

Abstract
‘Epigenetics’ has been defined as the study of ‘mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence’. Chromatin modifications are major carriers of epigenetic information that both reflect and affect the transcriptional states of underlying genes. Several histone modifications are key players that are responsible for classical epigenetic phenomena. However, the mechanisms by which cells pass their histone modifications to daughter cells through mitotic division remain to be unveiled. Here, we review recent progress in the field and conclude that epigenetic modifications are not precisely maintained at a near-mononucleosome level of precision. We also suggest that transcription repression may be maintained by a buffer system that can tolerate a certain degree of fluctuation in repressive histone modification levels. This buffer system protects the repressed genes from potential improper derepression triggered by chromatin modification-level fluctuation resulting from cellular events, such as the cell-cycle-dependent dilution of the chromatin modifications and local responses to environmental cues.
Keywords: epigenetics, inheritance, transcription, chromatin modification
1. Introduction
‘Epigenetics’ is defined as ‘the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence’ [1]. Epigenetic information is largely provided by components of chromatin, including DNA methylation, histone modifications and histone variants [2]. By definition, epigenetic information should be inherited during mitotic divisions. To date, the best such example is the maintenance of CpG DNA methylation, in which the maintenance DNA methyltransferase DNMT1 copies the methylation pattern from a template strand to a newly synthesized DNA strand during/after semi-conservative DNA replication [3–8]. Histone modifications are also believed to be major carriers of epigenetic information. However, how the epigenetic information carried by histone modifications or histone variants is inherited during mitotic cell division remains an interesting but not fully resolved question [9–14]. In this review, we focus on the inheritance of epigenetic information carried by histone modifications.
2. Which histone modifications are likely to be epigenetic?
Histone modifications include acetylation, phosphorylation, methylation, ubiquitination and crotonylation [15,16]. Many histone modifications clearly affect the transcription process [17] and reflect the transcriptional states of underlying genes [18]. However, histone modifications are not necessarily ‘epigenetic modifications’, although they are often referred to as such, simply because they are involved in transcription [19]. By definition, one characteristic feature of epigenetic information is heritability. However, the types of histone modification that are indeed inherited during mitotic divisions are largely unknown. Nonetheless, a few criteria may help us to clarify the candidate modifications that are epigenetic.
To qualify as a primary epigenetic mark that can be transmitted during mitotic division, a mark must be relatively stable. Among histone modifications with known half-life, histone lysine methylation appears to be the most stable. The half-lives of different histone lysine methylation states range from several hours to days [20]; by contrast, the half-lives of histone acetylation and phosphorylation are in the range of minutes [21,22].
Another critical criterion for a primary epigenetic mark is that the existing mark should be able to, at least in part, direct or guide the reestablishment of a new mark on newly deposited histones. This guided information duplication is a common feature shared by other heritable systems, such as DNA-encoded genetic information and CpG DNA methylation-encoded epigenetic information. Determining whether epigenetic histone modifications actually possess such a guided information duplication system and the accuracy of this system are challenging tasks.
3. H3K9 and H3K27 methylation are the best candidates for epigenetic histone modifications
Among the different histone lysine methylation states, H3K9 and H3K27 methylation appear to be the most likely to be epigenetic, not only because they are key regulators of classic epigenetic phenomena, including position effect variegation [23–25], Polycomb silencing [26–29] and X inactivation [30,31], but also because they have been reported to possess certain characteristics of a prototype-guided information duplication system. In fission yeast, H3K9 methylase Clr4 and H3K9 methylated-histone-binding protein Swi6 have been shown to spread along heterochromatin [32], suggesting the existence of a ‘copy and paste’ mechanism. In mammals, the Clr4 and Swi6 orthologues Suv39h1/h2 and HP1 may share similar characteristics [24,25]. Interestingly, another H3K9 methyltransferase G9a [33] that mediates the majority of H3K9 dimethylation (H3K9me2) in euchromatic regions [34] interacts with DNMT1 and localizes to replication foci, which suggests a role in mediating the mitotic inheritance of H3K9me2 [35]. The H3K27 methylase complex PRC2 has also been reported to localize to replication foci [36]. Moreover, the PRC2 subunit EED recognizes H3K27me3 [36,37] and facilitates the allosteric activation of the PRC2 complex [37]. All these findings suggest that H3K9 and H3K27 methylation are the best candidates for epigenetic marks among histone modifications.
