

Abstract
In recent times, the epigenetic study of pluripotency based on cellular reprogramming techniques led to the creation of induced pluripotent stem cells. It has come to represent the forefront of a new wave of alternative therapeutic approaches in the field of stem cell therapy. Progress in drug development has saved countless lives, but there are numerous intractable diseases where curative treatment cannot be achieved through pharmacological intervention alone. Consequently, there has been an unfortunate rise in incidences of organ failures, degenerative disorders and cancers, hence novel therapeutic interventions are required. Stem cells have unique self-renewal and multilineage differentiation capabilities that could be harnessed for therapeutic purposes. Although a number of mature differentiated cells have been characterized in vitro, few have been demonstrated to function in a physiologically relevant context. Despite fervent levels of enthusiasm in the field, the reality is that other than the employment of haematopoietic stem cells, many other therapies have yet to be thoroughly proven for their therapeutic benefit and safety in application. This review shall focus on a discussion regarding the current status of stem cell therapy, the issues surrounding it and its future prospects with a general background on the regulatory networks underlying pluripotency.
Keywords: stem cells, pluripotency, induced pluripotent stem cells, embryonic stem cells
1. Introduction: cellular reprogramming and half a century of stem cell therapy
In the past century, pharmacological advancements have dramatically improved the clinical outlook for patients suffering from all sorts of diseases. However, there are limitations to this approach; beyond a certain point, it becomes palliative only and no longer curative. Attempts at identifying the regulatory networks underlying pluripotency began in 1952, with cellular reprogramming experiments studying the nuclear transfer (NT) of amphibian nuclei [1]. Together with cell-fusion experiments involving embryonal carcinoma cells (ECCs) [2] and embryonic stem cells (ESCs) [3], it was demonstrated that the nuclear epigenetic modifications of somatic cells acquired during differentiation are reversible and can be rearranged to a configuration that supports a pluripotent state. At that time, ‘pluripotency’ was defined along the lines of ‘an unlimited capacity to give rise to all cells of the embryo’. Researchers had not yet begun to consider the true therapeutic impact of this statement. A decade later in 1963, Till & McCulloch [4] demonstrated the presence of repopulating cells in mouse bone marrow (BM). This was to be the first demonstration of stem cells and the birth of regenerative medicine as it is known today. In later years, the cells were identified and characterized as haematopoietic stem cells (HSCs) and since that first demonstration, numerous other somatic stem cell (SSC) populations have been identified [5].
The connection between cellular reprogramming and the clinical application of stem cells was not immediately obvious. In many ways, they are two sides of the same coin so to speak. It was not until difficulties were encountered in handling SSCs in vitro, and the realization that some tissues had no resident pool of regenerating stem cells, did researchers began to consider the necessity of searching for a cell higher up in the hierarchical order of cell potency. The two were thus inextricably linked. Some thought the answer came in 1998, with the first in vitro establishment of human embryonic stem cells (hESCs) [6]. Not only were hESCs found to be pluripotent, they were to become a vital tool in further dissecting the intricacies of the regulatory networks that underlies pluripotency. However, ESCs also became something of an ethical landmine, as the derivation of these cells required the destruction of human embryos [7]. In 2006, the first generation of induced pluripotent stem cells (iPSCs) took place, which appeared to be ‘ES cell-like’ but without the need for instigating an ethical debate [8]. The focus in medicine, in theory at least, began shifting towards how to harness the regenerative powers of stem cells with the hope of perhaps curing intractable diseases in transplantation settings. Not only that, scientists and clinicians dared to dream and began to explore the possibilities of replacing damaged cells either at a singular level or even as whole organs. It is natural therefore to ask what are stem cells? What makes stem cells unique and how can they be used to treat patients?
2. Stem cells overview
In reviewing stem cells, one of their key defining characteristics are their ability to self-renew. ‘Self-renewal’ is the ability to undergo cycles of mitotic division while maintaining the same undifferentiated state as the parent cell [9]. This is particularly important in tissues where there is a resident pool of stem cells that are responsible for maintaining the lifelong homeostasis of that tissue, including HSCs [10] and neural stem cells (NSCs) [11] and epithelial stem cells (EpSCs) [12]. In the clinical situation, a potential therapeutic strategy could be to aim for the replacement or the induction of these stem cells so that when injury occurs, they can reconstitute the tissue system in question or facilitate the natural mechanisms of repair. The other defining characteristic of stem cells, and perhaps the one that has most captured the imagination of so many is the characteristic of cell potency. ‘Potency’, from the Latin potens, meaning having power, is a rather appropriate way to characterize stem cells because it is this power that science is trying to harness. This refers to the capacity of stem cells in being able to differentiate into different cell types (figure 1). As shown in the figure, stem cells can be assigned to a hierarchical order based on their degree of potency. At the top, cells of the morula are deemed to be totipotent in that they can differentiate into all tissue types. ESCs and iPSCs are pluripotent cells that are one magnitude short of totipotency because they cannot differentiate into placental tissue. Multipotent cells can include resident tissue-specific stem cells that are more lineage-restricted. Oligopotent cells are usually committed progenitors such as the common myeloid and common lymphoid progenitors (CMP and CLP) that are restricted towards differentiating into an even smaller subcategory of cells (myeloid and lymphoid, respectively).
Figure 1.
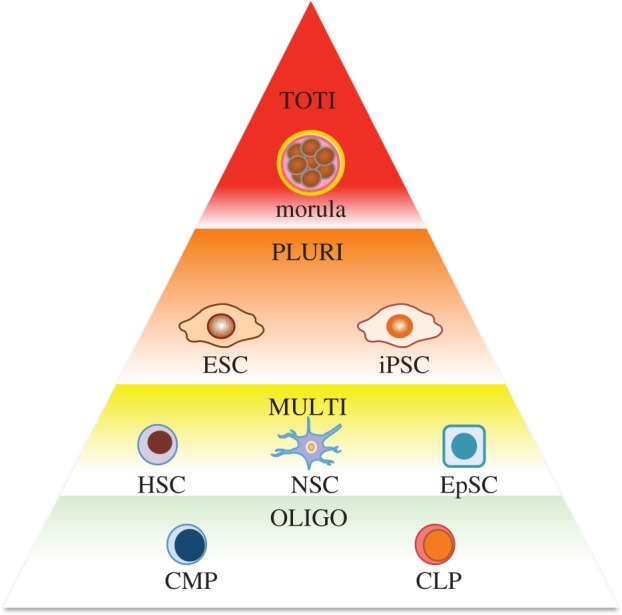
A pyramidal hierarchy of cell potency. Sitting atop the pyramid (red), cells of the morula are the most potent cells that can differentiate into all tissue types. ESCs and iPSCs occupy the next level, as they cannot differentiate into the placenta (orange). Tissue resident stem cells (HSCs, NSCs, EpSCs) can differentiate into multiple cell types restricted to that lineage (yellow). At the base are cells with more limited differentiation potential (CMP, CLP) usually committed progenitors (light green). TOTI, totipotent; PLURI, pluripotent; MULTI, multipotent; OLIGO, oligopotent; ESC, embryonic stem cell; iPSC, induced pluripotent stem cell; HSC, haematopoietic stem cell; NSC, neural stem cell; EpSC, epithelial stem cell; CMP, common myeloid progenitor; CLP, common lymphoid progenitor.
In broad terms, stem cells are categorized into ‘somatic’ or ‘embryonic’ stem cells. SSCs, also know as adult stem cells, are generally multipotent and able to differentiate into any cell of a specified lineage. Notable examples include HSCs, NSCs, EpSCs in the skin [13], cornea [14], gut [15] and mesenchymal stem/stromal cells (MSCs). Their anatomical location is a reflection of the critical roles that they possess in the development, maintenance and repair of specified tissues and organs [16]. These locations are often loosely referred to as the ‘niche’, but strictly speaking, the name has a much stronger emphasis on the surrounding micro-environment and its constituent supporting and regulatory cells derived from which are extrinsic signals that can strongly influence the functions of the residing stem cell [17]. This is an important concept to keep in mind regarding SSCs because clinical strategies can be targeted at replenishing the resident cell population either through transplantation or through endogenous manipulation to positively enhance cell functions [18]. The BM, host niche for HSCs, remains the best defined and, although incompletely so, it is one of the most readily accessible niches of all. The BM–HSC relationship was the basis for the first stem cell experiments. NSCs are found in the dentate gyrus of the hippocampus and the lateral ventricle wall of the olfactory bulb [19]. These represent very sensitive parts of the brain, thereby making the prospective isolation of NSCs rather difficult and treacherous. Of the EpSCs, these are a located within the epithelia overlying external tissue surfaces such as the skin, gut and cornea. The epithelia in these tissues are subject to high rates of cell turnover; thus the role of resident EpSCs is crucial in maintaining homeostasis. For MSCs, their in vivo physiological functions remain unclear much less having a defined niche. Recent studies indicate that when used in a transplant setting, it is the MSCs themselves that act as supporting cells through the paracrine and anti-inflammatory effects that these cells have [20]. Use of the term niche is probably inappropriate, given the lack of understanding of the surrounding micro-environment hosting MSCs. Generically, they are referred to purely by their source of derivation for example adipose stem cells (ASCs), as in MSCs derived from adipose tissue. However, not all tissues in the body contain a resident pool of stem cells, a notable example being the endocrine pancreas [21]. Damage to, or the defective function of these tissues can only be treated by a regimen of pharmaceutical drugs where applicable or whole organ transplantation from a HLA matched donor.
Occupying a higher order of cell potency than SSCs, the first derivation of mouse embryonic stem cells (mESCs) took place in 1981 [22,23] and then of hESCs in 1998 [6]. These are derived either from cells of the morula or from the inner cell mass (ICM) of the blastocyst stage embryo (figure 2). ESCs are pluripotent cells capable of differentiating into all cell types except for the placenta. This differentiation capacity can be assessed by testing the ability of the cells to partake in tissue development. Upon injection into a blastocyst, ESCs can contribute efficiently to the formation of all adult tissues including the germline [23]. For this reason, ESCs have become the gold standard with which all in vitro cultivated pluripotent cells are judged against. In the case of mESCs, it is possible to assess pluripotency by carrying out the teraploid complementation assay, which is currently the most stringent assay of testing developmental potential of its kind [24]. For ethical reasons, the same assay cannot be carried out using hESCs where the evaluation of the developmental potency of human pluripotent cells is limited to the teratoma formation assay. This assays for the spontaneous potential of the cells in differentiating into the three germ layers and hence it is not as stringent. Still, under defined conditions, hESCs can differentiate into transplantable neural precursors [25], functional hepatocytes [26], haematopoietic progenitors [27] and other tissue lineages. As such, their therapeutic potentials are clear to see, and efforts are ongoing to exploit this.
