

Abstract
Fundamental to genomic imprinting in mammals is the acquisition of epigenetic marks that differ in male and female gametes at ‘imprinting control regions’ (ICRs). These marks mediate the allelic expression of imprinted genes in the offspring. Much has been learnt about the nature of imprint marks, the times during gametogenesis at which they are laid down and some of the factors responsible especially for DNA methylation. Recent work has revealed that transcription and histone modifications are critically involved in DNA methylation acquisition, and these findings allow us to propose rational models for methylation establishment. A completely novel perspective on gametic DNA methylation has emerged from epigenomic profiling. Far more differentially methylated loci have been identified in gametes than known imprinted genes, which leads us to revise the notion that methylation of ICRs is a specifically targeted process. Instead, it seems to obey default processes in germ cells, giving rise to distinct patterns of DNA methylation in sperm and oocytes. This new insight, together with the identification of proteins that preserve DNA methylation after fertilization, emphasizes the key role played by mechanisms that selectively retain differential methylation at imprinted loci during early development. Addressing these mechanisms will be essential to understanding the specificity and evolution of genomic imprinting.
Keywords: genomic imprinting, DNA methylation, histone modifications, germ cells, epigenomics
1. Introduction
It is customary to expect that genes from both parents contribute equally to embryo development. This expectation is contravened by genomic imprinting, an epigenetic mechanism in mammals in which a subset of genes exhibits differential activity of the maternal and paternal allele [1]. The non-equivalence of the parental genomes in mice was demonstrated nearly 30 years ago, with the finding that embryos constructed to contain only maternal or paternal diploid genome complements failed to develop to term [2,3]. Separate experiments in which translocations were used to generate mice with uniparental inheritance of specific chromosomes (or chromosome segments) identified regions for which biparental inheritance is essential for normal development and viability [4]. Then, over 20 years ago, the first imprinted genes were discovered and molecular evidence was provided that one allele was transcriptionally silenced strictly according to parental origin [5–7]. During the ensuing years, a variety of dedicated screens and chance discoveries identified more than 100 genes with imprinted, mono-allelic expression in the mouse and human, and in several other placental mammals [8,9]. The advent of high through-put genome-wide profiling techniques is likely to lead to a definitive list of imprinted genes in the future.
Many imprinted genes have been shown to play important roles in development and growth; others have phenotypic effects only after birth, particularly in relation to behaviour [10–13]. The majority of imprinted genes are arranged in chromosomal clusters, which are similarly organized in mice and humans [14]. DNA methylation is essential in the process of genomic imprinting. Each of the 16 known imprinted gene clusters is thought to be controlled by an ‘imprinting control region’ (ICR), an essential regulatory sequence that is marked by DNA methylation on one of the two parental alleles [15]. Depending on the ICR, this DNA methylation imprint is acquired during oogenesis or during spermatogenesis. Because the allelic methylation has its origin in the germline, these differentially methylated regions (DMRs) are often referred to as germline DMRs (gDMRs; we shall use this more general term throughout in preference to ‘ICR’, which is reserved for elements that have been demonstrated functionally to control imprinting). To understand genomic imprinting, it is essential to explore the mechanisms involved in the gametic establishment and post-fertilization maintenance of methylation at gDMRs. When these processes are perturbed during development, or perhaps as a consequence of assisted reproduction technologies, this can give rise to complex diseases in humans [16].
Soon after the discovery of the first imprinted genes, epigenetic differences between their parental alleles began to be identified. A region in the imprinted Igf2r gene was described (an intronic CpG island) at which there was a difference in DNA methylation in the sperm and oocyte, and at which methylation remained specifically on the maternally derived allele throughout development [17]. This was the first description of a gDMR. It was subsequently shown that this element controls mono-allelic expression and DNA methylation of the Igf2r promoter, and imprinted expression of additional genes in cis, through the action of an imprinted, non-coding transcript [18]. This, and similar findings, gave rise to the concept that imprinting was controlled through the establishment of germline DNA methylation marks in one or the other germline (figure 1). Germline DMRs have since been identified and characterized in most imprinted gene clusters and isolated imprinted genes [19], and all functionally defined ICRs have this property [15]. The essential role of DNA methylation in ensuring mono-allelic expression emerged also from knock-out studies on the maintenance DNA methyltransferase DNMT1 [20]. Later, it was shown that the DNA methyltransferase-like protein DNMT3L was required, particularly for establishment of methylation at gDMRs in mouse oocytes [21], and DNMT3A was demonstrated to be the active enzyme responsible [22,23]. With the identification of discrete imprinting elements and factors involved in gametic methylation, much effort was expended on determining the sequence properties of gDMRs and searching for specific sequence motifs that would be responsible for their methylation. Although attributes such as tandem repeats were implicated [24], and some evidence for ‘imprinting signals’ was provided by transgenic experiments [25,26], the existence of discrete sequence elements for the specification of gDMR methylation remains in doubt [19]. This may explain the rather limited success of purely sequence-based predictions of imprinted genes [27,28].
Figure 1.
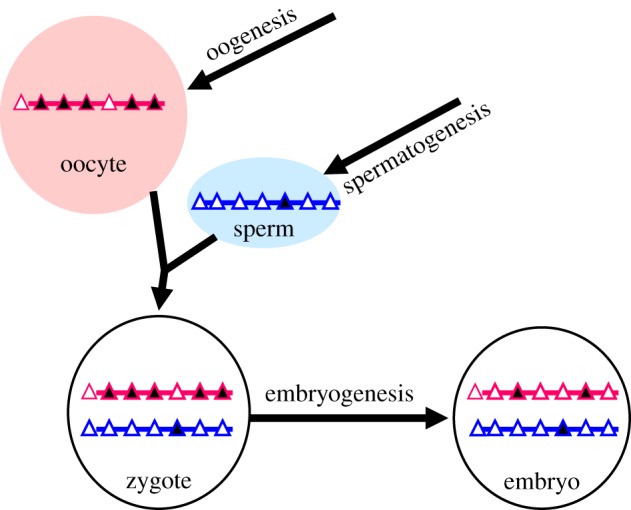
Establishment and post-fertilization maintenance of imprinted DNA methylation. In the schematic presentation, the maternal and the paternal genomes are coloured red and blue, respectively. Triangles represent CpG islands, or CpG-rich sequences: black filling indicates DNA methylation, open triangles indicate absence of methylation. More than a thousand CpG islands become de novo methylated during oogenesis. Conversely, several hundred CpG-rich sequences acquire methylation during spermatogenesis, mostly at intergenic positions. Following fertilization and during early embryonic development, only at a few per cent of these sequence elements is the maternally or paternally derived DNA methylation selectively maintained. Although still poorly understood, it is through early embryonic maintenance mechanisms that the specificity of these germline differentially methylated regions (gDMRs) comes about. The gDMRs that are maintained during development correspond to ‘imprinting control regions’ (ICRs) of known or yet-to-be discovered imprinted domains.
