

Abstract
A palladium-catalyzed hydroalkylation reaction of protected allylic alcohols using alkylzinc bromide reagents is reported. This account includes numerous allylic, homoallylic and bishomoallylic alcohol derivatives, all with uniform selectivity of >20:1 for the anti-Markovnikov product. The reaction features the ability to deliver enantiomerically-enriched alcohols in unfunctionalized regions, which results from the catalyst avoiding β-hydride elimination at the allylic position.
The remote functionalization of organic molecules is a preeminent challenge in chemical synthesis.1 Common isolated functional groups often displayed in natural products are alcohols, which are generally installed biosynthetically via oxidation processes.2 In contrast to this elegant enzymatic solution, the preparation of compounds containing remote chiral alcohols often requires the use of classical methods including organometallic additions to preformed chiral epoxides3 or enantioselective dialkylzinc additions to aldehydes,4 which each have fundamental synthetic limitations. Additionally, enantioselective ketone reductions (such as the CBS reduction or various transfer hydrogenation reactions) are not capable of effectively discriminating the prochiral faces of dialkyl ketones generally required for installation of a remote chiral alcohol in natural product synthesis.5 With a limited number of methods available for accessing these important structural motifs, we became interested in developing an alternative synthetic strategy. Specifically, we wished to expand our recently reported Pd-catalyzed anti-Markovnikov hydroalkylation of allylic amine derivatives to allylic alcohols.6 The successful development of this reductive cross-coupling reaction would provide an attractive unconventional method for the preparation of compounds containing remote chiral alcohols (A, Figure 1). Moreover, this approach utilizes the appealing features of cross-coupling technologies7 in that the starting materials, allylic alcohol derivatives (enantiomerically enriched and racemic) and alkylzinc halides, can be easily prepared incorporating diverse and useful functionality.
Figure 1.

a) Remote chiral alcohol accessed through a Pd-catalyzed hydroalkylation reaction of readily accessible allylic alcohol derivatives. B) Proposed catalytic cycle.
Conceptually, the proposed method relies on two distinct mechanistic processes to facilitate the hydroalkylation reaction. The first is the production of a Pd-hydride intermediate (E, Figure 1), which then mediates the hydridometallation of the alkene (B → F). Based on our previous efforts in Pd-catalyzed alkene hydroalkylation reactions, a reliable method to access the necessary Pd-hydride is to use a sacrificial equivalent of an alkylzinc halide, which undergoes transmetallation (C → D) followed by β-hydride elimination (D → E). The second distinct process is alkene insertion into the Pd-hydride (B → F) followed by the transmetallation/reductive elimination sequence with a second equivalent of the alkyl zinc halide (F → G → A).8 Several complications could arise including alkene isomerization via a consecutive β-hydride elimination/reinsertion sequence, which is attributable to the instability of Pd-alkyl species.7 This could also result in a mixture of cross-coupling product isomers and potentially erode the enantiomeric excess if an optically enriched substrate is used. In our previous report using allylic amine derivatives, generally good selectivity is observed for the anti-Markovnikov product albeit with some unexplained site-selectivity loss for certain substrates. This is a consequence of indiscriminate insertion of the alkene, both 1,2 and 2,1 insertion, into the Pd-hydride and an actively functioning Pd-alkyl rearrangement based on the analysis of isotopic labelling experiments.6 While a single example of an allylic ester was reported, only a modest yield was observed and it was unclear if these substrates would be generally compatible with the desired reaction class as allylic esters are well-known precursors for π-allyl Pd-chemistry.9 We report herein the optimization and scope of a Pd-catalyzed anti-Markovnikov hydroalkylation reaction of both allylic and homoallylic alcohols with alkylzinc halides to access remote alcohol derivatives. Additionally, key mechanistic insight is garnered into the preferred formation of the anti-Markovnikov product.
To optimize the reaction conditions, modest changes were found to enhance the isolated yield of the product. Specifically, under our previously reported hydroalkylation conditions, a 69% GC yield of the anti Markovnikov product of 1a was achieved (entry 1, Table 1). A slight increase in catalyst loading to 7 mol % led to a 74% yield (entry 2). The reaction conditions were further simplified with the removal of 3 Å molecular sieves, which gave a 79% yield (entry 3). We also found the optimal temperature of the reaction to be 0 °C (entries 3–5). It should be noted that >20:1 selectivity is observed for the formation of the linear product, as depicted.
Table 1.
Reaction Optimization.