Figure 2.
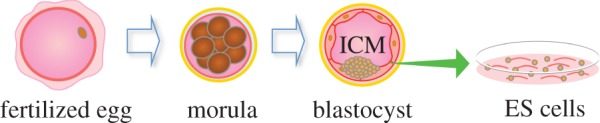
Derivation and establishment of embryonic stem cells (ESCs). Zygotes are maintained until reaching the morula or blastocyst stages of development. Cells from the intra cellular mass (ICM) are extracted and maintained in vitro on a feeder layer of cells.
However, there are some major obstacles that have precluded the successful clinical application of ESCs. The pluripotent potential of these cells makes them therapeutically attractive but by the same token, when this potential becomes dysregulated, undifferented ESCs can form teratomas in vivo [28]. The ramifications could be severe particularly in cell-replacement therapy should even one undifferentiated cell enter the patient. Another point for consideration is that there is the risk of eliciting an immune response in the recipient, given the mismatch in major histocompatibility complex (MHC) class I antigens [29]. Unfortunately, the most negatively publicized of these challenges is that the in vitro establishment of ESCs involves destruction of the embryo. Some have cried ‘the destruction of a life’. Criticism has ranged from public condemnation by the Vatican [30] to calls for restraint by the then US president George W. Bush [31]. These may not have any direct scientific or clinical relevance but some mention is warranted, given that this is an obstacle that has continued to remain insurmountable. Therefore, the application of ESCs for research or therapeutic purposes is likely to face continued restraint in developing further progress, particularly in translating into clinical trials.
In any case, none of this public negativity should serve to diminish the enormous impact that ESCs have had on studying the transcriptional regulation of pluripotency and of stem cell therapy. From a cellular reprogramming perspective, ESCs were a critical tool in achieving the seminal breakthrough of induced pluripotency [8] along with the technique of somatic cell nuclear transfer (SCNT) [32] and the study of pluripotency-associated transcription factors. In a cell-fusion experiment with ESCs and adult thymocytes, it was demonstrated that the epigenotype of the somatic cells could be reset to a configuration that supported a pluripotent state [33]. This dominant reprogramming activity exhibited by ESCs can in part be attributable to the raised expression of master regulatory transcription factors of pluripotency such as Oct3/4, Nanog and Sox2 [34]. The work of Takahashi and Yamanaka took this idea further and identified the combination of Oct3, Sox2, Klf4 and c-Myc from a pool of 24 predominantly ES-cell specific genes. In 2007, they generated the first human iPSCs by retrovirally expressing this combination in dermal fibroblasts [35,36]. These are considered to be ESC-like pluripotent cells. When subjected to the teratoma assay, human iPSCs can also contribute to the formation of tissue from all three germ layers.
The landmark discovery of iPSCs had fuelled enthusiasm to fervent levels and since then, research in the stem cell therapy field has grown almost exponentially. There were, and still are, many reasons to feel excitement. Initially at least, iPSCs seemed identical to ESCs but its establishment did not necessitate the destruction of an embryo (figure 3). This would permit ongoing research to continue without any lingering ethical constraints. From an epigenetic standpoint, it seemed incredible that the overexpression of a simple set of genes can induce a whole genome-wide wave of epigenetic modifications that one can assume must take place to restore a differentiated somatic cell to a pluripotent state. The initial two reports of iPSC generation used integrating vector systems to reprogramme fibroblasts. Since then, induced pluripotency has also been achieved using non-integrational adenoviruses [37], and even DNA-free Sendai viruses [38]. Different somatic targets were also selected for reprogramming, including haematopoietic cells, hepatic progenitor cells [39] and adipocytes, which all led to the generation of viable iPSCs displaying the same set of characteristics as do ESCs. So the framework for a new therapeutic strategy was set. Stem cell therapy can become more personalized [40]. This is because patient autologous iPSCs can be derived from somatic cells isolated directly from the patient. In theory at least, differentiated cells could be transplanted without encountering issues relating to immune rejection. Those with an inherited genetic disorder could have their autologous iPSCs genetically modified and differentiated into functional cells to then repair damaged tissues, for example, in the treatment of primary immunodeficiencies (PIDs) [41]. Such cell-replacement-type therapies are not the only applications of iPSCs. They can also be used to recapitulate a disease phenotype for the purposes of disease modelling [42]. This has profound promise in applications such as in vitro drug screening and the elucidation of disease pathophysiological development. Unfortunately, the therapeutic promise of iPSCs is not reflected in terms of actual numbers of ongoing clinical trials. There are some reservations, given that similar to ESCs, iPSCs retain the ability for teratoma formation in vivo. To date, there are no ongoing clinical trials employing the use of iPSCs, and similar to ESCs, unless the safety profile of these cells can be firmly established, for the immediate future at least, it may be challenging to bring iPSCs into clinical trial (see figure 4 for a comparison between ESCs, iPSCs and SSCs and the categories of interventional open trials that they have been employed in).
Figure 3.

Generation and applications of induced pluripotent stem cells (iPSCs). A somatic cell source is isolated from the patient. Pluripotency-associated genes are delivered to reprogramme the cell. Applications of iPSCs include cell therapy and studies of pathogenesis.
Figure 4.

A comparison of stem cell characteristics and major categories of interventional trials. (a) Table comparing stem cell characteristics between embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs) and somatic stem cells (SSCs). (b) Bar graph comparing the relative numbers of open trials of the major stem cell categories (www.clinicaltrials.gov and modified from [43]). MSC, mesenchymal stem cell.
Of all the open FDA-approved clinical trials, some 75 per cent are for HSCs but only 3 per cent involving ESCs and none involving iPSCs [43]. Those that make use of SSCs, despite the exclusion of MSCs, account for nearly 90 per cent. This pattern is informative in a number of ways and one could speculate upon the meaning of it. The tiny proportion of trials employing pluripotent stem cells may be a reflection of the difficulty in convincing the relevant regulatory body that the risks of tumorigenicity following transplantation can be thoroughly contained. In this regard, it may be unreasonable to question the lack of iPSC-based therapies in such a manner because they are still a relatively new discovery. Bear in mind it took an entire decade to pass between the first in vitro derivations of hESCs to the first clinical trial in 2011, which proved to be a monumental exercise in overcoming regulatory hurdles. Should iPSCs-based therapies eventually reach clinical trial, it is likely that these cells will be subjected to the same set of tight regulations as ESCs. As mentioned previously, there are numerous different ways in which induced pluripotency could be accomplished, but there are also discrepancies between institutions in how iPSCs are maintained [44]. Combined with existing technical limitations, it has not yet been possible to have a clear epigenetic profile or even a molecular definition of these cells. These are questions that must be addressed in order to be able to firmly establish their safety and to promote their use clinically. In the following sections, specific features of each category of stem cells shall be discussed in more detail alongside examples of potential applications and ongoing trials.
3. Somatic stem cells
Compared with the few ESC-related clinical trials that have taken place, SSC-based therapies very much dominate the current landscape. They may be considered as being more advantageous in that they are lineage-restricted and can theoretically be transplanted into patients with less prior ex vivo manipulation. However, in most cases, the identity of SSCs is not yet clearly defined whether in terms of phenotypic appearance or immunophenotype. Therefore, the isolation of a purified population of these cells can be technically challenging. Another issue lies in having sufficiently large enough numbers of cells to treat the disease in question. SSCs generally have more limited self-renewal capacity, whose precise mechanism remains to be clearly identified. This means that these cells undergo only a limited number of self-renewing divisions before differentiating into more committed cell progenitors [45]. In the case of HSCs, it has not been possible to expand them in vitro without a loss in potency. This represents a limitation of their regenerative potentials. Another limiting factor is that not all tissues possess an endogenous pool of stem cells. A notable example is the endocrine pancreas in diabetes [46]. For other categories such as NSCs, the endogenous stem cell may not spontaneously regenerate injured areas in response to damage. With EpSCs, there has been some clinical success in transplanting ‘sheets’ of epithelia following in vitro cultivation [47,48]. These tend to act as a temporary scaffold because epithelial tissue is subject to high rates of cell turnover and the multi-layered ‘sheets’ are eventually replaced by epithelia derived from the resident tissue EpSCs of the recipient. On the other hand, MSCs are adherent and expandable cells in culture. They show promise in that cells can be harvested in large numbers in the case of ASCs. Indeed, they are multipotent, but differentiation occurs only following considerable manipulation by cytokines in vitro, the extent of which may extend beyond what is clinically acceptable for therapeutic purposes. Here we discuss the more important clinical aspects of notable examples of SSCs.
(a). Haematopoietic stem cells
The field began with an investigation into the haematopoietic system following the seminal work of Till & McCulloch. Some may even argue that more so than the much-publicized ESCs and iPSCs, HSCs have defined the stem cell therapy field like no other. These cells exemplify all the properties characteristic of a so-called stem cell. Part of the simplistic elegance of the haematopoietic system is that cells can exist and function at a singular level unlike other systems where complex cell-to-cell interactions may be required. For these reasons, it has been possible to isolate highly purified populations of HSCs [49] and to use them to demonstrate at least in part, the hierarchy of mammalian development in relation to stem cells, which may also be applicable to other cellular systems. The regenerative powers of HSCs were epitomized in a very elegant and understated fashion by demonstrating that the transplantation and successful engraftment of only one single HSC is capable of reconstituting haematopoiesis in a recipient [50,51]. At the top of the haematopoietic hierarchy are the multipotent long-term HSCs (LT-HSCs) so called because they can reconstitute haematopoiesis upon transplantation into a primary recipient and subsequently into a secondary recipient [52]. These can either self-renew to produce another LT-HSC or become a short-term HSCs (ST-HSCs), which retains the multilineage potential but is no longer able to reconstitute long-term haematopoiesis [49]. Descending down the hierarchical system, HSCs eventually become committed progenitors of the major haematopoietic lineages lymphoid, myeloid and erythroid. Finally, committed progenitors become terminally differentiated cells [53].