However, whether these modifications are faithfully inherited during mitotic division remains unclear. This inquiry can be broken down to two specific questions: (i) are these marks quickly reestablished during S phase in a replication-dependent manner? and (ii) do newly deposited histones acquire the same amounts of modification as the old histones?
(a). H3K9me3 and H3K27me3 are not fully reestablished during S phase
Histone modifications are often studied with antibody-based techniques, such as western blot analysis. During the cell cycle, the expected global level changes of epigenetic histone marks are within twofold, a relative modest change that is difficult to be accurately measured using antibodies. Despite these challenges, it has been reported that H3K27me3 is reestablished during G1 phase using western blot analysis [38].
More recently, stable isotope labelling with amino acids in cell culture (SILAC)-based quantitative mass spectrometry [39] has been increasingly used to quantify histone modifications [20,40–43]. Using this technology, it has been reported that H4K20 and H3K79 methylation are gradually established in a replication-independent manner [40,42].
Using [13C6, 15N4] l-arginine (abbreviated as R10)-labelled histones as external reference, we recently determined the relative abundance of histone modifications, including H3K9 and H3K27 methylation at different stages of the cell cycle, using an Orbitrap mass spectrometer that offers a wide linear dynamic range and that enables discrimination between acetyl and tri-methyl groups [44]. Interestingly, although enzymes mediating H3K9me2 and H3K27me3 can associate with the replication foci [35,36], the global levels of these marks together with H3K9me3 and previously reported H3K79me2 [42] are not fully reestablished during S phase [44]. Instead, the global levels of higher methylations states (di-, tri-methylation) are reduced to about 70 per cent [44], because newly synthesized histones lack modifications that are normally associated with chromatic histones [45]. The levels of H3K9me2 are fully recovered shortly after S phase [44], which suggests that the replication-dependent reestablishment of this mark reported previously [35] is the major contributor. By contrast, the levels of H3K9me3 and H3K27me3 are reestablished with much slower kinetics. These modifications appear to be gradually reestablished throughout the cell cycle and the overall levels of these modifications are fully recovered during G1 phase [44].
(b). Newly deposited histones do not possess comparable levels of H3K9me3 and H3K27me3 when compared with the old histones even after one entire round of the cell cycle
The next question is whether newly deposited histones possess comparable levels of histone marks compared with the old histones. Comparable levels would suggest that the enzymatic machinery that reestablishes the histone marks might be able to distinguish the newly deposited histone from the old histones. Furthermore, this machinery might use pre-existing marks on the old histones to guide the establishment of new marks specifically onto the newly deposited histones.
SILAC again is the best approach available to address the above-mentioned question, because SILAC not only discriminates newly deposited histones from the old ones, but also allows the detection and quantification of histone modifications simultaneously. This approach has already been successfully used in studying H4K20 and H3K79 methylation [40,42].
H3K9me2 on newly deposited histones reaches a level comparable to that in the old histones shortly after S phase [44]. This implies that the replication-dependent reestablishment of this mark reported previously [35] is the primary mechanism responsible for this modification.
However, despite being considered the most epigenetic marks, the levels of H3K9me3 and H3K27me3 on the newly deposited histones are far lower than the levels on the old histones [44]. This finding, together with recovery of the levels of these modifications during G1 phase [44], suggests that old histones continue to be methylated throughout the cell cycle in order to maintain the overall levels of these modifications. Indeed, histones with these methylation states containing both old and new methyl groups have been detected [20,44]. In sum, the overall levels of epigenetic modifications are maintained during mitotic division, but a precise mechanism that faithfully maintains the modification pattern at a near-mononucleosome resolution appears to be absent.