Nevertheless, considerable progress has been made over the past few years in our understanding of gDMRs and the factors involved in their differential DNA methylation. This includes the discovery of interactions between chromatin status—post-translational modifications of histones—and the de novo methylation machinery [29]. Furthermore, recent research links the acquisition of DNA methylation imprints at gDMRs to transcriptional events [30,31] and, at one gDMR, to small RNAs [32]. Major advances have come from mouse and human genetics in identifying critical cis-acting sequences and trans-acting factors. Although explanations for the mechanisms involved are still incomplete, we may be near a unifying explanation for methylation establishment at gDMRs in female germ cells. In male germ cells, in which fewer gDMRs become methylated, there is less evidence so far of a common mechanism. Our ability to obtain genome-wide methylation profiles from gametes is shedding new light on the nature of the gametes' methylome, the extent to which there is specific targeting of methylation to gDMRs and the key role played by maintenance factors in selecting imprinted sequences from the larger set of germline-acquired methylation differences (figure 1).
Here, we consider recent discoveries that mechanistically link transcriptional events and chromatin modifications to the establishment of DNA methylation in female and male germ cells. Our review also discusses the recent discovery that germline establishment of DNA methylation occurs at many more regions than the limited number of gDMRs at imprinted loci [33–35]. This provides a novel framework in which to address the specificity of genomic imprinting, which seems to be determined by maintenance processes acting after fertilization, during the early stages of development.
2. Acquisition of maternal and paternal DNA methylation marks
Gametes originate from germ-cell precursors that are specified in the epiblast of the early post-implantation embryo prior to sex determination. Male and female gametogenesis follow very different paths, however, culminating in the production of highly differentiated gametes with distinct epigenomes (not least because there is a wholesale replacement of histones by protamines in mature sperm [36]). Indeed, recent genome-wide profiling studies in the mouse have revealed substantially different patterns of DNA methylation in sperm and oocytes [33–35]. These global differences are an important backdrop against which to consider the establishment of DNA methylation at imprinted gDMRs. It has been considered that most of the DNA methylation imprints are acquired in the female germline [1,37]. Of the gDMRs so far described in imprinted domains, 16 acquire methylation in oocytes (maternal gDMRs) and only three during spermatogenesis (paternal gDMRs) [19,38,39].
From their specification as a small founder population in the epiblast (embryonic day e7.25 in the mouse), primordial germ cells (PGCs) proliferate and migrate to the genital ridge, the precursor of the gonads. Shortly after their arrival (by e11.5), they undergo extensive DNA demethylation. The extent of this demethylation has been demonstrated recently by shotgun bisulphite sequencing (BS-Seq) and MeDIP-array analysis [40,41]. The exact mechanism of demethylation is still open to question, but does not concern us here. Its consequence is that at the onset of gametogenesis, the germ-cell genome is highly demethylated, aside from specific interspersed repeats, such as retrotransposons of the intracisternal-A particle class [40,41]. This includes full erasure of pre-existing parental-allele-specific methylation at DMRs [42,43].
In female embryos, germ cells enter the first stages of meiosis from e13.5 and then arrest in diplotene of prophase I. These primary oocytes remain quiescent until after birth. Soon after birth, they become incorporated into primordial follicles, and with follicle activation, oocytes enter a growth phase, such that over a two- to three-week period, their size increases from approximately 10 to greater than 70 µm, to become fully grown, germinal vesicle stage oocytes. It is during this period, initiating around the transition from the activated (primary) follicle to secondary (early anthral) follicle stage, that methylation at gDMRs is established [44,45]. Further hormonal stimulation after puberty results in resumption of meiosis I, germinal vesicle breakdown and ovulation. Methylation acquisition appears to be a progressive process that is dependent on oocyte size [45]. In addition, the onset and kinetics of methylation acquisition differ between gDMRs, such that different domains acquire imprinting competence at different rates during oocyte growth [45,46]. It is important to note, therefore, that gDMR methylation (and likely most CpG island, gene body and interspersed repeat methylation) occurs in non-dividing cells. This makes the oocyte a particularly interesting system in which to study mechanisms of de novo methylation, because there is no competing demethylation or the modification of methylation patterns by maintenance processes in these cells. The latter might account for why there is a particularly high incidence of non-CpG methylation in oocytes [19,34], because non-CpG methylation would be purged through the process of DNA replication (the maintenance methyltransferase DNMT1 restores only the ‘symmetrical’ DNA methylation at CpG dinucleotides following replication). Also, methylation is established late in gamete production, minimizing the possibility of acquiring mutations that could arise because of the susceptibility of 5-methylcytosine (5mC) to deamination.
The timing and cell biology of de novo methylation in male germ cells is different in several respects [47]. Methylation of gDMRs initiates in prospermatogonia (also known as gonocytes), which are arrested in mitotic G1, and occurs prior to the onset of meiosis. It commences in the foetal testis, and is largely complete around birth [31,48]. Between the onset of de novo methylation and the production of mature sperm, there are multiple rounds of cell division, such that initially determined methylation patterns may be modified through maintenance, and there is greater opportunity for methylation errors to accumulate or mutations to arise through deamination. Whether there are additional periods of DNA methylation (i.e. further acquisition of methylation at gDMRs or de novo methylation of additional sequences) is not known, largely because of the limited profiling that has been done at intervening stages and because mutations of Dnmt3a, Dnmt3b or Dnmt3L cause severe meiotic defects during spermatogenesis [49,50].
Perhaps as a reflection of the different cell biology of DNA methylation in female and male germ cells, the properties of maternal and paternal gDMRs differ [19,38,47]. Maternal gDMRs have a sequence composition that equates to that of CpG islands; they are also associated with transcription start sites (of coding transcripts or non-coding RNAs). Even so, they are relatively unusual for CpG islands in that they are predominantly intragenic in location ([30] and see below). Many maternal gDMRs contain tandemly reiterated sequences [51]. Although such features were proposed to have a role in targeting de novo methylation [24], there is no compelling evidence from functional studies that this is the case [52]. Structural studies have revealed that DNMT3A and its co-activator DNMT3L form a tetrameric complex, which has given rise to the proposition that DNA sequences with a regular spacing of CpGs (8–10 bp apart) would be preferred targets dictated by the steric constraints of the two catalytic sites in the complex [53]. Although an 8–10 bp periodicity of CpG was found preferentially in maternal gDMRs compared with a control set of CpG islands in one study [53], this has been disputed as a general, discriminating property of gDMRs [19,33]. Furthermore, maternal gDMRs can be very extensive CG-rich domains, with tandem repeat features occupying only a small proportion, in which all CpGs irrespective of their spacing become methylated [19]. In contrast to maternal gDMRs, the three characterized paternal gDMRs (the H19 DMD, Dlk1-Gtl2 IG-DMR and Rasgrf1 DMR) are all intergenic CG-rich elements.
It has generally been assumed that the establishment of imprinted gDMRs occurs through specific targeting of methylation to these sequences. However, systematic analysis of gDMR methylation and recent genome-wide methylation profiling in male and female gametes call into question this long-held assumption and offer a novel perspective. First, maternal gDMR methylation in oocytes might better be regarded as a default process, such that the protection from methylation of these elements in spermatogenesis may be the ‘active’ part of the mechanism. This is because maternal gDMRs are methylated in oocytes contiguously with the methylation of flanking, intragenic sequences, whereas in sperm they appear as unmethylated islands similar to most CpG islands generally (figure 2) [19,34,54]. Second, genome-wide profiling has revealed that many more CpG islands are fully methylated in oocytes and sperm than the known gDMRs [33–35]. However, most of these CpG islands lose much of their gamete-derived methylation during pre-implantation development [33–35,55]. Therefore, the methylated status of imprinted gDMRs might be better viewed as the combination of general methylation processes in oocytes or sperm, with a specific selection for maintenance of methylation in the embryo, especially during the genome-wide demethylation of the maternal and paternal genomes that occurs in pre-implantation embryos (figure 1). This maintenance might indeed have a sequence-based component, for example, through methylation-dependent, sequence-specific binding of factors such as ZFP57 that protect against demethylation [56–58].