The normal environment for HSCs is the BM, but it can also be derived from peripheral blood or cord blood. Bone marrow transplants (BMTs) have been successful for over 30 years. It has become by far the most successful and widely performed stem cell therapy, becoming standardized treatment for hematological diseases such as leukaemia, aplastic anaemia and immunodeficient disorders [54]. Despite having such a wide range of indications, its prognostic outcome is yet very poor with nearly 50 per cent 5-year overall mortality. BMT can either be autologous (self) or allogeneic (from donor). Allogeneic transplantation can be problematic because similar to organ transplantation, both the donor and the recipient have to be human leucocyte antigen (HLA) matched [55]. So for all its elegance and simplicity, the issue of donor availability can also be a limiting factor to the therapeutic potential of HSCs. Furthermore, a vigorous myeloablative pretreatment also know as ‘conditioning’ precludes the use of HSC transplantation for autoimmune or other non-fatal diseases that can be treated otherwise [56]. There are various theories on the matter but in general it is believed that the conditioning procedure can lead to overt, or excess damage to the immediate environment and the cells that constitute the BM ‘niche’. Consequently, this may compromise HSCs functions such as engraftment within the niche or the ability of cells to maintain its ‘stemness’, in other words, the typical characteristics of a stem cell. Efforts are ongoing into optimizing the conditions to facilitate the outcomes of BMT.
Putting these challenges aside, as mentioned previously, there are numerous ongoing examples of clinical trials involving HSCs. Standard BMT does not involve modification of the HSCs found within the BM before transplantation. In other cases, HSCs are taken from the peripheral blood and isolated by the identification of the surface CD34+ antigen. Strictly speaking, this antigen alone does not confer the identity of true LT-HSCs, but rather a heterogeneous population of LT-HSCs, ST-HSCs and committed progenitors are isolated. A viral vector can then used to correct or compensate for a defective gene before transplanting into patients of the genetically modified cells. Collectively, the transplantation of gene-modified HSCs is called gene therapy (GT) [57]. Some of the most notable examples involve the treatment of PIDs. These include severe combined immunodeficiency (SCID) [58], chronic granulomatous disease (CGD) [59] and Wiskott–Aldrich syndrome [60]. There has been some notable successes in the GT field but unfortunately, a number of clinical trials have been marred by incidences of insertional mutagenesis owing to activation of an oncogene leading to clonal expansion or frank leukaemia [61]. Additionally, harvesting HSCs from the BM is an invasive procedure that carries with it a certain degree of morbidity and mortality. The other alternative is to stimulate cell release from the BM by granulocyte colony stimulating factor, but some believe that this procedure can compromise the quality and function of the cells after it enters the peripheral blood. Currently, it is not possible to expand HSCs while retaining a steady population of LT-HSCs that are capable of reconstituting haematopoiesis upon serial transplantation. Efforts are still ongoing to understand the milieu of factors and conditions within the BM that allows HSCs to self-renew.
(b). Neural stem cells
Neurodegenerative disorders or injury involving the central and peripheral nervous systems have also raised considerable interest owing to the devastation they wreak on the quality of life that patients experience. Well-published examples include Parkinsons' disease (PD), amyotrophic lateral sclerosis (ALS), Alzheimer's disease (AD) and stroke. NSCs are the second most common stem cells that are currently used in clinical trials. However, where HSCs may be considered as a symbol of elegance and simplicity, the organization of neuronal tissue architecture is perhaps the most complex in the body. More so than other organs, the brain and other neuronal components are composed of a wide range of highly specified sub cell types (neuronal and non-neuronal) with a degree of interconnectedness that requires computational power to model. This level of complexity of structural organization makes it even more challenging to understand the precise disease pathophysiology or to formulate effective treatments. In examples such as PD and ALS, broadly speaking, the disease phenotype can be traced to the defective function of a single type of neuron (dopaminergic and motor). On the other hand, AD is characterized by neuronal loss in the cerebral cortex and other subcortical areas. In a stroke, or cerebrovascular accident, there is cell death (neuronal and non-neuronal) of entire areas [62]. Therefore, the challenges involved in treatments include not only generating enough numbers of cells, but also regeneration of the correct cell type. NSCs have been obtained from foetal and adult brain, and can be expanded in culture. In the adult brain, they are located in the dentate gyrus of the hippocampus and in the lateral ventricle wall of the olfactory bulb [19]. Despite the existence of NSCs in the adult brain, unlike haematopoiesis, neurogenesis may not be a lifelong process, as it appears to occur primarily during development until adulthood, when the roles of NSCs become much less clear [63]. NSCs show developmental-stage-dependent differentiation potentials. Early NSCs can make more neurons than glial cells, but late NSCs tend to make glial cells but not neurons. To date, neural differentiation from NSCs has been demonstrated for both neurons and glial cells albeit to a fairly limited repertoire. Unlike HSCs however, injections would have to be given invasively to specific areas of the brain. Combined together, these factors severely restrict the clinical potential of NSCs.
For more complex CNS disorders, to achieve optimum recovery, direct replacement of dead cells or tissue reconstruction may be required. However, given the complexity of CNS disorders in mind, this may be a difficult strategy to attempt. With current strategies, scientists are leaning towards neuroprotection, rather than towards neuroregeneration, based on the idea of tissue plasticity. This can be defined as the innate response which SSCs have at being able to adapt and reprogramme their functionality in response to pathophysiological changes to the environment in which they are subjected to [64]. Another aspect is that certain categories of NSCs may display latent activity, where, for example, dormant cells become physiologically active upon injury. When applied to NSCs, there is evidence to suggest that following the injection of NSCs into a site of damage, there is a concerted release of neuroprotective molecules that includes immunomodulatory substances, neutrotrophic growth factors and stem cell regulators, which combine to promote repair of the damaged region. Similar behaviour has also been demonstrated for HSCs and MSCs. Until such times that the anatomical arrangement of neural tissue is better understood as well as the function and derivation of these cells, for the moment at least NSC treatment will be aimed at neural protection. The transplantation of cultured human NSCs have been attempted for Batten's disease, Pelizaeus–Merzbacher disease, chronic spinal cord injuries, strokes, amyotropic lateral sclerosis and Parkinson's disease. Current clinical trials involving NSCs concern largely that of a phase-1 safety issue.
(c). Epithelial stem cells
Epithelial stem cells refer broadly to the resident stem cell population residing within epithelial tissue. In brief, epithelia are densely packed cells connected by tight junctions and desmosomes that form continuous sheets over tissue surfaces. Their functions include physical protection against external environments, selective absorption of water and nutrients, as well as producing and secreting glandular secretions. The functions of epithelia are dependent on the tissue that it overlies [13]. Notable examples include the skin epidermis, the gut epithelium and the corneal epithelium. Unlike other somatic tissues, epithelia are usually subject to relatively high rates of cell turnover, and hence the role of EpSCs is crucial for maintaining tissue homeostasis in replacing damaged or dead cells.
Among the most well-studied EpSCs are the epidermal stem cells, which include the basal, bulge and sebaceous gland stem cells. The epidermis is made up of numerous pilo-sebaceous units with each unit consisting of a hair follicle, a sebaceous gland and the more superficial interfollicular epidermis (IFE). Under normal physiological conditions, collectively these epidermal stem cells are responsible for the homeostatic regulation of maintaining the cellular layers of the IFE and for overseeing rounds of hair growth [65]. There is evidence to suggest the existence of some form of functional overlap between the different stem cell populations. For example, sustained activation of the WNT pathway can convert basal stem cells into hair follicle stem cells [66]. Additionally, there also appears to be some plasticity in their functions in response to changes in the host environment. For example, IFE and sebaceous gland stem cells from donor mice became hair follicle cells following transplantation [67]. Bulge cells, normally responsible for the maintenance of the hair follicle, can also contribute to IFE repair upon injury. In any case, these innate mechanisms of repair are limited to incidences where the cut or injury is relatively small such that the wound site can be sufficiently covered by granulation tissue on top of which new epithelium is built.
For more severe cases of skin trauma, epidermal stem cells are particularly important because these formed the basis for the first demonstration in vitro of highly expandable stem cells that could subsequently be applied to regenerate tissue. The first demonstration occurred in 1984 by Howard Green and colleagues in treating two patients with extensive skin damage caused by third-degree burns [68]. Keratinocytes, a generic name for epithelial cells in a multi-layered epithelium, were derived from patients without sorting, subsequently expanded in vitro and grown to a sheet, which was then transplanted over the sites of skin damage. As a continuation of this, the concept of cultivating keratinocytes in vitro was extended for the treatment of the genetic disease called junctional epidermolysis bullosa caused by a defect with the Laminin 5-β3 gene. It affects the ability of the basal layer of keratinocytes to adhere to the basal membrane causing blistering of the skin. The strategy involved using a retroviral vector to introduce Laminin 5-β3 cDNA into cultivated epidermal cells. This rescued the adherent properties of the cells and following subsequent transplantation successful engraftment was observed and the patient was cured [69].
Another tissue to have attracted a lot of attention in stem cell biology and regenerative medicine is the intestinal epithelium. This is a lining covering the luminal surface of the gut extending through the small to large intestines. It is composed of numerous functional units of crypts and villi. Crypts are invaginations that appear to ‘burrow’ into the lamina propria layer of the gut, which runs in continuation with the ‘finger-like’ projections of the villi [15]. The crypt-villus unit has been described as one of the simplest representations of a self-renewing entity. It is much simpler in structural organization in comparison with epidermal epithelia. Gut stem cells are found at the base of each crypt flanked by paneth cells and undergo self-renewal to produce more vigorously proliferating transient activating (TA) cells. These cells divide further and extend upwards along the walls of the crypt. At regular intervals, paneth cells insert into this vertically rising columnar procession. Upon reaching the villus junction, differentiation is initiated whereby TA cells become enterocytes or absorptive/secretive cells. Differentiated cells continue to be pushed upwards along the walls of the villus, where upon reaching the tip, cells undergo apoptotic death and shedding [70]. This regenerative process is very similar to what occurs in the IFE of the epidermis. However, as the intestinal epithelium is only one single-cell-thick, it is possible to observe the processes in a precise sequential order. This makes it much easier to identify and observe the basic cellular events encompassing tissue renewal.
Most recently, there has been exciting development where, using the Lgr5 protein as a marker to identify gut stem cells under defined culture conditions, single sorted cells became so-called gut organoids [71]. These are three-dimensional structures that are composed of gut stem cells as well as other cell types that can be found along the crypt–villus axis. In a group of recipient mice, injury was artificially induced to the colon. Following the delivery of donor-derived organoids through an enema, damaged areas were regenerated. Under histological examination, the regenerated areas displayed the appearance of normal gut epithelia. Most of all, it appeared that the transplanted cells of the organoids supported long-term epithelial regeneration by remaining engrafted after more than six months [72]. It was also demonstrated during the same study that these ‘gut organoids’ could be produced in large numbers from relatively few gut stem cells. These results hold promise for the treatment of diseases such as Crohn's disease, inflammatory bowel syndrome or other disorders of the gastrointestinal tract where there can be chronic damage to the gut epithelium [73]. Although it remains to be demonstrated whether these organoid-regenerated portions are fully functional, it remains an elegant demonstration of the clinical potential of EpSCs without the inevitable complications of using pluripotent cells.