4. A buffer model that unifies faithful gene silencing and imprecise epigenetic inheritance
A series of recent findings suggest that cells lack a precise general mechanism for the mitotic inheritance of histone modification-based epigenetic information. The findings that histone lysine methylation states are not required to exist symmetrically within each nucleosome [43] and that canonical histone (H3–H4)2 tetramers undergo conservative segregation [46] are inconsistent with some models of an epigenetic inheritance mechanism with mononucleosome resolution [47]. Moreover, old histones continue to acquire new methyl groups in the next cell cycle and possess much greater levels of higher methylation states than newly deposited histones [44]. This finding further contradicts the possibility of a general epigenetic inheritance mechanism with a near-mononucleosome level of precision.
Nevertheless, genes repressed by H3K27me3 or H3K9me3 have not been reported to be derepressed during S phase, which indicates that the transcriptional states of target genes that associate with these epigenetic marks are indeed faithfully maintained. Therefore, an important question concerns how faithful gene repression could be maintained without a precise mitotic inheritance mechanism for the epigenetic marks. We previously proposed a buffer model that unifies the imprecise reestablishment of modifications and faithful maintenance of transcriptional repression [44]. We elaborate below.
(a). Is epigenetic repression mediated by a few nucleosomes or a domain?
The term ‘repressive modifications’ has been widely used because of the association of these modifications with repressed transcriptional states and the requirement of these modifications for keeping their target genes in the repressed states. However, we want to emphasize that genes are not directly repressed by the repressive modifications themselves but by downstream events that help fold the underlying genes into non-permissive higher-order chromatin structures [48]. In other words, histone modification-based epigenetic repression is unlikely to be mediated by a few important nucleosomes, such as nucleosomes next to the transcription start sites. Instead, scores of nucleosomes within a domain that spans tens of kilobases are more likely organized into a non-permissive higher-order chromatin structure that ultimately represses gene expression. Indeed, repressive modifications often span a relatively wide region of chromatin, rather than being localized to a few nucleosomes [49–51].
If gene repression is achieved by folding scores of nucleosomes into higher-order chromatin structure, rather than by repressive modifications on a few critical nucleosomes, then the precise restoration of histone marks at near-mononucleosome level is neither practical [44] nor necessary for the faithful repression of the target genes. In other words, nucleosomes are the basic units for chromatin but are not necessarily the basic units for gene repression.
(b). Heterogeneous nature of histone methylation marks within the repressed domain
Lysine residues can be mono-, di- and tri-methylated. Increasing numbers of genome-wide histone modification profiling experiments using methylation state-specific antibodies have established a clear consensus within the chromatin field, which holds that distinct histone methylation states are often enriched at different regions of chromatin for specific functions [52]. However, these distinct methylation states are merely enriched and not uniformly distributed in their corresponding regions. SILAC-based studies clearly demonstrated that higher methylation states are gradually established over the lower methylation states for most histone lysine methylation sites [20,40–42,44]. Therefore, histone lysine methylation within the repressed domains that span scores of nucleosomes must be heterogeneous or all four forms—me0, me1, me2 and me3—must coexist, although certain forms might be dominant. Such heterogeneity is required for the reported gradual maturation of higher methylation states [40–42,44]. Such heterogeneity is also consistent with the observation that the levels of histone modifications fluctuate during the cell cycle [44].
(c). Bimodal effect of repressive histone marks on transcription
If histone modifications are indeed heterogeneous within the repressed domains and their levels fluctuate during the cell cycle, then why does such fluctuation not derepress the target genes? A simple explanation is that the effect of repressive histone marks on transcription is not linear, but bimodal (figure 1). We propose that a relatively low abundance of repressive marks, such as 20–30% of H3K27me3 within a region of scores of nucleosomes, is sufficient to abrogate transcriptional activity within the domain (figure 1). Throughout the cell cycle, the actual levels of H3K27me3 within such regions may fluctuate without affecting the transcriptional states of the region, so long as they are maintained above the threshold.