Figure 2.

Gene-body methylation in oocytes revealed by whole genome bisulphite sequencing. The panel at the top represents the consensus annotation of transcripts at the imprinted Gnas locus. The locations of CpG islands are shown as open boxes, above the CpG density plot. Below, the methylation level determined from whole-genome bisulphite sequencing in wild-type oocytes, Dnmt3L−/− oocytes, sperm and blastocysts is plotted. Each dot on the displays represents the average methylation level of an individual CpG. The location of the gDMRs at Nespas/Gnasxl and exon 1A are marked by the pink columns. As expected, the gDMRs are unmethylated in sperm and have intermediate levels of methylation in blastocyst DNA. The DMR at Nesp55 (which acquires methylation on the paternal allele only after implantation) and the CpG island (CGI) at the canonical Gnas promoter are unmethylated in all samples. Note that gDMR methylation in oocytes is contiguous with the high level of gene body methylation; the whole domain is within a transcription unit in oocytes determined by activity of the Nesp55 promoter (boxed in the upper panel). All DNA methylation within the domain, not just at the gDMRs, depends upon the presence of DNMT3L in oocytes, as revealed by the very low level of methylation throughout the domain in the Dnmt3L−/− oocyte track. Reproduced, with very minor additional annotation, from Kobayashi et al. [34].
3. Chromatin organization at the time of imprint acquisition
There has been considerable interest in whether specific histone modifications could prevent, or facilitate, de novo DNA methylation. This could be one of the features contributing to the specificity of gDMR establishment in male versus female germ cells. In vitro evidence indicates that methylation at lysine-4 of histone H3 (H3K4) could interfere with the establishment of cytosine methylation. Specifically, lysine-4 methylation blocks association of DNMT3L with the N-terminal part of histone H3, and would thus prevent DNMT3A–DNMT3L complexes from accessing DNA [29]. Association of DNMT3A itself with core histone H3 is also attenuated by lysine-4 methylation [59]. In embryonic stem (ES) and other embryonic cell types, chromatin at most CpG islands is enriched in H3K4 di- and trimethylation [60–62]. This chromatin configuration correlates with promoter activity and could prevent acquisition of aberrant DNA methylation. Recent studies indicate that similar mechanisms could be acting at gDMRs in germ cells in the germline where they are protected against the acquisition of DNA methylation [31,63].
All maternal gDMRs are CpG islands that comprise promoters and are fully protected against acquisition of DNA methylation in male germ cells. For several maternal gDMRs, it has been shown that they have biallelic enrichment of H3K4 trimethylation and promoter activity in male foetal germ cells at the time of imprint establishment [31]. This local enrichment of H3 lysine-4 methylation persists to postnatal stages of spermatogenesis, at least at some maternal gDMRs, possibly providing continuous protection against acquisition of DNA methylation [64]. In spermatozoa, about 1 per cent and 10 per cent of the genome remains associated with histones in mice and humans, respectively [65,66]. Imprinted loci are among the exceptional regions of the genome that retain nucleosomal organization in mature sperm. Although further research is required, this finding raises the intriguing possibility that maternal gDMRs could still be marked by H3K4 methylation in spermatozoa. Whether such a chromatin state is maintained after fertilization and helps prevent de novo DNA methylation in the early embryo remains to be explored.
H3K4 methylation status does have an important role in specifying DNA methylation in female germ cells. The H3K4-specific demethylase KDM1B (also known as AOF1 and LSD2) is expressed highly in growing oocytes, during the time frame that DNA methylation is acquired. Its ablation by gene targeting induces a global accumulation of H3K4me2/3 in oocytes and prevents imprint acquisition at several maternal gDMRs. Although normal DNA methylation establishment did occur at others [63], whether there was persistence of H3K4 methylation at these gDMRs in Kdm1b-deficient oocytes remains to be shown. Nevertheless, this important study strongly suggests that H3K4 methylation needs to be removed, at least at a subset of maternal gDMRs, before DNA methylation can be acquired in growing oocytes. In agreement with this hypothesis, chromatin immunoprecipitation (ChIP) studies on normal oocytes midway through the growth phase show that there is relative depletion of H3K4me3 at CpG islands destined for DNA methylation [33].
Conversely, other histone modifications could promote the acquisition of DNA methylation at gDMRs in germ cells. In Neurospora crassa [67], H3 lysine-9 trimethylation (H3K9me3) guides de novo DNA methylation, but there is no evidence yet that it does the same in mammals. While in somatic cells, the DNA-methylated alleles of gDMRs are consistently enriched in H3K9me3, in male germ cells, this repressive mark is absent at paternal gDMRs at the time of imprint acquisition [31,64]. The same was found for another repressive histone mark, H4 lysine-20 trimethylation (H3K20me3) [31,64]. These repressive histone modifications do therefore not signal DNA methylation acquisition in male germ cells, at least not at gDMRs. Symmetrical dimethylation on arginine-3 of histone H4 (H4R3me2s) has been reported to facilitate de novo DNA methylation by DNMT3A at the globin genes in erythroid cell progenitors [68]. It is not known whether H4R3me2s contributes to acquisition of DNA methylation in germ cells as well. ChIP studies on male foetal germ cells showed high levels of this modification at all gDMRs analysed as well as at non-imprinted gene loci [31,69]. Although H2A/H4R3me2s could promote DNMT3A recruitment in male germ cells [69], its presence on both maternal and paternal gDMRs indicates that it does not provide the signal to discriminate which gDMRs should become methylated [31]. Perhaps the most promising histone modification to serve as a permissive mark for DNA methylation is trimethylation of lysine-36 of H3 (H3K36), and this is discussed further below.
Besides the involvement of H3K4me3 described earlier, the link in vivo between chromatin configuration and de novo DNA methylation remains to be fully explored. This is not helped by our lack of knowledge of the histone modification profiles of germ-cell genomes at the onset of de novo methylation, but the technical challenges in undertaking genome-wide ChIP in small populations of germ cells are considerable. Future studies that target genes encoding histone modifying factors in mice will likely provide important new evidence, unless deficiencies of these factors severely impair germ-cell development prior to the methylation phase. It seems unlikely, however, that imprint acquisition is governed solely by histone modifications: conceptually, there must be an earlier event that dictates where the relevant histone modifications are laid down. In somatic cells, the well-studied histone modifications have broad distribution patterns and are not confined to specific loci. Whether, at the time of DNA acquisition, the same holds true in germ cells remains to be seen. A broad occurrence in germ cells of facilitating histone marks could, however, explain why there are so many methylation differences between male and female germ cells, many more than gDMRs at the known imprinted loci [33,34]. Although histone modifications undoubtedly contribute to the process, DNA methylation acquisition is likely provided by locus-specific signals in addition to, or which instruct, chromatin organization.