Regeneration of the cornea is another example where there has been experience of clinical success. The cornea is a multi-layered avascular tissue with optical functions such as the maintenance of visual acuity and acting as a physical barrier to protect the inner eye. On its outer surface is a layer of corneal epithelium, which extends in continuation into an area of the adult cornea, called the limbus [74]. This is a transitional area between the corneal and conjunctival epithelium, where so-called limbal stem cells are found. These are believed to be the primary stem cell population involved in corneal regeneration. Allogeneic corneal transplantation, also known as keratoplasty, is already an established therapy to treat damage to the cornea [75]. The success of such a therapy appears to depend on whether the limbus remains intact following injury. If damage to the cornea is extensive and does indeed extend to the limbus, then there may be a need, for example, to supplement it with a transplantation of limbal stem cells that are firstly derived from the opposite uninjured eye and then expanded by in vitro culture [76]. This clinical strategy has generated some interest, as the use of cultured limbal epithelial cells is already an established part of the treatment of ocular surface diseases.
(d). Mesenchymal stem/stromal cells
The study of MSCs began with a misunderstanding over their nomenclature and functions. They were initially named mesenchymal stem cells, as they were derived from BM stroma and that the expanded progeny cells possessed osteo- and chondrogenic potential [77]. It was inaccurate to call these ‘mesenchymal’, which by definition refers to cells of the haematopoietic lineage that MSCs are not capable of differentiating into. In addition, the earliest studies of MSCs were not clonal in experimental design and hence without demonstrating that the cells could self-renew, it was perhaps inappropriate to call them stem cells. Nowadays they are more correctly defined as stromal cells as some, but not all, MSCs can be defined as stem cells [78]. They have been characterized as fibroblastic, adherent cells that are expandable in culture and are positive for markers including CD90 and CD105 [79]. Worth noting is that there are no single characteristic marker that will allow for the prospective isolation of a highly purified population of these cells. For this reason, the precise identity of MSCs continues to remain under debate as do comparisons between different studies. Nonetheless MSCs can be derived from BM aspirates or in larger quantities from adipose tissue. They are also found in amniotic fluid, Wharton's jelly of the umbilical cord, umbilical cord blood, dental pulp of deciduous teeth and skeletal muscle [80].
Compared with pluripotent ESCs or iPSCs, MSCs have a greater biosafety profile and lower risk of tumorigenicity [81]. MSCs are believed to be multipotent for chondrogenic, osteogenic and adipogenic differentiation [82]. However, it is important to emphasize that demonstration of this multipotent differentiation potential occurred following extreme manipulation of in vitro culturing environments [83]. In vivo, there has been no firm suggestion to indicate that MSCs partake in the repair of these tissues; hence, their normal physiological roles remain unclear. Unfortunately, the ‘stem cell’ part of the name has continued to stick, leading to the often incorrect and erroneous assumption that whatever beneficial effects MSCs may have are provided through cell replacement. Gradually, there is increasing evidence to suggest that MSCs can secrete paracrine signals or act as immune suppressors to stimulate tissue recovery. Yet, at the same time, there is also contradicting evidence to suggest that this is not the case [84]. Despite this ongoing uncertainty, numerous MSC-based therapies have made it to clinical trial, perhaps in part due to the fact that they are much less immunogenic compared with other types of stem cell. The range of clinical trials that are ongoing include those attempting the treatment of bone and cartilage repair, myocardial infarct occlusive vascular disease, liver cirrhosis and strokes. Whether it is actually able to exert such effects, attempts have also been made to harness the suspected immunomodulatry effects of MSCs in treating graft versus host disease and autoimmune diseases such as multiple sclerosis, systemic lupus erythematosis and Crohn's disease. Again, the outcome has been mixed, and therapeutic benefits difficult to determine. The fact of the matter is probably that a number of the clinical trials that have taken place should not have been allowed to do so in the first place. For future MSC-therapies, the precise characterization of MSCs, better understanding of therapeutic mechanism and careful assessment of the efficacy of clinical trials are necessary.
4. Embryonic stem cells
Two clinical trials involving ESCs have so far taken place. In 2011, California-based venture company Geron Corporation started a first-in-man phase 1 clinical study, using ES cell-derived oligodendrocytes to treat patients with spinal cord injury. Unfortunately, the trial was discontinued for unknown business decisions. There has been no formal publication on the findings and no insight on either the efficacy of the proposed treatment or its safety. What was apparent from the trial was the stringent implementation of regulatory protocols, which some considered as ‘oversight’ before the trial was eventually approved. The other clinical trial involved the transplantation of ESC-derived retinal epthelium in patients with Stargardt's macular dystrophy and dry age-related macular degeneration. The company Advance Cell Technologies sponsored the trial, but clinical benefits were difficult to determine owing to the short four-month duration and that each trial contained only one patient. These are two of only a very limited number of examples of ESC-based therapies.
Much of the focus on ESCs has been largely on that of its potential clinical applications. However, as seen in the examples mentioned earlier, there remain considerable difficulties in assessing the efficacy of ESC-based therapies along with overcoming regulatory hurdles. Perhaps what has been overlooked slightly is the crucial role that ESCs have played as a comparative control alongside iPSCs in studies of the regulatory mechanisms that underlie pluripotency and differentiation. ESCs are considered to be derivatives of naturally occurring ICM cells and can pass stringent assays for assessing contribution to development. Therefore, ESCs are considered as reliable in vitro representations of mammalian development suitable for the aforementioned purposes. For example, techniques such as global gene-expression analysis and bisulphite genomic sequencing have been carried out to identify differences between ESCs and iPSCs during differentiation in terms of gene-expression profiles and epigenetic marks [40]. These types of comparative experiment were to prove very informative. The first of these insightful pieces of information concerns the concept of ‘somatic’ or ‘epigenetic memory’ [85]. These are residual configurations of the epigenome that were retained following the reprogramming process. By broad definition, ‘epigenome’ refers to the precise types of DNA methylation and histone modification, the combinatory effect of which can influence gene expression. Reprogramming usually results in the demethylation of most previously methylated regions. Methylation can have a repressive effect on gene expression, depending on its loci. This is not a permanent modification but as long as the epigenetic features that make up the ‘somatic memory’ remain, there are implications upon the propensity of somatic cells to be reprogrammed or that of iPSCs to undergo differentiation. For example, a failure to demethylate pluripotency-associated genes has been proposed as a mechanism that explains the partial reprogramming in iPSCs or cases where reprogramming efficiency is low. It is also possible that retention of this epigenetic memory may give preferential bias in differentiation into a certain lineage. This was observed in cases where haematopoietic differentiation was deemed to be less efficient from iPSCs that were derived from fibroblasts and neural progenitors [85]. These preferential biases may be avoided through either a second round of reprogramming (secondary reprogramming) [85] or through long-term culture in vitro that can remove the remaining traces of residual methylation. The repression may also be lifted by the treatment of cells with demethylating agents. On a functional level, these repressive effects of the epigenome on either reprogramming or cellular differentiation will need to be carefully considered when attempting to use iPSCs for clinical purposes.
Another important observation arising from similar studies is that during the reprogramming process, there is potential for the inadvertent capturing or retention of genomic regions from the somatic state that are prone to repression by methylation. These captured regions continue to be transferred to differentiated progenies; hence, pluripotency reprogramming can lead to some kind of a secondary accumulating effect. Studies that compared the DNA methylomes of human ESCs and iPSCs initially identified these regions as areas of aberrant iPSC-specific differential methylation [40]. Eventually, these were termed differentially methylated regions (DMRs). It was also found that this occurred at a higher frequency in iPSCs, and it appears to be a permanent defect that affects the actual genomic structure and not just the epigenome; hence it could not be erased through passaging [86]. Current evidence seems to identify the accumulation of DMRs as a stochastic process that is not caused by aberrant or random mutation. What is known is that the frequency of DMRs in ESCs and progenitor somatic cells are both lower compared with the frequency in iPSCs. It suggests that accumulation does not begin until after the cell moves away from the somatic state and yet it does not seem to be caused by intrinsic or extrinsic factors arising from being in a pluripotent state either as frequency in ESCs is also low. Therefore, the reprogramming process could be the cause of this DMR phenomenon. The functional consequences are unclear, but one could hypothesis that there may be a profound effect on the expression of genes in the proximity of the DMR region.
In general, ESCs have a relatively established role as a comparative control in relation to iPSCs. This role is supported by the assertion that ESCs are derivatives of the naturally occurring ICM as opposed to being a product of artificial reprogramming. However, it may be questionable to consider ESCs as ‘direct derivatives’ of the ICM, considering the methodology of ESC derivation that involves removal of cells from the natural in vivo environment without clear knowledge of the effect that it may have. Additionally, there is also the discrepancy in derivation between ESCs and iPSCs, which could have implications in accounting for the inherent heterogeneity within each population. Although both are characterized as pluripotent cells, only iPSC derivation methods account for sub-clonal distinction from an original donor source. Therefore, questions may be targeted at addressing the degree of ‘equivalence’ not only between cells of the ICM and ESCs, but also that between ESCs and iPSCs. Firstly, let us consider the methods of ESC derivation. The natural procession of embryonic development in vivo is likely to be in part, a product of dynamic cellular interactions including that between the ICM and the trophectoderm (TE). This is the extra-embryonic layer that surrounds the ICM but for the in vitro derivation of ESCs, TE removal can enhance the efficiency of ESC establishment and facilitate its maintenance [87]. It remains unclear what the exact consequences might be of disrupting this dynamism. There are, however, several lines of evidence; some more speculative than others at suggesting differences between ICM cells and ESCS. For example, critical genes involved in developmental signalling pathways, including transforming growth factor beta (TGFβ), insulin growth factor and the MAP kinase pathways were identified to be uniquely expressed in ICM cells but not in ESCs [88]. Other circumspective indications include findings to suggest that ESCs are thought to have a higher degree of global DNA methylation [89,90] and have longer telomere lengths [89]. Additionally, ERas, a transforming oncogene, is highly expressed in mouse ESCs but not in embryos [91]. Taken together, these findings highlight the need for closer scrutiny of the resemblance between ICM cells and ESCs.