| ||||
|---|---|---|---|---|
| entry | x | temp | additive | % yielda |
| 1 | 6 | 0°C | 3 Å MS | 69 |
| 2 | 7 | 0°C | 3 Å MS | 74 |
| 3 | 7 | 0°C | – | 79 |
| 4 | 7 | rt | – | 54 |
| 5 | 7 | −10 °C | – | 61 |
Measured by GC using an internal standard. Each entry had a >20:1 ratio of linear:branched product isomers, as determined by GC.
Using these conditions, we began examining the substrate scope of allylic alcohol derivatives. Our standard benzoyl-protected substrate 1a reacted to give a 73% isolated yield of the anti-Markovnikov product (2a, Table 2). Various acyl protecting groups are well-tolerated in this reaction such as para-methoxybenzoyl and 1-naphthoyl groups, which gave 72% and 65% yields, respectively (2b, 2c). Allylic acetates also gave excellent selectivity, albeit with lower yields (2d, 2e). It should be noted that these substrates are common precursors of π-allyl intermediates,9 but products resulting from these reactions are not observed. Next, the substitution at the carbon bearing the protected alcohol was varied: incorporation of a larger group, such as a cyclohexyl ring, resulted in 72% yield (2f), and substrates containing benzyloxyl (2g) and benzyl (2h) groups both led to good yields for the linear products. Next, a variety of alkylzinc reagents were successfully incorporated including an alkyl chloride (2i), a bulky methylcyclohexyl group (2j), a pivalate ester (2k), a TBDPS-protected alcohol (2l), a benzoyl group (2m) and a morpholine-derived amide (2n).
Table 2.
Pd-Catalyzed hydroalkylation of protected allylic alcohols with alkylzinc reagents.

|
Each entry had a >20:1 ratio of linear:branched product isomers, as determined by GC. Each entry represents the isolated yield on 0.5 mmol scale. Naphth = 1-naphthoyl, PMP = para-methoxyphenyl.
8 equiv of R1ZnBr was used.
A hypothesized origin of the high anti-Markovnikov selectivity for the hydroalkylation reaction may be the coordinating ability of the ester (or the amide in our previous report).6, 10 Therefore, a substrate containing a tert-butyl ether, which is unlikely to participate in coordination with the catalyst, was evaluated and afforded >20:1 selectivity for the linear product, albeit in 35% yield (2o). This result strongly suggests that the selectivity for the anti-Markovnikov product does not arise from coordination of the Lewis base, although enhanced yields are generally observed when these groups are incorporated. This improvement may be due to superior binding of substrates with Lewis bases as an equivalent of unfunctionalized alkene, derived from the alkylzinc reagent, may be competitively coordinating to the catalyst.
The competence of the hindered substrate leading to 2o and other non-coordinating allylic substituents,6 all of which deliver high levels of anti-Markovnikov selectivity, suggest that the selectivity is not dictated by the electronic nature of the allylic group.11 To probe this hypothesis, a protected homoallylic alcohol, lacking substitution at the allylic position, was submitted to the reaction (Figure 2). Consistent with a model that includes a steric bias, the reaction of substrate 3a afforded a 2.8:1 mixture of anti-Markovnikov (4a) and Markovnikov (4b) products in low yield (<20%). Additionally, significant isomerization to the internal alkene was also observed.12
Figure 2.

Benzoyl-protected homoallylic alcohol 3a illustrating substitution in the allylic position is necessary for high yields and selectivity.
This observation encouraged us to evaluate a homoallylic alcohol derivative with methyl substitution at the allylic position, which is a product of aldehyde crotylation, a common motif in synthesis. Submission of this substrate afforded a 46% yield of the anti-Markovnikov product (6a, Table 3) with no observed Markovnikov addition. The remainder of the mass balance was unreacted starting material and a small amount (<10%) of alkene isomerization. Again, this is consistent with a steric bias influencing product distribution. Therefore, several substrates with alkyl substitution at the allylic site were tested including a TBDPS-protected homoallylic alcohol with geminal dimethyl substitution (6b) (incapable of β-hydride elimination). Additionally, benzoyl-protected homoallylic and bishomoallylic alcohols, both with dimethyl substitution at the allylic site, resulted in high yields, indicating the placement of the protected alcohol is not important for selectivity (6c, 6d).
Table 3.
Hydroalkylation of substrates with methyl substitution in the allylic position.

