7.9 Hz), 0.87 (9H, s), 0.90 (9H, s), 0.94 (9H, q, J = 7.9 Hz), 1.18 (6H, s), 2.18 (1H, dd, J = 12.5, 7.9 Hz), 2.42 (3H, m), 2.86 (1H, d, J = 13.6 Hz), 4.42 (2H, m), 4.93 (1H, s), 4.96 (1H, s), 5.92 (1H, d, J = 11.1 Hz), 6.19 (1H, d, J = 11.1 Hz); 13C NMR (100 MHz, CDCl3) δ −5.1, −4.9, −4.9, 6.8, 7.1, 18.2, 24.8, 2 × 25.8, 25.8, 27.7, 28.9, 29.1, 29.9, 31.0, 34.9, 38.7, 45.1, 45.2, 47.5, 52.3, 53.9, 71.9, 72.3, 73.4, 106.3, 113.8, 122.5, 132.9, 143.6, 153.0; MS (EI) m/z (relative intensity) 468 (50), 366 (61), 337 (35), 219 (54), 173 (64), 73 (100); exact mass (ESI) calculated for C43H82O3Si3Na 753.5470, measured 753.5450.
Removal of the silyl protecting groups in the vitamin 39 was performed analogously to the process described above for 38. The crude product was purified on a Waters silica Sep-Pak cartridge (30→55% ethyl acetate/hexane) to give 10 mg of the crude compound 9. The vitamin was further purified by HPLC (acetonitrile/water/methanol 7:18:75, 4 mL/min, 9.4 mm × 25 cm Zorbax Eclipse XDB-C18 column; Rt = 10.35 min) to give 18,19,21-trinorvitamin D analog 9 in 77% yield; UV (EtOH) λmax = 243, 251, 260 nm; 1H NMR (500 MHz, CDCl3) δ 1.09 (6H, s), 2.30–2.36 (2H, m), 2.58 (1H, dd, J = 13.2, 3.8 Hz), 2.81 (1H, dd, J = 13.2, 4.4 Hz), 2.86 (1H, d, J = 13.8 Hz), 4.48 (2H, m), 5.09 (1H, s), 5.11 (1H, s), 5.97 (1H, d, J = 11.2 Hz), 6.35 (1H, d, J = 11.2 Hz); 13C NMR (125 MHz, CDCl3) δ 24.7, 25.3, 27.7, 28.8, 29.0, 29.2, 30.9, 34.7, 38.0, 44.0, 45.0, 45.9, 52.2, 53.8, 70.8, 71.1, 71.7, 107.7, 113.0, 124.2, 130.7, 145.8, 152.0; MS (EI) m/z (relative intensity) 388 (M+, 61), 370 (42), 352 (20), 337 (20), 285 (100); exact mass calculated for C25H40O3 388.2977, measured 388.2962.
5.25. Biological in vitro studies
VDR binding, HL-60 differentiation and 24-hydroxylase transcription assays were performed as previously described.31
5.26. Biological in vivo studies: bone calcium mobilization and intestinal calcium transport
Male, weanling Sprague-Dawley rats were purchased from Harlan (Indianapolis, IN). The animals were group housed and placed on Diet 11 (0.47% Ca) + AEK oil for one week followed by Diet 11 (0.02% Ca) + AEK oil for three weeks. The rats were then switched to a diet containing 0.47% Ca for one week followed by two weeks on a diet containing 0.02% Ca. Dose administration began during the last week on 0.02% Ca diet. Four consecutive intraperitoneal doses were given approximately 24 h apart. Twenty-four hours after the last dose, blood was collected from the severed neck, and the serum calcium level was determined as a measure of bone calcium mobilization. The first 10 cm of the intestine was also collected for the intestinal calcium transport analysis using the everted gut sac method.32 Statistical analyses were conducted in all in vivo bone calcium mobilization studies. ANOVA followed by pairwise comparisons using Tukey’s test was done in vehicle; 260 pmol and 780 pmol dose groups for each of the studies.
Acknowledgments
The work was supported in part by funds from the Wisconsin Alumni Research Foundation. We would like to express our gratitude to Dr. Wanda Sicinska for her valuable comments concerning the X-ray crystallographic data of a 2MD complex with the rVDR and a coactivator peptide. A special thanks to Jean Prahl and Jennifer Vaughan for carrying out the in vitro studies and to Heather Neils and Xiaohong Ma for conducting the in vivo studies.
This study made use of the National Magnetic Resonance Facility at Madison, which was supported by the NIH Grants P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS). Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF(DMB-8415048, OIA- 9977486, BIR-9214394), and the USDA.