Another important point to consider is that there is a fundamental difference between how ESCs and iPSCs are derived. In brief, ESCs are usually cells of the ICM plated onto mitotically inactivated mouse embryonic fibroblasts (MEFs). Under the appropriate culture conditions, cells expand and aggregate to form colonies, which are subsequently expanded further. With the derivation of iPSCs, somatic cells are reprogrammed and similarly plated onto MEFs. The crucial difference is that there is usually a subcloning step involved where once iPSC colonies appear, they are individually transferred onto fresh MEFs and expanded separately. Therefore, iPSCs are often referred to as ‘clones’ of a given cell line, whereas ESCs are purely referred to in relation to the original embryo from which it is derived without further clonal distinction. It is therefore reasonable to suspect that there could be heterogeneity within each ESC line that may become apparent for example in the efficacy and efficiency of differentiation into a specified lineage [92]. There is also the possibility that certain clones within a population of colonies may be afforded some kind of growth advantage, which means that after a certain number of passages, only a few dominant ones will remain. This is speculative, however, and can only be determined if there is also a subcloning step involved sufficiently early during ESC derivation. Therefore, returning to the question of equivalence between ESCs and iPSCs, the simple answer would be ‘neither’. Current opinions seem to confer on the notion that these are two distinctly different classes of cells each with considerable amounts of inherent heterogeneity but at the same time, there is also significant overlapping similarities between the two [40]. While the degree of resemblance between ICM cells and ESCs has yet to be clearly defined, for the moment at least, ESCs remain the closest in vitro recapitulation of human embryonic development. Nonetheless, a more cautious approach is warranted when interpreting results of epigenetic studies involving ESCs, where it must be considered in the appropriate context particularly in comparison with iPSCs.
5. Induced pluripotent stem cells
With the challenges faced in the application of ESCs, the stem cell therapy field appeared to be stuck at a bottleneck as the search began for an alternative pluripotent cell source. In humans, this was achieved in 2007 with the derivation of hiPSCs [35]. Since then, much research has been undertaken using these cells but thus far, there are no ongoing iPSC-based clinical trials. Already in the previous section, there has been a discussion on some of the technical limitations relating to reprogramming by defined factors and differentiation from a pluripotent state that had been discovered in comparative studies alongside ESCs. In addition, one can imagine that iPSC-based therapy would encounter similar regulatory obstacles to those that were encountered in bringing forward their ESC counterpart. It is therefore worth foreseeing what these might be and how clinical translation may be accelerated. The first thing to consider is that as arrogant as it may sound, dare one say it, perhaps induced pluripotency came about too easily. Earlier attempts at cellular reprogramming to pluripotency were achieved either through SCNT or cellular fusion with ECCs and ESCs. Both of these techniques were difficult to perform and are limited by even lower levels of efficiency than reprogramming by defined factors [93,94]. SCNT has always been associated with some negative social and ethical issues. With cellular fusion, the resulting hybrid cell is tetraploid, thus precluding its application for clinical purposes. The ectopic co-expression of defined transcription factors to achieve pluripotency is considerably simpler on many levels and much more reproducible. Numerous other viral and non-viral methods of co-expressing sets of defined factors in different somatic cells have also been attempted and were successful. It led some researchers to think that perhaps the difficult part in achieving cellular therapy for patients is over. The truth is there are other similarly difficult and equally as important challenges to overcome in addition to pluripotency reprogramming.
One of the main difficulties in iPSC-based therapies may well be in the directed differentiation from pluripotent to functional somatic cells. This is possible in vitro by manipulating the culture conditions using cytokines critical to the development of that specific tissue. However, the in vivo development of cells and tissue systems is a product of dynamic interactions between cells and its environment. The supplementation of cytokines represents only part of this dynamism and it remains a tremendous technical challenge to fully replicate this in vitro. Many differentiated cells have been tested for characteristic functions but very rarely in a physiologically relevant context to comprehensively prove that they are functionally comparable to their in vivo counterparts. Therefore, casting aside all other considerations for the moment, a reasonable question to ask might therefore be whether transplanted cells are able to partake in the homeostatic regulation of tissue functions in vivo? There are numerous such examples, one of them being the differentiation from pluripotent cells into transplantable β-pancreatic cells for the treatment of diabetes [95]. Although there are now established protocols for producing these cells, none has yet been able to demonstrate the same regulated secretion of insulin in response to glucose stimulation as observed in vivo [96]. What this example highlights is that basic functional tests are sufficient for characterization purposes, but more comprehensive assessments will be necessary, perhaps involving transplantation into an animal host, to predict in vivo functioning [97]. Also applicable to both ESCs and iPS cells, there is inherent instability of the genome over long-term culture [98]. Whether it is reprogramming or differentiation into functional somatic tissue, both processes takes place in relatively harsh culture environments that subject cells to stressful conditions on its genome, which could lead to aberrant gene mutations. Therefore, to make sure that cells are clinically safe for transplantation, it may be necessary to screen each and every clone for signs of epigenetic aberrations or gene mutations [99]. These are all important considerations to keep in mind during the ongoing efforts of generating not only transplantable, but also physiologically functional cells.
One of the much-trumpeted features of iPSCs is that they can give rise to more personalized therapy to patients. This can take the form of disease modelling, where autologous iPSCs are generated for applications such as the study of disease pathophysiology, drug screening and toxicology [100]. For example, patient autologous iPSCs can be generated from those with an inherited disease. The genetic defect would be transferred to differentiated cells, which may be reflected as an observable disease phenotype or be detectable at a molecular level. These concepts may be applicable to monogenic diseases caused by a single mutation to a given gene but perhaps not in more complicated circumstances where iPSCs may not be able to faithfully recapitulate the disease phenotype. Fragile X syndrome, the most common form of inherited mental retardation, is an epigenetic disorder occurring as a result of silencing of the FMR1 gene. The disease phenotype is the result of an accumulation in aberrant DNA methylation and repressive histone marks [101]. It was found that reprogramming to pluripotency could not restore the epigenetic aberrations, but in ESCs derived from embryos during preimplantation diagnosis, the FMR1 gene was expressed in its unrepressed form and that transcriptional repression occurred only after differentiation [102]. Another challenge relating to disease modelling is the issue of clonal variation. To simplify, assuming that reprogramming to pluripotency and subsequent differentiation has been achieved, clonal variation may exist in the propensity of cell to display the disease phenotype. There are a number of possible explanations, one of them being heterogeneity in the starting somatic cells. Again with reference to Fragile X syndrome, in one study, the fibroblasts obtained from a patient contained a mixture of cells displaying the functional FMR1 gene but also those where the gene was repressed to different extents [103]. This heterogeneity in the starting somatic cell population became apparent during the characterization of differentiated neurons and glial cells from patient autologous iPSCs. In this particular example, it demonstrates the ‘infidelity’ of the reprogramming process by demonstrating that induced pluripotency can be the result non-discriminately targeted reprogramming [104]. Together with the relative inefficiency of reprogramming and possible remnants of somatic memory, there are numerous factors, the combinative effect of which may lead to significant variation in the presentation of the disease phenotype. Therefore in attempting to model human diseases using iPSCs, it is important to consider the molecular basis of the disease. For more complicated examples such as Fragile X syndrome, it may be necessary to start reprogramming from an isogenic starting population of somatic clones.
6. Novel developments and future prospects of stem cell therapy
It is clear that there is much about the regenerative powers of stem cells that science may attempt to harness for clinical purposes. Of course, there are obstacles that must be overcome before this goal is realized with some challenges being more difficult than others. However, it must not dampen the infectious enthusiasm that is behind all the ongoing efforts in driving the field forward. Other than the examples of clinical trials that have been given, there are a number of ongoing developments that are worth noting, some of which will be highlighted here.
(a). In vitro differentiation of pluripotent cells into haematopoietic stem cells and blood products
The holy grail of the haematopoietic field continues to remain that of obtaining a continuous supply of cells that may be used in transplantation therapies or be manipulated ex vivo for gene therapy. Efforts of generating HSCs capable of long-term engraftment in vivo began soon after the first establishment of hESCs. Previously in a mouse model of sickle cell anaemia, it was found that haematopoietic progenitors generated from gene corrected iPSCs were capable of long-term reconstitution of the recipient haematopoietic system several weeks following transplantation, which rescued the disease phenotype [105]. It is important, however, to consider that this was in part achieved through ectopic HoxB4 expression, which enhances the engraftment potential of haematopoietic progenitors derived from pluripotent cells. Therefore, a considerable amount of genetic manipulation was required to achieve the results noted during the study. Unfortunately, the same level of success has not been matched in humans, where it has only been possible to generate haematopoietic progenitors from ESCs that produced transient benefits in a mouse model but were not transplantable in the long term [106]. This could be due to shortcomings in replicating the in vivo environment to support haematopoietic differentiation. For the most part, stem cell differentiation is a stochastic process. Relatively speaking, much is known about haematopoetic differentiation from HSCs to mature blood cells but less so before that. Perhaps revisiting the mechanisms at an epigenetic level [107] may help to identify the epigenome of LT-HSCs and to develop a strategy to arrest differentiation at that specific point during haematopoiesis.
Given the simplistic nature in the functioning of haematopoietic cells, there is hope in the development of ‘off-the-shelf’ products that are ‘ready to use’. Candidate products include those consisting of platelets (PLTs) and red blood cells (RBCs). Normally donated blood products have a limited shelf life, with packaged platelets lasting only around 5 days and RBCs approximately 42 days. Not only that, in all countries, donor availability continues to remain a problem. Pluripotent stem cell-derived PLTs or RBCs may potentially represent a safe and stable supply of these cells. Although the technology is not yet ready for clinical application, there have already been reports of erythrocytes [108] and platelet [109,110] derivation from both ESCs and iPS cells. It will, however, have to be achievable on a large enough scale to meet the numbers that are required clinically per transfusion. Nonetheless, success in generating these products would greatly benefit the treatment of patients with hematological disorders or to supply those requiring a blood transfusion.
(b). Direct lineage conversion
Direct lineage conversion or forward reprogramming is a technique that converts one cell type directly to another through the ectopic expression of defined factors without an intermediate pluripotency step. In other words, it is a one-step as opposed to a two-step process. This technique is not limited only to conversion between somatic cells, but it can also occur between different germ cells. There are notable advantages over iPS reprogramming for example where the dynamics of reprogramming are faster and the efficiency of reprogramming is much higher [111]. However, there may be difficulties with applications in cell transplantation owing to issues with scalability. More recently, it has been suggested that direct reprogramming could be applied in situ for the treatment of heart disease at areas of myocardial infarcted damage [112]. Theoretically at least, this technique seems promising, but there are some important questions that need to be addressed. First of all, there is the possibility that similar to differentiation from an induced pluripotent state, epigenetic memory can impact upon the efficiency of conversion between two cell types. Perhaps even residual amount of epigenetic memory retained from the previous state could affect the functioning of the newly converted cell [112]. Another critical but more clinically relevant question is why conversion between human cells is more difficult than in mice? To date, there have been direct reports of conversion into neurons [113] and multilineage blood progenitors [114] from fibroblasts. However, demonstrating the effective functionality of these converted cells would be quite a different matter. As mentioned earlier, clinical benefits of therapies treating neurodegenerative disorders can be difficult to assess and in the latter report, the converted blood progenitors are not LT-HSCs; hence they would not be able to reconstitute the haematopoietic system in a recipient. Nonetheless, direct conversion is a technique that seems promising not only in transplanting therapy, but also as a more simplified and direct method of studying the mechanisms of transcription factor action.