|
Each entry had a >20:1 ratio of linear:branched product isomers, as determined by GC. Each entry represents the isolated yield on 0.5 mmol scale.
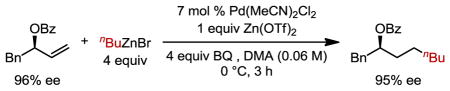
This data suggests that the high anti-Markovnikov selectivity observed might be a direct result of a steric effect whereby only the primary alkyl-Pd complex undergoes transmetallation, presumably under Curtin-Hammett control.6 Moreover, the secondary alkyl-Pd complex does not β-hydride eliminate toward the carbon containing the heteroatom, thereby leaving the adjacent C–H unperturbed (I′ → J′, Scheme 1a). However, it should be noted in the case of methyl substitution in the allylic position (6a, Table 3) a small amount of β-hydride elimination was observed in the form of internal alkene isomers. As a result, this reductive coupling can deliver highly enantiomerically-enriched protected alcohols. To showcase this process, an enantiomerically-enriched allylic benzoate with 96% ee was subjected to the reaction conditions and the product was found to retain its enantiomeric excess (Scheme 1b).
Scheme 1.

a) Proposed mechanism illustrating β-hydride elimination does not occur in the allylic position. b) Evaluation of retention of enantiomeric excess.
In summary, we have developed a Pd-catalyzed hydroalkylation reaction of protected allylic alcohols. This unconventional method is highlighted by the ability to access products with a stereocenter embedded in an unfunctionalized alkyl region common in natural products. This feature is a mechanistic consequence of the reaction, whereby β-hydride elimination does not occur at the heteroatom substituted allylic position. Additionally, homo- and bishomoallylic alcohols that contain methyl-substitution also afford the anti-Markovnikov product selectively. Future work will be focused on applying this catalyst system to synthetic goals as well as investigating alternative hydride sources and coupling partners in alkene hydrofunctionalization reactions.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (RO1GM3540).
Footnotes
Supporting Information Available Experimental procedures, full spectroscopic data, and chiral separations for new compounds are available free of charge via the internet at http://pubs.acs.org.
References
- 1.a) Bigi MA, Reed SA, White MC. J Am Chem Soc. 2012;134:9721. doi: 10.1021/ja301685r. [DOI] [PubMed] [Google Scholar]; b) White MC. Science. 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]; c) Davies HML, Du Bois J, Yu JQ. Chem Soc Rev. 2011;40:1855. doi: 10.1039/c1cs90010b. [DOI] [PubMed] [Google Scholar]; d) Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]; e) Che CM, Lo VKY, Zhou CY, Huang JS. Chem Soc Rev. 2011;40:1950. doi: 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]
- 2.a) Griggs ND, Phillips AJ. Org Lett. 2008;10:4955. doi: 10.1021/ol802041c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Son S, Fu GC. J Am Chem Soc. 2008;130:2756. doi: 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]; c) Suh YG, Kim SA, Jung JK, Shin DY, Min KH, Koo BA, Kim HS. Angew Chem Int Ed. 1999;38:3545. doi: 10.1002/(sici)1521-3773(19991203)38:23<3545::aid-anie3545>3.3.co;2-s. [DOI] [PubMed] [Google Scholar]; d) Trost BM, Ceschi MA, König B. Angew Chem Int Ed. 1997;36:1486. [Google Scholar]; e) Statsuk AV, Liu D, Kozmin SA. J Am Chem Soc. 2004;126:9546. doi: 10.1021/ja046588h. [DOI] [PubMed] [Google Scholar]; f) Katoh O, Sugai T, Ohta H. Tetrahedron: Asymmetry. 1994;5:1935. [Google Scholar]; g) Lewis JC, Coelho PS, Arnold FH. Chem Soc Rev. 2011;40:2003. doi: 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Dorling EK, Öhler E, Mulzer J. Tetrahedron Lett. 2000;41:6323. [Google Scholar]; b) Crimmins MT, King BW. J Am Chem Soc. 1998;120:9084. [Google Scholar]; c) He L, Byun HS, Bittman R. J Org Chem. 1998;63:5696. doi: 10.1021/jo971977y. [DOI] [PubMed] [Google Scholar]; d) Reddy PP, Yen KF, Uang BJ. J Org Chem. 2002;67:1034. doi: 10.1021/jo016216g. [DOI] [PubMed] [Google Scholar]; e) Bonini C, Chiummiento L, Lopardo MT, Pullez M, Colobert Fo, Solladié G. Tetrahedron Lett. 2003;44:2695. [Google Scholar]; f) Wipf P, Xu W. J Org Chem. 1993;58:825. [Google Scholar]; g) Gravestock MB, Knight DW, Lovell JS, Thornton SR. J Chem Soc, Perkin Trans. 1999;1:3143. [Google Scholar]; h) Alam M, Wise C, Baxter CA, Cleator E, Walkinshaw A. Org Process Res Dev. 2012;16:435. [Google Scholar]; i) Hanson RM. Chem Rev. 1991;91:437. [Google Scholar]