References and notes
- 1.Feldman D, Pike JW, Glorieux FH, editors. Vitamin D. 2. Elsevier Academic Press; Burlington: 2005. [Google Scholar]
- 2.Jones G, Strugnell SA, DeLuca HF. Physiol Rev. 1998;78:1193. doi: 10.1152/physrev.1998.78.4.1193. [DOI] [PubMed] [Google Scholar]
- 3.Norman AW, Bouillon R, Thomasset M, editors. Molecular Endocrinology and Clinical Applications. Walter de Gruyter; Berlin: 1994. Vitamin D, a Pluripotent Steroid hormone: Structural Studies. [Google Scholar]
- 4.Suda T, Shinki T, Takahashi N. Annu Rev Nutr. 1990;10:195. doi: 10.1146/annurev.nu.10.070190.001211. [DOI] [PubMed] [Google Scholar]
- 5.Suda T. Proc Soc Exp Biol Med. 1989;191:214. doi: 10.3181/00379727-191-42911. [DOI] [PubMed] [Google Scholar]
- 6.Ostrem VK, DeLuca HF. Steroids. 1987;49:73. doi: 10.1016/0039-128x(87)90080-8. [DOI] [PubMed] [Google Scholar]
- 7.Abe E, Miyaura C, Sakagami H, Takeda M, Konno K, Yamazaki T, Yoshiki S, Sudo T. Proc Natl Acad Sci USA. 1981;78:4990. doi: 10.1073/pnas.78.8.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemire JM. J Cell Biochem. 1992;49:26. doi: 10.1002/jcb.240490106. [DOI] [PubMed] [Google Scholar]
- 9.Evans RM. Science. 1988;240:889. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenfeld MG, Lunyak VV, Glass CK. Genes Dev. 2006;20:1405. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 11.Feldman D, Glorieux FH, Pike JW, editors. Vitamin D. Academic; New York: 1997. [Google Scholar]
- 12.Bouillon R, Okamura WH, Norman AW. Endocr Rev. 1995;16:200. doi: 10.1210/edrv-16-2-200. [DOI] [PubMed] [Google Scholar]
- 13.Perlman KL, Sicinski RR, Schnoes HK, DeLuca HF. Tetrahedron Lett. 1990;31:1823. [Google Scholar]
- 14.See, for example: Shimizu M, Miyamoto Y, Takaku H, Matsuo M, Nakabayashi M, Masuno H, Udagawa N, DeLuca HF, Ikura T, Ito N. Bioorg Med Chem. 2008;16:6949. doi: 10.1016/j.bmc.2008.05.043.Sicinski RR, Perlman KL, DeLuca HF. J Med Chem. 1994;37:3730. doi: 10.1021/jm00048a009.
- 15.Sicinski RR, Prahl JM, Smith CM, DeLuca HF. J Med Chem. 1998;41:4662. doi: 10.1021/jm9802618. [DOI] [PubMed] [Google Scholar]
- 16.Shevde NK, Plum LA, Clagett-Dame M, Yamamoto H, Pike JW, DeLuca HF. Proc Natl Acad Sci USA. 2002;99:13487. doi: 10.1073/pnas.202471299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plum LA, Fitzpatrick LA, Ma X, Binkley NC, Zella JB, Clagett-Dame M, DeLuca HF. Osteoporosis Int. 2006;17:704. doi: 10.1007/s00198-005-0036-3. [DOI] [PubMed] [Google Scholar]
- 18.Sicinska W, Rotkiewicz P, DeLuca HFJ. Steroid Biochem Mol Biol. 2004;89–90:107. doi: 10.1016/j.jsbmb.2004.03.102. (Presented at the 12th Workshop on Vitamin D (Maastricht, The Netherlands, 6–10 July 2003) [DOI] [PubMed] [Google Scholar]
- 19.Grzywacz P, Plum LA, Sicinski RR, Clagett-Dame M, DeLuca HF. Arch Biochem Biophys. 2007;460:274. doi: 10.1016/j.abb.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 20.Posner GH, Crawford K, Siu-Caldera M-L, Satyanarayana Reddy G, Sarabia SF, Feldman D, van Etten E, Mathieu C, Gennaro L, Vouros P, Peleg S, Dolan PM, Kensler TW. J Med Chem. 2000;43:3581. doi: 10.1021/jm000215j. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez B, Perez JAM, Granja JR, Castedo L, Mourino A. J Org Chem. 1992;57:3173. [Google Scholar]
- 22.Huffman JW, Liddle J, Duncan SG, Jr, Yu S, Martin BR, Wiley JL. Bioorg Med Chem. 1998;6:2383. doi: 10.1016/s0968-0896(98)80014-x. [DOI] [PubMed] [Google Scholar]
- 23.De los Angeles Rey M, Martinez-Perez JA, Fernandez-Gacio A, Halkes K, Fall Y, Granja J, Mourino A. J Org Chem. 1999;64:3196. doi: 10.1021/jo982393e. [DOI] [PubMed] [Google Scholar]
- 24.Sicinski RR, DeLuca HF. Bioorg Med Chem Lett. 1995;5:159. [Google Scholar]
- 25.Sicinski RR, Perlman KL, Prahl J, Smith C, DeLuca HF. J Med Chem. 1996;39:4497. doi: 10.1021/jm950745t. [DOI] [PubMed] [Google Scholar]
- 26.Akhtar M, Barton DHR. J Am Chem Soc. 1964;86:1528. [Google Scholar]
- 27.Cuvigny T, Larcheveque M, Normant H. Bull Soc Chim Fr. 1974:1174. [Google Scholar]
- 28.Vanhooke JL, Benning MM, Bauer CB, Pike JW, DeLuca HF. Biochemistry. 2004;43:4101. doi: 10.1021/bi036056y. [DOI] [PubMed] [Google Scholar]
- 29.Biodesigner, Molecular Modeling and Visualization Program. http://www.pirx.com.
- 30.Yamada S, Yamamoto K. Curr Top Med Chem. 2006;6:1255. doi: 10.2174/156802606777864881. [DOI] [PubMed] [Google Scholar]
- 31.Glebocka A, Sicinski RR, Plum LA, Clagett-Dame M, DeLuca HF. J Med Chem. 2006;49:2909. doi: 10.1021/jm051082a. [DOI] [PubMed] [Google Scholar]
- 32.Suda T, DeLuca HF, Tanaka Y. J Nutr. 1970;100:1049. doi: 10.1093/jn/100.9.1049. [DOI] [PubMed] [Google Scholar]