4. The role of transcription in imprint acquisition
Given the inadequacy of sequence-based models of gDMR methylation, especially for maternal gDMRs, the possibility that transcription is an additional requirement in specifying sites of methylation has been advanced. Several lines of evidence support a possible involvement of transcription, at least at maternal gDMRs. First, as mentioned earlier, most maternal gDMRs can be considered to be CpG islands that are located within genes. An intragenic location for some gDMRs might not be obvious from canonical gene structures. However, many of the associated genes are found to be transcribed from alternative, upstream transcription start sites in growing oocytes, placing the gDMRs within active transcription units at the time of DNA methylation establishment [30]. This property has now been shown to extend to a high proportion of CpG islands that become methylated in oocytes [33]. It is also worth noting that intragenic CpG islands are frequently methylated in somatic cells [70,71], suggesting that gDMR methylation in oocytes obeys general principles of gene body methylation. A further piece of evidence is provided by imprinted retrogenes. This class of imprinted gene has arisen from retrotransposed genes that have integrated within transcription units and acquired CG-rich gDMRs, often causing isoform-specific imprinted expression of the gene into which they have inserted [28]. The human retinoblastoma locus, RB1, contains an intronic CpG island that provides an alternative first exon to the gene. This CpG island has maternal-allele-specific DNA methylation and arose following retrotransposition from a locus on chromosome 9. Retrotransposition events from the same locus into intergenic locations on another chromosome have not resulted in generation of new DMRs [72]. Third, microdeletions in cis but distinct from gDMRs in two imprinted gene syndromes provide evidence that sequence elements in addition to the gDMRs are essential for methylation. In Angelman syndrome, a large number of such microdeletions have now been catalogued, resulting in the delineation of an essential element positioned 30 kb upstream of the SNRPN DMR that controls imprinting of the Angelman/Prader-Willi syndrome domain in 15q11-13 [73]. Although the molecular function of this genetically defined element has remained obscure until recently, multiple, alternative promoters for SNRPN upstream of this element had been described including, in the case of the mouse locus, a promoter active in oocytes [74,75]. In pseudohypoparathyroidism type 1b (PHP1b), a disorder caused by disrupted imprinting of the GNAS locus, loss of maternal allele methylation in this locus occurs in association with two genetic defects in cis: deletions or rearrangements of the NESP55 upstream promoter region [76] or a recurrent microdeletion in the neighbouring STX16 gene [77]. The mechanism underlying loss of methylation caused by NESP55 deletion can now be understood from functional studies (see below), but the role played by the element in STX16 remains unclear.
While the observations above constitute circumstantial evidence, functional studies have indeed demonstrated a requirement for transcription. In the mouse Gnas locus, deletion of the Nesp55 promoter or insertion of a transcription termination cassette downstream of the Nesp55 exons results in impaired methylation establishment of the maternal gDMRs in this locus (figure 3) [30,78,79]. At the Snrpn locus, a BAC transgenic experiment showed that methylation of the Snrpn DMR depended upon upstream promoters active in oocytes [80]. The latter report offers a novel explanation for the imprinting errors in Angelman syndrome. It may be anticipated that methylation defects in additional imprinted gene syndromes may be caused by disruption of promoter or enhancer elements required to drive transcription events necessary for methylation establishment. These observations lead to a model in which maternal gDMRs correspond to CpG islands associated with silent promoters that are located within active transcription units (figure 3) [30,81]. However, although transcription through CpG island-like features may be essential (and this does need to be tested at additional loci), it is not sufficient. Transcription from Nesp55 leads to methylation of the intragenic gDMRs, but the intragenic CpG island at the Gnas promoter remains fully unmethylated, perhaps protected because it is active during oocyte growth (figure 3) [30]. If a combination of promoter inactivity and transcriptional read-through is an underlying mechanism, why should it apply specifically to imprinted loci? The finding that ‘the somatic promoter for Dnmt3L becomes methylated in oocytes and is downstream of the oocyte-specific promoter’ suggests that the mechanism is more general [82].
Figure 3.
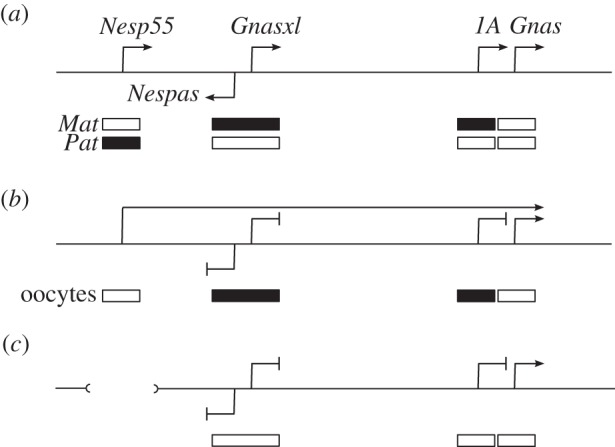
The role of transcription in promoting methylation of gDMRs in the Gnas locus. Schematic of the mouse Gnas locus. (a) The alternative transcription start sites, associated with the promoters for Nesp55, Gnasxl, exon 1A and Gnas, are indicated by the arrows; on the antisense strand, the promoter for the non-coding antisense transcript Nespas is indicated. Below the line, the DMRs and the CpG island at the Gnas promoter are indicated as boxes, with methylation on the maternal (Mat) or paternal (Pat) alleles shown by the filled boxes. (b) In growing oocytes, prior to or during de novo methylation, the DMR-associated promoters at Nespas, Gnasxl and 1A are not expressed, while the Gnas promoter is. In addition, the Nesp promoter is expressed, placing the Nespas/Gnasxl and 1A DMRs and Gnas promoter CpG island within an active transcription unit. Repression of the Nespas, Gnasxl and 1A promoters may be necessary for de novo methylation, whereas activity of the Gnas promoter may protect it from methylation. (c) Deletion of the Nesp55 promoter region or insertion of a transcription termination cassette downstream of the Nesp55 exon to ablate transcription through the DMRs results in failure to establish DNA methylation at the DMRs in oocytes [30,78,79]. Not drawn to scale: the distance between the Nespas and Gnas promoters is approximately 45 kb.