- 4.a) Hatano M, Miyamoto T, Ishihara K. J Org Chem. 2006;71:6474. doi: 10.1021/jo060908t. [DOI] [PubMed] [Google Scholar]; b) Knochel P, Singer RD. Chem Rev. 1993;93:2117. [Google Scholar]; c) Pu L, Yu HB. Chem Rev. 2001;101:757. doi: 10.1021/cr000411y. [DOI] [PubMed] [Google Scholar]; d) Bolm C, Hildebrand JP, Muñiz K, Hermanns N. Angew Chem Int Ed. 2001;40:3284. doi: 10.1002/1521-3773(20010917)40:18<3284::aid-anie3284>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; e) Hatano M, Miyamoto T, Ishihara K. Adv Synth Catal. 2005;347:1561. [Google Scholar]; f) Blaser HU. Chem Rev. 1992;92:935. [Google Scholar]; g) Knochel P, Jones P. Organozinc Reagents: A Practical Approach. Oxford University Press; New York: 1999. [Google Scholar]; h) Noyori R, Kitamura M. Angew Chem Int Ed. 1991;30:49. [Google Scholar]; i) Soai K, Niwa S. Chem Rev. 1992;92:833. [Google Scholar]; j) Xu Q, Wang G, Pan X, Chan ASC. Tetrahedron: Asymmetry. 2001;12:381. [Google Scholar]; k) Knochel P. Handbook of Functionalized Organometallics. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]; l) Noyori R. Asymmetric Catalysis in Organic Synthesis. Wiley; New York: 1994. [Google Scholar]
- 5.a) Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; b) Kawanami Y, Murao S, Ohga T, Kobayashi N. Tetrahedron. 2003;59:8411. [Google Scholar]; c) Zanotti-Gerosa A, Hems W, Groarke M, Hancock F. Platin Met Rev. 2005;49:158. [Google Scholar]
- 6.a) DeLuca RJ, Sigman MS. J Am Chem Soc. 2011;133:11454. doi: 10.1021/ja204080s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Urkalan KB, Sigman MS. J Am Chem Soc. 2009;131:18042. doi: 10.1021/ja908545b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Frisch AC, Beller M. Angew Chem Int Ed. 2005;44:674. doi: 10.1002/anie.200461432. [DOI] [PubMed] [Google Scholar]; c) Ishiyama T, Abe S, Miyaura N, Suzuki A. Chem Lett. 1992:691. [Google Scholar]; d) Zhou J, Fu GC. J Am Chem Soc. 2003;125:12527. doi: 10.1021/ja0363258. [DOI] [PubMed] [Google Scholar]; e) Netherton MR, Dai C, Neuschütz K, Fu GC. J Am Chem Soc. 2001;123:10099. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]; f) Netherton MR, Fu GC. Angew Chem Int Ed. 2002;41:3910. doi: 10.1002/1521-3773(20021018)41:20<3910::AID-ANIE3910>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]; g) Menzel K, Fu GC. J Am Chem Soc. 2003;125:3718. doi: 10.1021/ja0344563. [DOI] [PubMed] [Google Scholar]; h) Bissember AC, Levina A, Fu GC. J Am Chem Soc. 2012;134:14232. doi: 10.1021/ja306323x. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Stokes BJ, Opra SM, Sigman MS. J Am Chem Soc. 2012;134:11408. doi: 10.1021/ja305403s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.At the end of the cross-coupling sequence, Pd(0) is converted back to Pd(II) using benzoquinone as the oxidant. Zn(OTf)2 likley acts as a Lewis acid to facilitate this transformation.
- 9.For a review, see: Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w.
- 10.Weiner B, Baeza A, Jerphagnon T, Feringa BL. J Am Chem Soc. 2009;131:9473. doi: 10.1021/ja902591g. [DOI] [PubMed] [Google Scholar]
- 11.Vinylcyclohexane is selectively functionalized albeit in <30% yield.
- 12.Internal alkenes and 1,1 disubstituted alkenes were also submitted to the reaction conditions, which resulted in mainly unreacted starting material.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.