(c). Embryonic stem cell- or induced pluripotent stem cell-derived organs in xenogenic animals via blastocyst complementation
As mentioned previously, most current therapeutic strategies in stem cell therapy target diseases that are amenable to cell transplantation only. Certain chronic diseases will ultimately lead to organ failure. In the current climate, there is an absolute shortage of donor organs. The differentiation potential of pluripotent cells (ESCs or iPSCs) have been well documented at the cellular level. However, the generation of a whole organ is considerably more difficult. This is because organogenesis is a complex process that involves the establishment of complex cell–cell interactions and recapturing of the conditions that naturally occur in vivo during embryonic development. A solution may be to use a xenogenic host. The Nakauchi laboratory was the first to demonstrate the generation of functionally normal rat pancreas into a Pdx1−/− (pancreatogenesis-disabled) mouse [115]. Wild-type rat iPSCs were injected into the blastocysts of Pdx1−/− mice resulting in a chimaeric animal with a pancreas entirely derived from the rat iPSCs. Transplantation of mature β cells from the chimaeric mice into a rat with streptozocin-induced diabetes was able to reinstate glucose regulatory capacity. Attempts are currently ongoing at replicating this strategy in larger mammals. There may be further technical and ethical challenges in the future before this technology is ready for clinical application, but it is certainly one of the most enterprising strategies at addressing donor organ shortages.
7. Premature clinical translation and risks of progress retardation
For purposes that are pure and others that are morally questionable, there is great eagerness in translating stem cell therapies into clinical practice. In some regards, the initial public furore stirred up by the media regarding the therapeutic potentials of ESCs and iPSCs may have been somewhat dampened a little. The mammoth regulatory hurdles faced by Geron [116] and Advanced Cell Technologies [117] were well documented. Clear evidence of clinical benefits from these trials remains elusive, certainly not enough to satisfy the ever-hungry public and media. Unfortunately, this meant that there has been an alarming rise in the appearance of the so-called cell banks all around the world. These are generally private institutions advertising the therapeutic benefits of MSCs, harvesting them from cord blood or adipose tissue and falsely promising numerous cures ranging from neurodegenerative diseases to diabetes [118]. As mentioned in an earlier section, MSCs are ill-defined not only in the identity of these cells, but also in their therapeutic benefits. However, stem cell therapy in general has been so overly romanticized by the media that naive members of the public have been made vulnerable to dishonest businesses.
Again, these are issues that may not appear to be scientifically relevant, but there are real concerns that because of them, further progress in the field may become compromised. Currently, there is neither a concrete set of guidelines on how to classify stem cell-based products nor a consensus on how they should be classified. Unfortunately, this lack of agreement may have led to the appearance of fraudulent clinics and for every bit of negative publicity that they attract, it becomes more and more damaging to the scientific community in multiple folds. One might speculate that such cases contributed significantly to the ‘regulatory oversight’ encountered with the ESC trials [43]. Tough regulations are positive within reasonable proportions but not at the expense of stifling scientific progress. Already in a rather regrettable example, the European Court of Justice has formally banned the patenting of technologies derived from hESCs [119]. Although the decision was made on ethical grounds, it is quite reasonable to assume that such negative examples raised here would have influenced that decision. Therefore, unless the issue is urgently addressed, not only would further progress stall, there is a real risk of regression. It is clear that there needs to be tighter regulations by internal regulatory boards (IRBs) in all countries participating in stem cell research.
8. Conclusion
So where are we now? The field of stem cell therapy is currently riding on a wave of rapid growth and excitement. However, perhaps one should be inclined to call for some restraint while stressing the need to proceed with prudence. Many conceptual therapeutic ideas have been demonstrated only by ‘proof of principal’. Patience is required to allow for the completion of a comprehensive pre-clinical assessment into the safety and efficacy of these treatments before their eventual translation into the clinic. Even with HSC-based therapies, the area that has shown the most advancement, it took half a century from the first attempts at BMT before the field reached its current level of progress. The modern perception of stem cells is that of ‘the solution’. It appears to many that this is some kind of transplantable differentiated cell. However, until key questions have been satisfactorily addressed, for the present moment at least, the main application especially that of pluripotent cells will likely be as a ‘tool’ to understand with precision, in vivo developmental mechanisms or disease pathophysiological development. With a view to stimulate further advancements, the field now hopes to revisit the fundamentals of stem cell biology at an epigenetic level. Epigenetic studies into pluripotency, cellular reprogramming, lineage fate decisions and other such related areas have already provided the knowledge to define the field as it is known today. Ultimately, the dream is to achieve the ambitious goal of establishing safe and effective stem cell therapy.
References
- 1.Briggs R, King TJ. 1952. Transplantation of living nuclei from blastula cells into enucleated frogs’ eggs. Proc. Natl Acad. Sci. USA 38, 455–463 10.1073/pnas.38.5.455 (doi:10.1073/pnas.38.5.455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller RA, Ruddle FH. 1976. Pluripotent teratocarcinoma-thymus somatic cell hybrids. Cell 9, 45–55 10.1016/0092-8674(76)90051-9 (doi:10.1016/0092-8674(76)90051-9) [DOI] [PubMed] [Google Scholar]
- 3.Foshay KM, et al. 2012. Embryonic stem cells induce pluripotency in somatic cell fusion through biphasic reprogramming. Mol. Cell 46, 159–170 10.1016/j.molcel.2012.02.013 (doi:10.1016/j.molcel.2012.02.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Till JE, McCulloch CE. 1961. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res. 14, 213–222 10.2307/3570892 (doi:10.2307/3570892) [DOI] [PubMed] [Google Scholar]
- 5.Metcalf D. 2007. Concise review: hematopoietic stem cells and tissue stem cells: current concepts and unanswered questions. Stem Cells 25, 2390–2395 (doi:10.1634/stemcells.2007–0544) [DOI] [PubMed] [Google Scholar]
- 6.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. 1998. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 10.1126/science.282.5391.1145 (doi:10.1126/science.282.5391.1145) [DOI] [PubMed] [Google Scholar]
- 7.Evans M. 2011. Discovering pluripotency: 30 years of mouse embryonic stem cells. Nat. Rev. Mol. Cell Biol. 12, 680–686 10.1038/nrm3190 (doi:10.1038/nrm3190) [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 10.1016/j.cell.2006.07.024 (doi:10.1016/j.cell.2006.07.024) [DOI] [PubMed] [Google Scholar]
- 9.Weissman IL. 2000. Stem cells: units of development, units of regeneration, and units in evolution. Cell 100, 157–168 10.1016/S0092-8674(00)81692-X (doi:10.1016/S0092-8674(00)81692-X) [DOI] [PubMed] [Google Scholar]
- 10.Seita J, Rossi DJ, Weissman IL. 2010. Differential DNA damage response in stem and progenitor cells. Cell Stem Cell 7, 145–147 10.1016/j.stem.2010.07.006 (doi:10.1016/j.stem.2010.07.006) [DOI] [PubMed] [Google Scholar]
- 11.Martino G, Pluchino S. 2006. The therapeutic potential of neural stem cells. Nat. Rev. Neurosci. 7, 395–406 10.1038/nrn1908 (doi:10.1038/nrn1908) [DOI] [PubMed] [Google Scholar]
- 12.Van Keymeulen A, Blanpain C. 2012. Tracing epithelial stem cells during development, homeostasis, and repair. J. Cell Biol. 197, 575–584 10.1083/jcb.201201041 (doi:10.1083/jcb.201201041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanpain C, Horsley V, Fuchs E. 2007. Epithelial stem cells: turning over new leaves. Cell 128, 445–458 10.1016/j.cell.2007.01.014 (doi:10.1016/j.cell.2007.01.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinnamaneni N, Funderburgh JL. 2012. Concise review: stem cells in the corneal stroma. Stem Cells 30, 1059–1063 10.1002/stem.1100 (doi:10.1002/stem.1100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barker N, van de Wetering M, Clevers H. 2008. The intestinal stem cell. Genes Dev. 22, 1856–1864 10.1101/gad.1674008 (doi:10.1101/gad.1674008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakada D, Levi BP, Morrison SJ. 2011. Integrating physiological regulation with stem cell and tissue homeostasis. Neuron 70, 703–718 10.1016/j.neuron.2011.05.011 (doi:10.1016/j.neuron.2011.05.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones DL, Wagers AJ. 2008. No place like home: anatomy and function of the stem cell niche. Nat. Rev. Mol. Cell Biol. 9, 11–21 10.1038/nrm2319 (doi:10.1038/nrm2319) [DOI] [PubMed] [Google Scholar]
- 18.Wagers AJ. 2012. The stem cell niche in regenerative medicine. Cell Stem Cell 10, 362–369 10.1016/j.stem.2012.02.018 (doi:10.1016/j.stem.2012.02.018) [DOI] [PubMed] [Google Scholar]
- 19.Goritz C, Frisen J. 2012. Neural stem cells and neurogenesis in the adult. Cell Stem Cell 10, 657–659 10.1016/j.stem.2012.04.005 (doi:10.1016/j.stem.2012.04.005) [DOI] [PubMed] [Google Scholar]
- 20.Keating A. 2012. Mesenchymal stromal cells: new directions. Cell Stem Cell 10, 709–716 10.1016/j.stem.2012.05.015 (doi:10.1016/j.stem.2012.05.015) [DOI] [PubMed] [Google Scholar]