The generality of transcription-dependent methylation of gDMRs and CpG islands is supported by recent genome-wide methylation studies in mouse oocytes. Profiling CpG island methylation in mouse oocytes by reduced representation bisulphite sequencing demonstrated that very few CpG islands are methylated at the onset of oocyte growth (5 days post partum), but that approximately 7 per cent of CpG islands (1062 of those assessed) have become highly methylated in fully grown oocytes [33]. CpG island methylation, as with gDMRs, was found to depend upon both DNMT3A and DNMT3L. Analysis of sequence properties did not identify features that strongly discriminated methylated from unmethylated CpG islands. Instead, a high proportion of methylated CpG islands proved to be intragenically located, especially when evidence for active transcription units and alternative promoter use from mRNA-Seq data from growing oocytes was considered [33]. Whole genome bisulphite sequencing BS-Seq data corroborate the finding of CpG island methylation: with somewhat greater coverage of CpG islands, more than 2000 were identified to be methylated in oocytes [34]. This latter study also revealed the presence of high levels of gene body methylation, which is dependent on DNMT3L and correlates with transcriptional activity inferred from mRNA-Seq data (figure 2). These observations give rise to the notion that gDMR and intragenic CpG island methylation is part of the general process of gene body methylation occurring during oocyte growth (intragenic CpG islands becoming methylated unless otherwise protected). This goes back to our earlier proposition that maternal gDMRs are not specifically targeted for methylation so, instead, we also have to consider mechanisms that prevent their methylation in the other germline to account for germline differential methylation status. The great majority of CpG islands are unmethylated in sperm (the numbers of methylated CpG islands identified by Smallwood et al. [33] and Kobayashi et al. [34] range from 185 to 818). As discussed earlier, at several maternal gDMRs, it has been demonstrated that their associated promoters are active in male foetal germ cells and are marked by H3K4 trimethylation during the de novo methylation phase, consistent with protection against methylation [31]. It might also be pertinent that the known paternal gDMRs are intergenically located and are not transcribed in oocytes [30] and for this reason may not be subject to methylation during oogenesis.
With the likelihood that transcription plays a major role in de novo methylation of gDMRs in growing oocytes, is there evidence that a similar mechanism applies in the male germline? Although DNA methylation has now been comprehensively profiled in mature sperm (both in mouse and human [34,54]), this is many cell divisions after the onset of DNA methylation and no whole genome methylation profiling in prospermatogonia at the initial wave of DNA methylation has yet been reported to allow correlation of methylated sites with active transcription units. However, analysis of individual paternal gDMRs (the H19 DMD and Dlk1-Gtl2 IG-DMR) in male germ cells collected late in gestation has detected transcripts traversing the DMRs, which could be consistent with a transcription-dependent methylation mechanism analogous to that for maternal gDMRs in growing oocytes [31]. Other models, for example, that DMR-associated non-coding RNA could be involved in recruiting histone modifier complexes to guide DNA methylation, are also possible, but there is no evidence to support such a possibility at this time. There is substantial promoter activity in developing male germ cells and it would be interesting to explore the extent to which this broad transcriptional activity contributes in the extensive DNA methylation observed in sperm.
5. Integrating transcription and histone modifications in gDMR methylation
The new perspective of intragenic methylation in oocytes provides novel insights into possible mechanisms relating to transcription-dependent histone modifications and the recruitment of the DNMT3A/DNMT3L methyltransferase complex (figure 4). As described earlier, binding of both DNMT3A (the principal methyltransferase activity in growing oocytes [23,83]) and its essential co-activator DNMT3L is sensitive to the modification state of the N-terminal tail of histone H3. Histone tail binding by DNMT3L and DNMT3A is inhibited by methylation at lysine 4, while the PWWP domain of DNMT3A specifically binds trimethylated H3K36 [29,59,84]. Trimethylation of H3K36 is enriched in active transcription units [85], and the H3K36 methyltransferase SETD2 is associated with the elongating Ser2-phoshorylated form of RNA polymerase II [86]. The H3K4me2/3 demethylase KDM5B (also known as PLU-1/JARID1B) is recruited to gene bodies by H3K36 trimethylation in ES cells, likely via interaction with the chromodomain protein MRG15 [87]. Similarly, the H3K4me1/2 demethylase KDM1B (also known as LSD2 or AOF1) is associated with active gene bodies in human HeLa cells and is present in a complex that includes Ser2-phosphorylated RNA polymerase II and the H3K36 methyltransferase NSD3 [88]. Therefore, processes associated with transcription could directly set up a combination of histone modifications permissive to interaction of DNMT3A/DNMT3L (figure 4). Whether active gene bodies and gDMRs do indeed have these histone modifications in growing oocytes, and whether these specific factors are involved in DNA methylation, will have to await further experiments. Nevertheless, the histone modifications generally associated with transcription and the known interaction properties of DNMT3A and DNMT3L do provide the basis for a plausible model. As noted earlier, some CpG islands within active transcription units do not become DNA-methylated in oocytes. Most CpG islands in somatic cells are enriched in the binding of CXXC-domain proteins, which specifically bind unmethylated CpGs, including the H3K36 demethylase KDM2A and CFP1, which is associated with the SET1 H3K4 methylation complex [89]. Binding of such factors could protect a subset of intragenic CpG islands from DNA methylation in oocytes (figure 4); however, whether these factors do maintain CpG islands hypomethylated in oocytes and what dictates which CpG islands they bind to remains to be determined.
Figure 4.

A model for DNA methylation in oocytes incorporating transcription and histone modification state. At the top, an idealized transcription unit active in oocytes, defined by an upstream, oocyte-specific transcription start site (A), and two intragenic CpG island promoter regions, one (B) that is inactive in oocytes, and a second (C) that is expressed in oocytes. Directly below, individual CpG dinucleotides are indicated, with dense clusters of CpGs at CpG islands. CpG islands A and C are predicted to be bound by CxxC-containing proteins KDM2A and CFP1 that have H3K36 demethylase and H3K4 methyltransferase activities, respectively (the latter via recruitment of the SET1 complex). Transcription through the locus brings the H3K36 methyltransferase SETD2 and the H3K4 demethylases KDM5B and KDM1B, as these activities are associated with the elongating RNA polII complex. (Note that these predictions are based on the properties of these proteins established in somatic cells, only KDM1B has thus far been shown to be involved in gDMR methylation in oocytes. Other members of the respective protein families may perform these roles in oocytes.) The combination of these activities generates distinct states of H3K4 and H3K36 methylation within transcribed regions, active and inactive CpG island promoters and intergenic regions. This profile of H3K4 and H3K36 methylation is read by the DNMT3A/DNMT3L de novo DNA methyltransferase complex to establish methylation at gene bodies and silent, intragenic CpG islands and generate the DNA methylation profile indicated at the bottom. By this mechanism, CpG island B could become a gDMR; differential acquisition of DNA methylation of intragenic CpG islands B and C may be determined by promoter activity or other factors that promote binding of proteins such as KDM2A and CFP1 at CpG island C that are hostile to DNA methylation.