Figure 1.
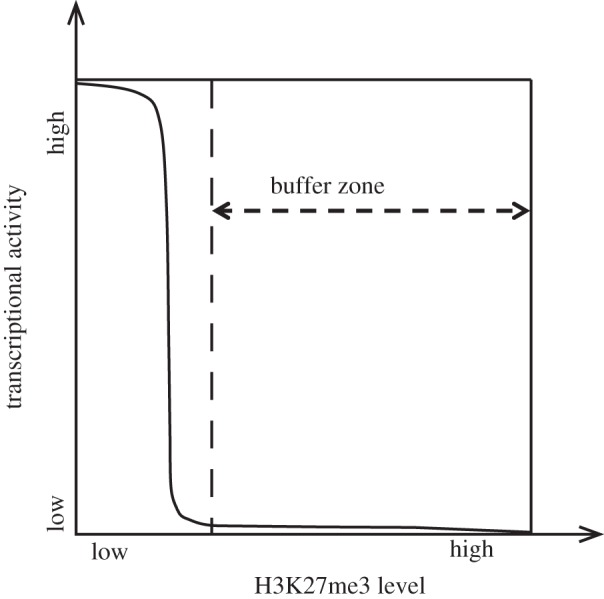
A buffer model for mitotic inheritance of histone modifications. Bimodal relationship between transcriptional activity and the levels of repressive histone marks creates a buffer zone that allows the fluctuation of repressive histone modifications without sacrificing the faithful maintenance of target gene repression. H3K27me3 was used as a representative example for repressive histone marks.
Because this system functions like a buffer, we termed it the buffer model, and the range that repressive marks are allowed to fluctuate is designated the buffer zone (figure 1).
We speculate that this buffer system not only protects the repressed status of genes from cell-cycle-dependent chromatin modification fluctuations, but also safeguards gene repression from certain assaults against the levels of chromatin modifications mediated by environmental cues.
(d). Reinforcement of silencing by combinatory chromatin modifications
In the preceding sections, we presented a simplified view for the role of histone lysine methylation in mitotic epigenetic inheritance. However, we also would like to point out that these repressive histone lysine methylation marks often function coordinately with other chromatin modifications. For example, H3K9me3, H4K20me3 and DNA methylation are all important components of pericentromeric constitutive heterochromatin in mammals [53–55]. Likewise, H3K27me3 and H2A mono-ubiquitylation also often function together to repress Polycomb targets in facultative heterochromatin [26,56–59]. It is highly possible that fluctuation of one modification can also be compensated by other cooperative modifications.
5. Pre-existing histone modifications are necessary but not sufficient to reestablish the epigenetic marks
Ideally, heritable information should be duplicated during mitotic division by a templated information copying mechanism, such as DNA replication for DNA sequence-encoded genetic information and maintenance DNA methylation for CpG methylation-encoded epigenetic information. For histone modifications, some of the histone methyltransferases such as Clr4 and PRC2 possess enzymatic activities that can amplify the pre-existing modifications, and these amplification activities are required for the maintenance of heterochromatin [32,37]. However, these systems apparently have much lower fidelity than other information duplication systems, such as the DNA replication system and the CpG DNA methylation maintenance system. Therefore, the inheritance of histone modification-mediated epigenetic information is not solely determined by the pre-existing modifications and their corresponding enzymes. Instead, other players including cis-elements of DNA and the transcription status of the region are also essential [18,60].