- 21.Grompe M. 2012. Tissue stem cells: new tools and functional diversity. Cell Stem Cell 10, 685–689 10.1016/j.stem.2012.04.006 (doi:10.1016/j.stem.2012.04.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin GR. 1981. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl Acad. Sci. USA 78, 7634–7638 10.1073/pnas.78.12.7634 (doi:10.1073/pnas.78.12.7634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans MJ, Kaufman MH. 1981. Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156 10.1038/292154a0 (doi:10.1038/292154a0) [DOI] [PubMed] [Google Scholar]
- 24.Stadtfeld M, Hochedlinger K. 2010. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 24, 2239–2263 10.1101/gad.1963910 (doi:10.1101/gad.1963910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. 2001. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 19, 1129–1133 (doi:10.1038/nbt1201–1129) [DOI] [PubMed] [Google Scholar]
- 26.Takayama K, et al. 2012. Efficient generation of functional hepatocytes from human embryonic stem cells and induced pluripotent stem cells by HNF4alpha transduction. Mol. Ther. 20, 127–137 10.1038/mt.2011.234 (doi:10.1038/mt.2011.234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chadwick K, Wang L, Li L, Menendez P, Murdoch B, Rouleau A, et al. 2003. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 102, 906–915 (doi:10.1182/blood-2003–03–0832) [DOI] [PubMed] [Google Scholar]
- 28.Ben-David U, Benvenisty N. 2011. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 11, 268–277 10.1038/nrc3034 (doi:10.1038/nrc3034) [DOI] [PubMed] [Google Scholar]
- 29.Swijnenburg RJ, et al. 2008. Immunosuppressive therapy mitigates immunological rejection of human embryonic stem cell xenografts. Proc. Natl Acad. Sci. USA 105, 12 991–12 996 10.1073/pnas.0805802105 (doi:10.1073/pnas.0805802105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenthal E. 2006. Excommunication is sought for stem cell researchers. NY Times (Print), A3. 1st July, 2006. [PubMed]
- 31.Tanne JH. 2006. Bush vetoes bill to expand stem cell research. Br. Med. J. 333, 216. 10.1136/bmj.333.7561.216-b (doi:10.1136/bmj.333.7561.216-b) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurdon JB, Wilmut I. 2011. Nuclear transfer to eggs and oocytes. Cold Spring Harb. Perspect. Biol. 3, pii: a002659. 10.1101/cshperspect.a002659 (doi:10.1101/cshperspect.a002659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T. 2001. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr. Biol. 11, 1553–1558 10.1016/S0960-9822(01)00459-6 (doi:10.1016/S0960-9822(01)00459-6) [DOI] [PubMed] [Google Scholar]
- 34.Masui S, et al. 2007. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat. Cell Biol. 9, 625–635 10.1038/ncb1589 (doi:10.1038/ncb1589) [DOI] [PubMed] [Google Scholar]
- 35.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 10.1016/j.cell.2007.11.019 (doi:10.1016/j.cell.2007.11.019) [DOI] [PubMed] [Google Scholar]
- 36.Yu J, et al. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 10.1126/science.1151526 (doi:10.1126/science.1151526) [DOI] [PubMed] [Google Scholar]
- 37.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. 2008. Induced pluripotent stem cells generated without viral integration. Science 322, 945–949 10.1126/science.1162494 (doi:10.1126/science.1162494) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. 2009. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 85, 348–362 10.2183/pjab.85.348 (doi:10.2183/pjab.85.348) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu H, Ye Z, Kim Y, Sharkis S, Jang YY. 2010. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology 51, 1810–1819 10.1002/hep.23626 (doi:10.1002/hep.23626) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinton DA, Daley GQ. 2012. The promise of induced pluripotent stem cells in research and therapy. Nature 481, 295–305 10.1038/nature10761 (doi:10.1038/nature10761) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booth C, Gaspar HB, Thrasher AJ. 2011. Gene therapy for primary immunodeficiency. Curr. Opin. Pediatr. 23, 659–666 10.1097/MOP.0b013e32834cd67a (doi:10.1097/MOP.0b013e32834cd67a) [DOI] [PubMed] [Google Scholar]
- 42.Colman A, Dreesen O. 2009. Pluripotent stem cells and disease modeling. Cell Stem Cell 5, 244–247 10.1016/j.stem.2009.08.010 (doi:10.1016/j.stem.2009.08.010) [DOI] [PubMed] [Google Scholar]
- 43.Daley GQ. 2012. The promise and perils of stem cell therapeutics. Cell Stem Cell 10, 740–749 10.1016/j.stem.2012.05.010 (doi:10.1016/j.stem.2012.05.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newman AM, Cooper JB. 2010. Lab-specific gene expression signatures in pluripotent stem cells. Cell Stem Cell 7, 258–262 10.1016/j.stem.2010.06.016 (doi:10.1016/j.stem.2010.06.016) [DOI] [PubMed] [Google Scholar]
- 45.He S, Nakada D, Morrison SJ. 2009. Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol. 25, 377–406 10.1146/annurev.cellbio.042308.113248 (doi:10.1146/annurev.cellbio.042308.113248) [DOI] [PubMed] [Google Scholar]
- 46.Lechner A, Habener JF. 2003. Stem/progenitor cells derived from adult tissues: potential for the treatment of diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 284, E259–E266 10.1152/ajpendo.00393.2002 (doi:10.1152/ajpendo.00393.2002) [DOI] [PubMed] [Google Scholar]
- 47.Shortt AJ, Secker GA, Notara MD, Limb GA, Khaw PT, Tuft SJ, Daniels JT. 2007. Transplantation of ex vivo cultured limbal epithelial stem cells: a review of techniques and clinical results. Surv. Ophthalmol. 52, 483–502 10.1016/j.survophthal.2007.06.013 (doi:10.1016/j.survophthal.2007.06.013) [DOI] [PubMed] [Google Scholar]
- 48.Fuchs E. 2008. Skin stem cells: rising to the surface. J. Cell Biol. 180, 273–284 10.1083/jcb.200708185 (doi:10.1083/jcb.200708185) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weissman IL, Shizuru JA. 2008. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood 112, 3543–3553 (doi:10.1182/blood-2008–08–078220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okabe M, et al. 2009. Definitive proof for direct reprogramming of hematopoietic cells to pluripotency. Blood 114, 1764–1767 10.1182/blood-2009-02-203695 (doi:10.1182/blood-2009-02-203695) [DOI] [PubMed] [Google Scholar]
- 51.Osawa M, Hanada K, Hamada H, Nakauchi H. 1996. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 273, 242–245 10.1126/science.273.5272.242 (doi:10.1126/science.273.5272.242) [DOI] [PubMed] [Google Scholar]
- 52.Orkin SH, Zon LI. 2008. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644 10.1016/j.cell.2008.01.025 (doi:10.1016/j.cell.2008.01.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seita J, Weissman IL. 2010. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2, 640–653 10.1002/wsbm.86 (doi:10.1002/wsbm.86) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Copelan EA. 2006. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 354, 1813–1826 10.1056/NEJMra052638 (doi:10.1056/NEJMra052638) [DOI] [PubMed] [Google Scholar]
- 55.Little MT, Storb R. 2002. History of haematopoietic stem-cell transplantation. Nat. Rev. Cancer 2, 231–238 10.1038/nrc748 (doi:10.1038/nrc748) [DOI] [PubMed] [Google Scholar]
- 56.Doulatov S, Notta F, Laurenti E, Dick JE. 2012. Hematopoiesis: a human perspective. Cell Stem Cell 10, 120–136 10.1016/j.stem.2012.01.006 (doi:10.1016/j.stem.2012.01.006) [DOI] [PubMed] [Google Scholar]
- 57.Greenberger JS. 2008. Gene therapy approaches for stem cell protection. Gene Ther. 15, 100–108 10.1038/sj.gt.3303004 (doi:10.1038/sj.gt.3303004) [DOI] [PubMed] [Google Scholar]
- 58.Gaspar HB, et al. 2011. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci. Transl. Med. 3, 97ra80. 10.1126/scitranslmed.3002716 (doi:10.1126/scitranslmed.3002716) [DOI] [PubMed] [Google Scholar]
- 59.Kang HJ, et al. 2011. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Mol. Ther. 19, 2092–2101 10.1038/mt.2011.166 (doi:10.1038/mt.2011.166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Galy A, Thrasher AJ. 2011. Gene therapy for the Wiskott–Aldrich syndrome. Curr. Opin. Allergy Clin. Immunol. 11, 545–550 10.1097/ACI.0b013e32834c230c (doi:10.1097/ACI.0b013e32834c230c) [DOI] [PubMed] [Google Scholar]
- 61.Hacein-Bey-Abina S, et al. 2003. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419 10.1126/science.1088547 (doi:10.1126/science.1088547) [DOI] [PubMed] [Google Scholar]
- 62.Lindvall O. 2012. Why is it taking so long to develop clinically competitive stem cell therapies for CNS disorders? Cell Stem Cell 10, 660–662 10.1016/j.stem.2012.04.004 (doi:10.1016/j.stem.2012.04.004) [DOI] [PubMed] [Google Scholar]
- 63.Fuentealba LC, Obernier K, Alvarez-Buylla A. 2012. Adult neural stem cells bridge their niche. Cell Stem Cell 10, 698–708 10.1016/j.stem.2012.05.012 (doi:10.1016/j.stem.2012.05.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martino G, Bacigaluppi M, Peruzzotti-Jametti L. 2011. Therapeutic stem cell plasticity orchestrates tissue plasticity. Brain 134, 1585–1587 10.1093/brain/awr115 (doi:10.1093/brain/awr115) [DOI] [PubMed] [Google Scholar]
- 65.Blanpain C, Fuchs E. 2006. Epidermal stem cells of the skin. Annu. Rev. Cell Dev. Biol. 22, 339–373 10.1146/annurev.cellbio.22.010305.104357 (doi:10.1146/annurev.cellbio.22.010305.104357) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nguyen H, Merrill BJ, Polak L, Nikolova M, Rendl M, Shaver TM, Pasolli HA, Fuchs E. 2009. Tcf3 and Tcf4 are essential for long-term homeostasis of skin epithelia. Nat. Genet. 41, 1068–1075 10.1038/ng.431 (doi:10.1038/ng.431) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arwert EN, Hoste E, Watt FM. 2012. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 12, 170–180 10.1038/nrc3217 (doi:10.1038/nrc3217) [DOI] [PubMed] [Google Scholar]
- 68.Green H. 2008. The birth of therapy with cultured cells. Bioessays 30, 897–903 10.1002/bies.20797 (doi:10.1002/bies.20797) [DOI] [PubMed] [Google Scholar]
- 69.Mavilio F, et al. 2006. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 12, 1397–1402 10.1038/nm1504 (doi:10.1038/nm1504) [DOI] [PubMed] [Google Scholar]
- 70.Radtke F, Clevers H. 2005. Self-renewal and cancer of the gut: two sides of a coin. Science 307, 1904–1909 10.1126/science.1104815 (doi:10.1126/science.1104815) [DOI] [PubMed] [Google Scholar]
- 71.Sato T, et al. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 10.1038/nature07935 (doi:10.1038/nature07935) [DOI] [PubMed] [Google Scholar]