6. DMR methylation induced by small RNAs
One paternal gDMR, the ICR for Rasgrf1, depends on a quite distinct mechanism that involves small RNAs of the Piwi-interacting RNA (piRNA) class. The Rasgrf1 gDMR is also unusual in that it is the only one known to require not only DNMT3A, but also DNMT3B [50], the de novo methyltransferase essential for methylation of various interspersed repeats in the male germline. The gDMR, which is greater than 20 kb upstream of the Rasgrf1 promoter, is an extensive element adjacent to a tandem repeat comprising 40 copies of a 41 base motif. A region containing the tandem repeat has been demonstrated to be essential for establishing methylation in the gDMR in male germ cells [90]. The gDMR also contains a copy of a RMER4B repeat, a solo long terminal repeat-type retrotransposon present in several thousand copies in the mouse genome. From sequence libraries of 23–31-nt small RNAs from foetal testis, multiple small RNAs were identified that exactly or closely matched the RMER4B sequence in the gDMR; these small RNAs were absent in mutants for MitoPLD, a protein involved in primary piRNA production [32]. Spermatogonia from MitoPLD mutants also had strongly reduced methylation of a core region of the gDMR, including the RMER4B sequence (these mutants had normal methylation of the two other paternal gDMRs). A closely related RMER4B element is located in a piRNA cluster on another chromosome (chromosome 7). Evidence was presented that the Rasgrf1 tandem repeat region serves as a promoter for a transcript that traverses the RMER4B element and is targeted by piRNAs produced from the chromosome 7 piRNA cluster. This process results in the production of secondary piRNAs and induction of the piRNA ping-pong amplification cycle [32]. piRNAs have been implicated as guides in de novo methylation of transposable elements specifically in male germ cells [91], although the underlying mechanism is unclear. The question whether additional gDMRs exist that depend on DNMT3B and the piRNA pathway will require genome-wide profiling of methylation in spermatogonia from mutants. Given the critical role of piRNAs in silencing transposable elements in male germ cells, there are likely to be other sites of differential DNA methylation between sperm and oocytes dependent on the piRNA pathway. This finding reiterates the possibility that selection for imprinting could act on any gDMR, irrespective of the mechanism by which it is established, as long as a selective advantage would accrue to ensure maintenance of the allelic DNA methylation after fertilization.
7. Parental imprints independent of DNA methylation?
Unexpected findings in a number of experimental studies have indicated that imprinting mechanisms other than CpG methylation could be acting in mammalian germ cells as well. Although the precise nature of such DNA methylation-independent marks remains enigmatic, these observations potentially identify pathways acting in parallel to germline DNA methylation. Perhaps the best documented example is imprinted X chromosome inactivation [92]. In the mouse, X inactivation is subject to imprinting during early pre-implantation development and in the extra-embryonic lineages [93,94]. In these situations, the non-coding RNA Xist is expressed from, and triggers inactivation of, the paternal X chromosome, whereas the maternally inherited, active X chromosome is marked such that it does not express the Xist gene in the early embryo [95]. Remarkably, this maternal germline-specific repression mechanism does not require DNA methylation because it still occurs in the absence of the de novo DNA methyltransferases during oogenesis [96]. Several autosomal domains in the mouse comprise genes that appear to be imprinted in the extra-embryonic lineage (placenta) only. A carefully conducted study in the mouse placenta recently identified two new placenta-specific imprinted loci, Ano1 and Gab1, at which no evidence for allelic DNA methylation was found [97]. Similarly, the Sfmbt2 gene, which exhibits paternal-allele-specific expression specifically in early embryos and extra-embryonic tissues, appears to lack a gDMR [98]. These intriguing observations raise the question whether certain autosomal loci might be controlled by a germline-derived imprint distinct from DNA methylation.
Knockout studies on Dnmt3L have provided unexpected findings as well. DNMT3L-deficient females are phenotypically normal but do not establish DNA methylation imprints in their oocytes. Absence of maternal DNA methylation imprints gives rise to aberrant imprinted gene expression in embryos produced by Dnmt3L−/− females [21,22]. Unexpectedly, however, in some embryos from DNMT3L-deficient females, normal allelic DNA methylation was detected at the Snrpn and Peg3 gDMRs and was associated with the expected histone modifications at these DMRs in somatic cells [99,100]. However, whether this observation reflects stochastic methylation establishment even in DNMT3L-deficient oocytes or allele-specific de novo methylation after fertilization is not clear, owing to the conflicting reports of whether these mutant oocytes have residual methylation of the Snrpn DMR [21,34,83]. Failure in methylation of the Snrpn DMR also occurs in oocytes from female mice lacking the Krüppel-associated box (KRAB)-containing zinc finger protein ZFP57 [57]. The Snrpn DMR was unique among five maternal gDMRs tested in depending on ZFP57 for de novo methylation in oocytes. Surprisingly, however, in about half the offspring from Zfp57−/− female mice, the Snrpn DMR regained methylation and did so specifically on the maternal allele. This finding suggests that zygotically provided ZFP57 can read an epigenetically distinct state of the maternal allele of the DMR, which might be carried over from the oocyte, but which must persist until after implantation, because DMR methylation is still absent in e3.5 blastocysts [57]. The mechanistic basis for this recovery of methylation is currently unclear, in particular because ZFP57 binding to DMRs appears to be methylation-dependent [58].
When ectopically inserted into the genome in transgenic studies, the H19 ICR, which is a paternal gDMR at its endogenous location, does not acquire DNA methylation in male germ cells. However, in some of the offspring of transgenic males, CpG methylation was found to be acquired on the paternal allele at around the time of implantation [101,102]. Thus, upon paternal transmission, the ectopically inserted DMR had become marked in sperm so that it could attract DNA methylation during embryonic development.
What parental mark other than DNA methylation could be involved in these cases and could be somatically maintained, at least during early development? As concerns the female germ line, several nuclear and cytoplasmic proteins are known to influence imprint establishment at single or multiple ICRs (table 1). For most such proteins, whether they are directly involved in the DNA methylation pathway or bring about marks other than DNA methylation is not known. Given that the oocyte genome remains nucleosomally organized throughout oogenesis, chromatin at maternal gDMRs could acquire a state permissive to DNA methylation establishment. If such a configuration were maintained after fertilization, in exceptional cases, this could allow DNA methylation establishment even after fertilization. As discussed earlier, recent studies show that at imprinted loci, the paternal genome remains nucleosomally organized, at least in part, in mature spermatozoa [65,66]. Defined chromatin states could thus facilitate early-embryonic acquisition of allelic DNA methylation on the paternal chromosome, as observed in the H19 DMR transgenic studies.
Table 1.
Proteins involved in establishment and maintenance of DNA methylation at gDMRs.