6. Comparison between epigenetic inheritance mediated by histone lysine methylation and epigenetic inheritance mediated by the sir proteins in budding yeast
There is no repressive histone lysine methylation marks in budding yeast Saccharomyces cerevisiae. Nevertheless, budding yeast possess the most studied heterochromatin-like structures at the silent mating-type loci and telomere regions mediated by the SIR complex [61]. Sir2, an NAD-dependent histone deacetylase that mainly deacetylates histone H4 lysine 16 [62] forms a complex with Sir4 and Sir3 [61,63,64], a protein preferentially binds to nucleosome with deacetylated H4K16 [65]. SIR complex is first recruited to nucleation site though silencer binding protein [61] and then spread to neighbouring regions via its activities in deacetylating H4 histones and binding to deacetylated H4 histones [62,65].
Although mediated through a different modification, this silencing mechanism in budding yeast shares certain similarities with epigenetic inheritance mediated by histone lysine methylations. First, silencing in yeast is also achieved through the formation of regional compacted heterochromatic environments containing several kilobases of hypoacetylated heterochromatic regions [60]. Second, pre-existing modifications can partially guide the establishment of epigenetic states of newly assembled chromatin [60,62,65].
7. Concluding remarks
The proposed buffer model was based on experimental observations. It is, however, worth asking a more philosophic question: why is the epigenetic system not granted the same fidelity that the genetic system possesses?
To answer this question it is necessary to know the basic tasks of the epigenetic system. Multicellular organisms are required to achieve two simple tasks: (i) cells should be able to alter their fate to generate distinct cell types for different functions during differentiation and (ii) cells and their progeny should be able to maintain their fate when further differentiation is not required at the post-mitotic stages or during proliferation. To fulfil these tasks, the epigenetic system must simultaneously possess the characteristics of plasticity and inheritability. Plasticity allows the transformation of one genome into hundreds of epigenomes and transcriptomes, whereas inheritability permits the maintenance of every single epigenome and its corresponding transcriptome. To achieve both tasks, the epigenetic system cannot afford to have the same fidelity as the genetic system, which inevitably sacrifices its plasticity.
Acknowledgements
Research in B.Z.'s laboratory is supported by grants (nos 2011CB812700, 2011CB965300) from the Chinese Ministry of Science and Technology and by the Howard Hughes Medical Institute International Early Career Scientist Programme.
References
- 1.Russo VEA, Martienssen RA, Riggs AD. 1996. Introduction. In Epigenetic mechanisms of gene regulation, Russo VEA, Martienssen RA, Riggs AD. (eds), pp. 1–4 New York, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- 2.Allis CD, Jenuwein T, Reinberg D. 2006. Overview and concepts. In Epigenetics, Allis CD, Jenuwein T, Reinberg D. (eds), pp. 23–56 New York: Cold Spring Harbor Laboratory [Google Scholar]
- 3.Holliday R, Pugh JE. 1975. DNA modification mechanisms and gene activity during development. Science 187, 226–232 10.1126/science.187.4173.226 (doi:10.1126/science.187.4173.226) [DOI] [PubMed] [Google Scholar]
- 4.Bestor TH. 1992. Activation of mammalian DNA methyltransferase by cleavage of a Zn binding regulatory domain. EMBO J. 11, 2611–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF. 1997. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277, 1996–2000 10.1126/science.277.5334.1996 (doi:10.1126/science.277.5334.1996) [DOI] [PubMed] [Google Scholar]
- 6.Hermann A, Goyal R, Jeltsch A. 2004. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 279, 48 350–48 359 10.1074/jbc.M403427200 (doi:10.1074/jbc.M403427200) [DOI] [PubMed] [Google Scholar]