- 72.Yui S, et al. 2012. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat. Med. 18, 618–623 10.1038/nm.2695 (doi:10.1038/nm.2695) [DOI] [PubMed] [Google Scholar]
- 73.Shaker A, Rubin DC. 2012. Stem cells: one step closer to gut repair. Nature 485, 181–182 10.1038/485181a (doi:10.1038/485181a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takacs L, Toth E, Berta A, Vereb G. 2009. Stem cells of the adult cornea: from cytometric markers to therapeutic applications. Cytometry A 75, 54–66 10.1002/cyto.a.20671 (doi:10.1002/cyto.a.20671) [DOI] [PubMed] [Google Scholar]
- 75.Daniels JT, Dart JK, Tuft SJ, Khaw PT. 2001. Corneal stem cells in review. Wound Repair Regen. 9, 483–494 10.1046/j.1524-475x.2001.00483.x (doi:10.1046/j.1524-475x.2001.00483.x) [DOI] [PubMed] [Google Scholar]
- 76.Pellegrini G, Rama P, Mavilio F, De Luca M. 2009. Epithelial stem cells in corneal regeneration and epidermal gene therapy. J. Pathol. 217, 217–228 10.1002/path.2441 (doi:10.1002/path.2441) [DOI] [PubMed] [Google Scholar]
- 77.Caplan AI. 1991. Mesenchymal stem cells. J. Orthop. Res. 9, 641–650 10.1002/jor.1100090504 (doi:10.1002/jor.1100090504) [DOI] [PubMed] [Google Scholar]
- 78.Horwitz EM, Dominici M. 2008. How do mesenchymal stromal cells exert their therapeutic benefit? Cytotherapy 10, 771–774 10.1080/14653240802618085 (doi:10.1080/14653240802618085) [DOI] [PubMed] [Google Scholar]
- 79.Mizuno H, Tobita M, Uysal AC. 2012. Concise review: adipose-derived stem cells as a novel tool for future regenerative medicine. Stem Cells 30, 804–810 10.1002/stem.1076 (doi:10.1002/stem.1076) [DOI] [PubMed] [Google Scholar]
- 80.da Silva Meirelles L, Chagastelles PC, Nardi NB. 2006. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 119, 2204–2213 10.1242/jcs.02932 (doi:10.1242/jcs.02932) [DOI] [PubMed] [Google Scholar]
- 81.Ra JC, et al. 2011. Safety of intravenous infusion of human adipose tissue-derived mesenchymal stem cells in animals and humans. Stem Cells Dev. 20, 1297–1308 10.1089/scd.2010.0466 (doi:10.1089/scd.2010.0466) [DOI] [PubMed] [Google Scholar]
- 82.Uccelli A, Moretta L, Pistoia V. 2008. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 8, 726–736 10.1038/nri2395 (doi:10.1038/nri2395) [DOI] [PubMed] [Google Scholar]
- 83.Pittenger MF, et al. 1999. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 10.1126/science.284.5411.143 (doi:10.1126/science.284.5411.143) [DOI] [PubMed] [Google Scholar]
- 84.Kebriaei P, Robinson S. 2011. Treatment of graft-versus-host-disease with mesenchymal stromal cells. Cytotherapy 13, 262–268 10.3109/14653249.2010.549688 (doi:10.3109/14653249.2010.549688) [DOI] [PubMed] [Google Scholar]
- 85.Kim K, et al. 2010. Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290 10.1038/nature09342 (doi:10.1038/nature09342) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lister R, et al. 2011. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68–73 10.1038/nature09798 (doi:10.1038/nature09798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen AE, et al. 2009. Optimal timing of inner cell mass isolation increases the efficiency of human embryonic stem cell derivation and allows generation of sibling cell lines. Cell Stem Cell 4, 103–106 10.1016/j.stem.2008.12.001 (doi:10.1016/j.stem.2008.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Niakan KK, Han J, Pedersen RA, Simon C, Pera RA. 2012. Human pre-implantation embryo development. Development 139, 829–841 10.1242/dev.060426 (doi:10.1242/dev.060426) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Varela E, Schneider RP, Ortega S, Blasco MA. 2011. Different telomere-length dynamics at the inner cell mass versus established embryonic stem (ES) cells. Proc. Natl Acad. Sci. USA 108, 15 207–15 212 10.1073/pnas.1105414108 (doi:10.1073/pnas.1105414108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamanaka S. 2012. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell 10, 678–684 10.1016/j.stem.2012.05.005 (doi:10.1016/j.stem.2012.05.005) [DOI] [PubMed] [Google Scholar]
- 91.Takahashi K, Mitsui K, Yamanaka S. 2003. Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature 423, 541–545 10.1038/nature01646 (doi:10.1038/nature01646) [DOI] [PubMed] [Google Scholar]
- 92.Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz-Gerald CS, Sato Y, Cowan CA, Chien KR, Melton DA. 2008. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 26, 313–315 10.1038/nbt1383 (doi:10.1038/nbt1383) [DOI] [PubMed] [Google Scholar]
- 93.Whitworth KM, Prather RS. 2010. Somatic cell nuclear transfer efficiency: how can it be improved through nuclear remodeling and reprogramming? Mol. Reprod. Dev. 77, 1001–1015 10.1002/mrd.21242 (doi:10.1002/mrd.21242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tat PA, Sumer H, Pralong D, Verma PJ. 2011. The efficiency of cell fusion-based reprogramming is affected by the somatic cell type and the in vitro age of somatic cells. Cell Reprogram. 13, 331–344 10.1089/cell.2011.0002 (doi:10.1089/cell.2011.0002) [DOI] [PubMed] [Google Scholar]
- 95.Hansson M, Madsen OD. 2010. Pluripotent stem cells, a potential source of beta-cells for diabetes therapy. Curr. Opin. Invest. Drugs 11, 417–425 [PubMed] [Google Scholar]
- 96.Naujok O, Burns C, Jones PM, Lenzen S. 2011. Insulin-producing surrogate beta-cells from embryonic stem cells: are we there yet? Mol. Ther. 19, 1759–1768 10.1038/mt.2011.165 (doi:10.1038/mt.2011.165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kroon E, et al. 2008. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 26, 443–452 10.1038/nbt1393 (doi:10.1038/nbt1393) [DOI] [PubMed] [Google Scholar]
- 98.Chen LW, Kuang F, Wei LC, Ding YX, Yung KK, Chan YS. 2011. Potential application of induced pluripotent stem cells in cell replacement therapy for Parkinson's disease. CNS Neurol. Disord. Drug Targets 10, 449–458 [DOI] [PubMed] [Google Scholar]
- 99.Baker M. 2009. Stem cells: fast and furious. Nature 458, 962–965 10.1038/458962a (doi:10.1038/458962a) [DOI] [PubMed] [Google Scholar]
- 100.Grskovic M, Javaherian A, Strulovici B, Daley GQ. 2011. Induced pluripotent stem cells—opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 10, 915–929 10.1038/nrd3577 (doi:10.1038/nrd3577) [DOI] [PubMed] [Google Scholar]
- 101.Bagni C, Greenough WT. 2005. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat. Rev. Neurosci. 6, 376–387 10.1038/nrn1667 (doi:10.1038/nrn1667) [DOI] [PubMed] [Google Scholar]
- 102.Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. 2010. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6, 407–411 10.1016/j.stem.2010.04.005 (doi:10.1016/j.stem.2010.04.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ, Cookson MR. 2011. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE 6, e26203. 10.1371/journal.pone.0026203 (doi:10.1371/journal.pone.0026203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Onder TT, Daley GQ. 2012. New lessons learned from disease modeling with induced pluripotent stem cells. Curr. Opin. Genet. Dev. (doi:10.1016/j.gde.2012.05.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hanna J, et al. 2007. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 318, 1920–1923 10.1126/science.1152092 (doi:10.1126/science.1152092) [DOI] [PubMed] [Google Scholar]
- 106.Wang L, Menendez P, Shojaei F, Li L, Mazurier F, Dick JE, Cerdan C, Levac K, Bhatia M. 2005. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J. Exp. Med. 201, 1603–1614 10.1084/jem.20041888 (doi:10.1084/jem.20041888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saleque S, Kim J, Rooke HM, Orkin SH. 2007. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 27, 562–572 10.1016/j.molcel.2007.06.039 (doi:10.1016/j.molcel.2007.06.039) [DOI] [PubMed] [Google Scholar]
- 108.Lu SJ, Feng Q, Park JS, Lanza R. 2010. Directed differentiation of red blood cells from human embryonic stem cells. Methods Mol. Biol. 636, 105–121 10.1007/978-1-60761-691-7_7 (doi:10.1007/978-1-60761-691-7_7) [DOI] [PubMed] [Google Scholar]
- 109.Takayama N, Nishikii H, Usui J, Tsukui H, Sawaguchi A, Hiroyama T, et al. 2008. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 111, 5298–5306 (doi:10.1182/blood-2007–10-117622) [DOI] [PubMed] [Google Scholar]
- 110.Takayama N, et al. 2010. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J. Exp. Med. 207, 2817–2830 10.1084/jem.20100844 (doi:10.1084/jem.20100844) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vierbuchen T, Wernig M. 2011. Direct lineage conversions: unnatural but useful? Nat. Biotechnol. 29, 892–907 10.1038/nbt.1946 (doi:10.1038/nbt.1946) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jayawardena TM, et al. 2012. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 110, 1465–1473 10.1161/CIRCRESAHA.112.269035 (doi:10.1161/CIRCRESAHA.112.269035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pang ZP, et al. 2011. Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223 10.1038/nature10202 (doi:10.1038/nature10202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Szabo E, Rampalli S, Risueno RM, Schnerch A, Mitchell R, Fiebig-Comyn A, Levadoux-Martin M, Bhatia M. 2010. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 468, 521–526 10.1038/nature09591 (doi:10.1038/nature09591) [DOI] [PubMed] [Google Scholar]
- 115.Kobayashi T, et al. 2010. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 142, 787–799 10.1016/j.cell.2010.07.039 (doi:10.1016/j.cell.2010.07.039) [DOI] [PubMed] [Google Scholar]
- 116.Strauss S. 2010. Geron trial resumes, but standards for stem cell trials remain elusive. Nat. Biotechnol. 28, 989–990 10.1038/nbt1010-989 (doi:10.1038/nbt1010-989) [DOI] [PubMed] [Google Scholar]
- 117.Schwartz SD, et al. 2012. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379, 713–720 10.1016/S0140-673660028-2 (doi:10.1016/S0140-673660028-2) [DOI] [PubMed] [Google Scholar]
- 118.Cyranoski D. 2012. Stem-cell therapy takes off in Texas. Nature 483, 13–14 10.1038/483013a (doi:10.1038/483013a) [DOI] [PubMed] [Google Scholar]
- 119.Smith A. 2011. ‘No’ to ban on stem-cell patents. Nature 472, 418. 10.1038/472418a (doi:10.1038/472418a) [DOI] [PubMed] [Google Scholar]