| protein | alternative name(s) | function | phenotype due to protein-deficiency | references |
|---|---|---|---|---|
| DNMT3A | — | de novo DNA methyltransferase | lack of imprint establishment in germ cells | [23] |
| DNMT3B | — | de novo DNA methyltransferase | impaired establishment of Rasgrf1 gDMR in male germ cells | [23,50] |
| DNMT3L | — | DNA methyltransferase-like protein | lack of imprint establishment in germ cells | [21,22,50] |
| MIWI2; MILI; MitoPLD | PIWIL4; PIWIL2; ZUC, PLD6 | piRNA pathway proteins | impaired establishment of Rasgrf1 gDMR in male germ cells | [32,103] |
| KDM1B | LSD2, AOF1 | H3 lysine-4 demethylase | defects in establishment of maternal gDMRs in oocytes | [63] |
| NLRP2; NLRP7 | — | cytoplasmic caterpillar family proteins, unknown molecular function | maternal mutations associated with hydatidiform moles affect methylation at multiple maternal DMRs (human); NLRP2 mutations found in a Beckwith–Wiedemann syndrome family with loss of methylation at the KvDMR | [104,105] |
| C6orf221 | (ECAT1) | cytoplasmic protein with N-terminal KH domain, interacts with NLRP7 | maternal mutations associated with hydatidiform moles affect methylation at multiple maternal DMRs (human). | [106] |
| DPPA3 | PGC7, STELLA | aberrant gain of DNA methylation at some maternal gDMRs in the zygote | [107] | |
| ZFP57 | — | KRAB domain zinc finger protein | somatic loss of DNA methylation at multiple gDMRs (mouse, human); absence of maternal imprint establishment at Snrpn gDMR (mouse) | [56–58] |
| KAP1 | TRIM28, TIF1β, KRIP1 | KRAB-associated protein 1, scaffold protein for heterochromatin factors | stochastic loss of DNA methylation at multiple gDMRs | [108] |
| DNMT1 | — | maintenance DNA methyltransferase | somatic loss of DNA methylation, including at imprinted loci | [109,110] |
| UHRF1 | NP95, ICBP90 | binds to hemimethylated DNA, recruits DNMT1 | somatic loss of DNA methylation, including at imprinted loci | [111] |
| H1 | — | linker histone | partial loss of DNA methylation at H19 gDMR and Dlk1-Gtl2 IG-DMR | [112] |
| MBD3; MTA2 | — | methyl CpG-binding domain protein-3, metastasis tumour antigen 2; components of the NuRD complex | somatic loss of DNA methylation at the H19 and Peg3 gDMRs | [113,114] |
| RBBP1; RBBP1L | — | RB binding proteins | somatic loss of DNA methylation at Snrpn gDMR | [115] |
| CTCF | — | zinc finger protein | aberrant gain of DNA methylation at the H19 gDMR during early development | [116] |
| ZFP42 | REX1 | zinc finger protein, with similarity to YY1 | aberrant acquisition of DNA methylation on paternal allele of Peg3 gDMR | [117] |
8. An essential role for imprint maintenance during early development
As discussed earlier, many CpG islands and other CpG-rich regions become DNA-methylated during gametogenesis, but it is only at a small fraction of these sequence elements that methylation persists throughout the pre-implantation period and during later stages of development (figure 1) [33–35,55,118]. Therefore, the cellular mechanisms that maintain germline-derived methylation during early development are the key to the specificity of genomic imprinting. They determine which gDMRs maintain their differential DNA methylation and, hence, can bring about parental-allele-specific gene expression during development. Despite the importance of this ‘selection’ process, the maintenance mechanisms acting specifically on imprinted gDMRs remain poorly understood. As concerns human disease, understanding how differential DNA methylation states are maintained at these exceptional sequence elements is of considerable importance. In most cases of imprinting-related diseases [16], epigenetic alterations that perturb imprinted gene expression are thought to arise in the early embryo. Studies on human imprinting disorders and mouse models have pinpointed several proteins that are involved in the maintenance of parental-allele-specific DNA methylation during embryonic development (table 1). Some of these proteins have a general role in the dynamics and maintenance of DNA methylation, while others may be specifically directed at imprinted loci. Moreover, DMR status requires activities that maintain methylation on the methylated allele as well as those that protect the unmethylated allele against aberrant gain of methylation during development.
The first few embryonic cell cycles are particularly critical for the maintenance of DNA methylation. Following fertilization, the paternal genome loses a substantial part of its DNA methylation through an active demethylation process. The maternal genome is largely resistant to this active demethylation, but gradually loses much of its DNA methylation during pre-implantation development [119]. Apart from the essential and general role of the maintenance DNA methyltransferase DNMT1 [109], several maternal proteins contribute to the protection against DNA demethylation after fertilization. DPPA3 (also called PGC7 or STELLA) is highly expressed during oogenesis, until the oocyte maturates, and persists in the pre-implantation embryo. Gene targeting studies in the mouse have shown that after fertilization up to the 2-cell stage, DPPA3 plays a crucial role in protecting the maternal genome against DNA demethylation. This includes methylation of the maternal gDMRs at Peg1, Peg3 and Peg10, but not those at Snrpn or Peg5. On the paternal genome, DPPA3 protects two paternally methylated gene loci against demethylation, H19 and Rasgrf1 [107]. Although DPPA3 contains a DNA-binding domain, its precise mode of action and specificity towards a subset of gDMRs, and whether this is linked to the presence of specific histone modifications, remain to be discovered. Possibly, some of the observed effects could be linked to the recently reported role of DPPA3 in chromatin condensation during oogenesis [120], which might set up histone modifications that protect against DNA demethylation. DPPA3 appears to have a general role in protecting the maternal genome against conversion of 5mC to 5-hydroxymethylcytosine (5hmC) [121], and so is best not viewed as a selective factor in gDMR maintenance.
A nuclear protein that appears to have the most extensive and specific role in protecting gDMRs against loss of methylation, and whose mechanism of action is perhaps best understood, is ZFP57. The first insight into the essential role of this KRAB-domain zinc finger protein came from studies on transient neonatal diabetes (TNDM), a disorder caused by aberrant imprinting of PLAGL1. Intriguingly, in some TNDM pedigrees, loss of DNA methylation was detected at multiple DMRs and was found to correlate with homozygous mutations in the ZFP57 gene [56]. A serendipitous observation from gene targeting in the mouse revealed that ZFP57 is essential in the somatic maintenance of DNA methylation at multiple gDMRs, as well as for de novo methylation of the Snrpn DMR [57]. In ES cells, ZFP57 is bound to almost all imprinted gDMRs studied, and to their DNA-methylated alleles only. The specificity of this interaction is mediated by a hexanucleotide sequence (TGCCGC) found at gDMRs and at only several tens of additional loci [58]. A plausible mechanism by which ZFP57 mediates the somatic maintenance of DNA methylation involves KRAB-associated protein 1 (KAP1, also known as TRIM28, TIF1B or KRIP1). Through its KRAB domain, ZFP57 brings its cofactor KAP1 to the chromatin, which in turn leads to the recruitment of repressive chromatin modifiers, including ESET/SETB1, which controls H3K9 trimethylation [58,122]. Furthermore, ZFP57/KAP1 complexes also interact with DNMTs and the hemimethylated DNA binding protein UHRF1 (also called NP95), which are both essential for the maintenance of DNA methylation as well (table 1). It has also recently been shown that KAP1 derived from the oocyte is essential for maintenance of gDMR methylation during the first cell divisions, prior to production of zygotic KAP1 [108]. In the absence of maternal KAP1, there is stochastic loss of methylation at several gDMRs, both maternal and paternal, but whether all gDMRs are equally likely to be affected remains to be seen. Further work should elucidate the precise in vivo links between these maintenance factors. It will be important to demonstrate whether the distribution of the TGCCGC motif alone can explain why only a limited number of the large set of CpG islands that arrive highly methylated from the oocyte and sperm genomes fully retain methylation during pre-implantation development, and whether KAP1 is part of this discrimination process.
Additional zinc finger proteins may also affect DMR methylation status, but in different ways and probably without the specificity towards gDMRs exercised by ZFP57. ZFP42 (also known as REX1) is a well-known stem-cell marker whose structure is similar to that of the more broadly expressed YY1 protein. ZFP42-deficient blastocysts show a degree of hypermethylation at two gDMRs, at Peg3 and at Gnas, both known to contain YY1 binding sites [117]. The multifunctional zinc finger protein CTCF plays diverse roles in gene regulation. In somatic cells, it associates with the unmethylated maternal allele of the H19 DMR. Mutation of all four CTCF binding sites in the H19 DMR leads to aberrant gain of DNA methylation on the maternal allele during post-implantation development [116,123]. In both cases, therefore, DNA binding by these sequence-specific zinc finger proteins protects against methylation of the unmethylated allele. In this regard, whole-genome methylome analysis in mouse ES cells identified that hypomethylation at regulatory regions of low CpG density (so-called ‘low methylated regions’) is conferred by binding of transcription factors, such as CTCF [124]. Together, these observations suggest that continual binding of such factors at the unmethylated copy of gDMRs could provide protection against methylation during development, but in a manner that is not specific to gDMRs.