- 7.Sharif J, et al. 2007. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912 10.1038/nature06397 (doi:10.1038/nature06397) [DOI] [PubMed] [Google Scholar]
- 8.Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. 2007. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317, 1760–1764 10.1126/science.1147939 (doi:10.1126/science.1147939) [DOI] [PubMed] [Google Scholar]
- 9.Henikoff S, Furuyama T, Ahmad K. 2004. Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet. 20, 320–326 10.1016/j.tig.2004.05.004 (doi:10.1016/j.tig.2004.05.004) [DOI] [PubMed] [Google Scholar]
- 10.Martin C, Zhang Y. 2007. Mechanisms of epigenetic inheritance. Curr. Opin. Cell Biol. 19, 266–272 10.1016/j.ceb.2007.04.002 (doi:10.1016/j.ceb.2007.04.002) [DOI] [PubMed] [Google Scholar]
- 11.Probst AV, Dunleavy E, Almouzni G. 2009. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 10, 192–206 10.1038/nrm2640 (doi:10.1038/nrm2640) [DOI] [PubMed] [Google Scholar]
- 12.Zhu B, Reinberg D. 2011. Epigenetic inheritance: uncontested? Cell Res. 21, 435–441 10.1038/cr.2011.26 (doi:10.1038/cr.2011.26) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan G, Zhu B. 2012. Histone variants and epigenetic inheritance. Biochim. Biophys. Acta 1819, 222–229 10.1016/j.bbagrm.2011.06.007 (doi:10.1016/j.bbagrm.2011.06.007) [DOI] [PubMed] [Google Scholar]
- 14.Xu M, Zhu B. 2010. Nucleosome assembly and epigenetic inheritance. Protein Cell 1, 820–829 10.1007/s13238-010-0104-0 (doi:10.1007/s13238-010-0104-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 10.1038/cr.2011.22 (doi:10.1038/cr.2011.22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan M, et al. 2011. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 10.1016/j.cell.2011.08.008 (doi:10.1016/j.cell.2011.08.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Carey M, Workman JL. 2007. The role of chromatin during transcription. Cell 128, 707–719 10.1016/j.cell.2007.01.015 (doi:10.1016/j.cell.2007.01.015) [DOI] [PubMed] [Google Scholar]
- 18.Henikoff S, Shilatifard A. 2011. Histone modification: cause or cog? Trends Genet. 27, 389–396 10.1016/j.tig.2011.06.006 (doi:10.1016/j.tig.2011.06.006) [DOI] [PubMed] [Google Scholar]
- 19.Ptashne M. 2007. On the use of the word ‘epigenetic’. Curr. Biol. 17, R233–R236 10.1016/j.cub.2007.02.030 (doi:10.1016/j.cub.2007.02.030) [DOI] [PubMed] [Google Scholar]
- 20.Zee BM, Levin RS, Xu B, LeRoy G, Wingreen NS, Garcia BA. 2010. In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 285, 3341–3350 10.1074/jbc.M109.063784 (doi:10.1074/jbc.M109.063784) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson V, Shires A, Chalkley R, Granner DK. 1975. Studies on highly metabolically active acetylation and phosphorylation of histones. J. Biol. Chem. 250, 4856–4863 [PubMed] [Google Scholar]
- 22.Chestier A, Yaniv M. 1979. Rapid turnover of acetyl groups in the four core histones of simian virus 40 minichromosomes. Proc. Natl Acad. Sci. USA 76, 46–50 10.1073/pnas.76.1.46 (doi:10.1073/pnas.76.1.46) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rea S, et al. 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 10.1038/35020506 (doi:10.1038/35020506) [DOI] [PubMed] [Google Scholar]
- 24.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120 10.1038/35065132 (doi:10.1038/35065132) [DOI] [PubMed] [Google Scholar]
- 25.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124 10.1038/35065138 (doi:10.1038/35065138) [DOI] [PubMed] [Google Scholar]
- 26.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 10.1126/science.1076997 (doi:10.1126/science.1076997) [DOI] [PubMed] [Google Scholar]
- 27.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. 2002. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111, 185–196 10.1016/S0092-8674(02)00975-3 (doi:10.1016/S0092-8674(02)00975-3) [DOI] [PubMed] [Google Scholar]
- 28.Muller J, et al. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208 10.1016/S0092-8674(02)00976-5 (doi:10.1016/S0092-8674(02)00976-5) [DOI] [PubMed] [Google Scholar]
- 29.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of Zeste protein. Genes Dev. 16, 2893–2905 10.1101/gad.1035902 (doi:10.1101/gad.1035902) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plath K, et al. 2003. Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135 10.1126/science.1084274 (doi:10.1126/science.1084274) [DOI] [PubMed] [Google Scholar]
- 31.Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. 2004. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303, 644–649 10.1126/science.1092727 (doi:10.1126/science.1092727) [DOI] [PubMed] [Google Scholar]
- 32.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. 2001. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110–113 10.1126/science.1060118 (doi:10.1126/science.1060118) [DOI] [PubMed] [Google Scholar]
- 33.Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. 2001. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 276, 25 309–25 317 10.1074/jbc.M101914200 (doi:10.1074/jbc.M101914200) [DOI] [PubMed] [Google Scholar]
- 34.Tachibana M, et al. 2002. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 10.1101/gad.989402 (doi:10.1101/gad.989402) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. 2006. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 20, 3089–3103 10.1101/gad.1463706 (doi:10.1101/gad.1463706) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K. 2008. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10, 1291–1300 10.1038/ncb1787 (doi:10.1038/ncb1787) [DOI] [PubMed] [Google Scholar]
- 37.Margueron R, et al. 2009. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767 10.1038/nature08398 (doi:10.1038/nature08398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aoto T, Saitoh N, Sakamoto Y, Watanabe S, Nakao M. 2008. Polycomb group protein-associated chromatin is reproduced in post-mitotic G1 phase and is required for S phase progression. J. Biol. Chem. 283, 18 905–18 915 10.1074/jbc.M709322200 (doi:10.1074/jbc.M709322200) [DOI] [PubMed] [Google Scholar]
- 39.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. 2002. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 10.1074/mcp.M200025-MCP200 (doi:10.1074/mcp.M200025-MCP200) [DOI] [PubMed] [Google Scholar]
- 40.Pesavento JJ, Yang H, Kelleher NL, Mizzen CA. 2008. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell Biol. 28, 468–486 10.1128/MCB.01517-07 (doi:10.1128/MCB.01517-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scharf AN, Barth TK, Imhof A. 2009. Establishment of histone modifications after chromatin assembly. Nucleic Acids Res. 37, 5032–5040 10.1093/nar/gkp518 (doi:10.1093/nar/gkp518). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sweet SM, Li M, Thomas PM, Durbin KR, Kelleher NL. 2010. Kinetics of re-establishing H3K79 methylation marks in global human chromatin. J. Biol. Chem. 285, 32 778–32 786 10.1074/jbc.M110.145094 (doi:10.1074/jbc.M110.145094) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Xiong J, Xu M, Chen S, Zhu B. 2011. Symmetrical modification within a nucleosome is not required globally for histone lysine methylation. EMBO Rep. 12, 244–251 10.1038/embor.2011.6 (doi:10.1038/embor.2011.6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu M, Wang W, Chen S, Zhu B. 2012. A model for mitotic inheritance of histone lysine methylation. EMBO Rep. 13, 60–67 10.1038/embor.2011.206 (doi:10.1038/embor.2011.206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G. 2006. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol. Cell 24, 309–316 10.1016/j.molcel.2006.08.019 (doi:10.1016/j.molcel.2006.08.019) [DOI] [PubMed] [Google Scholar]
- 46.Xu M, Long C, Chen X, Huang C, Chen S, Zhu B. 2010. Partitioning of histone H3–H4 tetramers during DNA replication-dependent chromatin assembly. Science 328, 94–98 10.1126/science.1178994 (doi:10.1126/science.1178994) [DOI] [PubMed] [Google Scholar]
- 47.Wu H, Zhu B. 2011. Split decision: why it matters? Front. Biol. 6, 88–92 10.1007/s11515-011-1040-y (doi:10.1007/s11515-011-1040-y) [DOI] [Google Scholar]
- 48.Li G, Reinberg D. 2011. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 21, 175–186 10.1016/j.gde.2011.01.022 (doi:10.1016/j.gde.2011.01.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwartz YB, Kahn TG, Nix DA, Li XY, Bourgon R, Biggin M, Pirrotta V. 2006. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat. Genet. 38, 700–705 10.1038/ng1817 (doi:10.1038/ng1817) [DOI] [PubMed] [Google Scholar]
- 50.Boyer LA, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353 10.1038/nature04733 (doi:10.1038/nature04733) [DOI] [PubMed] [Google Scholar]
- 51.Lee TI, et al. 2006. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125, 301–313 10.1016/j.cell.2006.02.043 (doi:10.1016/j.cell.2006.02.043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shilatifard A. 2006. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 75, 243–269 10.1146/annurev.biochem.75.103004.142422 (doi:10.1146/annurev.biochem.75.103004.142422) [DOI] [PubMed] [Google Scholar]
- 53.Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. 2004. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 1251–1262 10.1101/gad.300704 (doi:10.1101/gad.300704) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peters AH, et al. 2001. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337 10.1016/S0092-8674(01)00542-6 (doi:10.1016/S0092-8674(01)00542-6) [DOI] [PubMed] [Google Scholar]
- 55.Lehnertz B, et al. 2003. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13, 1192–1200 10.1016/s0960-9822(03)00432-9 (doi:10.1016/s0960-9822(03)00432-9) [DOI] [PubMed] [Google Scholar]
- 56.Simon JA, Kingston RE. 2009. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 10, 697–708 10.1038/nrm2763 (doi:10.1038/nrm2763) [DOI] [PubMed] [Google Scholar]
- 57.Tavares L, et al. 2012. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148, 664–678 10.1016/j.cell.2011.12.029 (doi:10.1016/j.cell.2011.12.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ku M, et al. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 4, e1000242. 10.1371/journal.pgen.1000242 (doi:10.1371/journal.pgen.1000242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kallin EM, Cao R, Jothi R, Xia K, Cui K, Zhao K, Zhang Y. 2009. Genome-wide uH2A localization analysis highlights Bmi1-dependent deposition of the mark at repressed genes. PLoS Genet. 5, e1000506. 10.1371/journal.pgen.1000506 (doi:10.1371/journal.pgen.1000506) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moazed D. 2011. Mechanisms for the inheritance of chromatin states. Cell 146, 510–518 10.1016/j.cell.2011.07.013 (doi:10.1016/j.cell.2011.07.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moretti P, Freeman K, Coodly L, Shore D. 1994. Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 8, 2257–2269 10.1101/gad.8.19.2257 (doi:10.1101/gad.8.19.2257) [DOI] [PubMed] [Google Scholar]
- 62.Imai S, Armstrong CM, Kaeberlein M, Guarente L. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 10.1038/35001622 (doi:10.1038/35001622) [DOI] [PubMed] [Google Scholar]
- 63.Moazed D, Kistler A, Axelrod A, Rine J, Johnson AD. 1997. Silent information regulator protein complexes in Saccharomyces cerevisiae: a SIR2/SIR4 complex and evidence for a regulatory domain in SIR4 that inhibits its interaction with SIR3. Proc. Natl Acad. Sci. USA 94, 2186–2191 10.1073/pnas.94.6.2186 (doi:10.1073/pnas.94.6.2186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. 1997. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 11, 83–93 10.1101/gad.11.1.83 (doi:10.1101/gad.11.1.83) [DOI] [PubMed] [Google Scholar]
- 65.Hecht A, Laroche T, Strahl-Bolsinger S, Gasser SM, Grunstein M. 1995. Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: a molecular model for the formation of heterochromatin in yeast. Cell 80, 583–592 10.1016/0092-8674(95)90512-X (doi:10.1016/0092-8674(95)90512-X) [DOI] [PubMed] [Google Scholar]