Other proteins contribute to the embryonic maintenance of DNA methylation at gDMRs as well. Methyl CpG-binding domain protein-3 (MBD3) and metastasis tumour antigen 2 (MTA2) are both part of the nucleosome remodelling and deacetylase (NuRD) chromatin remodelling complex. Knocking-down either MTA2 or MBD3 was found to lead to some reduction in methylation at the H19 DMR, but not the Peg3 or Snrpn DMRs, in pre-implantation embryos [113,114], suggesting that the NuRD complex contributes to gDMR maintenance with some degree of specificity, but the underlying mechanism is not known. Besides chromatin regulatory proteins, histone modifications could also contribute directly to the early somatic maintenance of differential DNA methylation at germline DMRs. Of particular interest is the consistent enrichment of H3K4me2 and H3K4me3 on the unmethylated allele at all gDMRs analysed. At maternally methylated gDMRs, this mark correlates with promoter activity. Because the de novo DNA methylation machinery is expressed highly in the early embryo, allelic enrichment of H3K4me2/3 could prevent acquisition of aberrant de novo methylation in embryonic cells, as discussed earlier for germ cells, because it would prevent binding of the DNMT3A- and DNMT3B/DNMT3L complexes to chromatin [29,59]. Whether other promoter-associated histone modifications directly prevent DNA methylation on the unmethylated alleles of gDMRs remains to be explored.
Proteins of the ten-eleven translocation (TET) family have been hypothesized to be involved in the control of CpG methylation in mammalian cells [125]. A few years ago, TET1 was shown to oxidize 5mC to 5hmC, thereby contributing to DNA demethylation [126]. TET proteins are expressed in different cell types, at highest levels in the zygote and in ES cells, and this could pinpoint a role in DNA demethylation. TET3 has been shown to be involved in the conversion of 5mC to 5hmC in the mouse zygote, particularly in the sperm-derived pronucleus [127], but a role specifically in controlling DNA methylation at CpG islands that acquire methylation in the germline has not been demonstrated. Proteins of this family could also help in maintaining the unmethylated allele of gDMRs unmethylated by removing aberrantly acquired 5mC.
9. Outlook
Two new concepts that challenge how we view imprinting have emerged from recent work. First, that methylation at gDMRs is not the result of specific targeting mechanisms, but is better viewed as part of the general processes of methylation in the female and male germlines (figures 2 and 4). This is illustrated particularly graphically in oocytes, in which gDMRs are seen to be part of gene body methylation (figure 3). This new perspective suggests that ‘imprint-specific factors’ do not exist in germ cells. Second, that many CpG-rich sequences become DNA-methylated in oocytes or sperm, but only a fraction of these gametic marks survive through the early stages of development (figure 1). Therefore, there must be a selection process in pre-implantation embryos that specifically protects gDMRs from demethylation and brings about the persistent parental-allele-specific DNA methylation that defines (most) imprinted domains. In the pre-implantation embryo and beyond, gDMR status is likely maintained by a combination of generic factors and, more excitingly, one or more imprinting specific factors, as exemplified by ZFP57. The latter has particularly interesting implications for the evolution of imprinting. This novel angle from which to consider the specificity of genomic imprinting raises interesting questions that could be addressed in future research. Is maintenance of gDMR methylation determined by specific DNA sequence motifs or by additional epigenetic marks conferred in the gametes? To properly address this second issue, it would be essential to know all imprinted genes. Recent mRNA-Seq analysis of gene expression in brain from reciprocal hybrid mice raised the possibility that there were ten-fold more potentially imprinted transcripts than previously recognized [128], but this conclusion has been disputed [129]. Integration of such mRNA-Seq data with methylation profiles of gametes [33–35] and allele-specific methylation in embryo or adult tissues [118] should provide a definitive list of imprinted loci and the developmental fate of gamete-derived methylation.
An important question relates to the chromatin (re)organization at gDMRs during gametogenesis. The histone modification status at gDMRs may be relevant not only by serving as a template for de novo DNA methylation, but might also influence the fate of these elements after fertilization, and could also contribute to imprinting phenomena apparently independent of DNA methylation. Besides the acquired CpG methylation, what other features of chromatin characterize gDMRs in the mature oocyte (maternal imprints) or sperm (paternal imprints)? Could additional features of chromatin help protect these exceptional gDMRs against loss of DNA methylation following fertilization? Could such a protection mechanism also be linked to the binding of non-histone proteins already in the gametes? Technically, this question is a not trivial to address. However, interesting insights have emerged relative to paternal imprints from studies showing that imprinted loci partially remain associated with histones in mature sperm [65,66]. At the protected paternal gDMRs, therefore, specific histone modifications could contribute to the maintenance of DNA methylation after fertilization.
Maternal factors that recognize specific gDMRs are likely to play a significant role as well. Above, we described the example of DPPA3, a protein that protects a subset of gDMRs after fertilization. The cell biology of the maturing oocyte can also impact on maintenance processes after fertilization. Several recent studies show that hormone-induced superovulation can affect maintenance of DNA methylation at both maternal and paternal gDMRs during pre-implantation development [130,131]. This maternal effect could be linked to altered protein accumulation in the oocyte as a consequence of superovulation, for example, by prematurely recruiting oocytes to ovulate. Histone modifications are likely to contribute to the early somatic maintenance of allelic methylation at gDMRs as well, and we discussed the possible role of H3K4 methylation. Studies have only just started to explore chromatin at imprinted loci in pre-implantation embryos [132], for which the available material is still limiting for currently used ChIP methods. When considering chromatin in the pre-implantation embryo, histone variants may also need to be considered. Although its role remains to be explored, enrichment of the H2A variant macroH2A1 on the DNA-methylated allele of several gDMRs in somatic cells was reported in one study [133]. Understanding the maintenance of allelic DNA methylation in the early embryo is particularly relevant in relation to in vitro culture of human embryos and other assisted reproduction technologies that may increase the risk of epigenetic abnormalities and give rise to imprinted-related diseases.
It is sometimes assumed that the maintenance of allelic methylation imprints is comparable between the different cells of the early embryo. This is not necessarily the case, particularly comparing the cells that eventually give rise to the embryo itself and those that form the extra-embryonic lineages. In vitro culture of pre-implantation embryos frequently affects the maintenance of DNA methylation imprints, particularly in the trophoblast cells that give rise to the placenta [134]. It remains to be discovered why these cells maintain methylation at gDMRs less tightly, and whether this could be linked to lower levels of expression of specific protein factors.
Finally, the recent discoveries raise the intriguing question as to how and why mechanisms evolved to maintain DNA methylation only at certain gDMRs in the early embryo. To what extent does this represent a conflict between the maternal and paternal interests, in accordance with one theory for the evolution of imprinting [135]? Some of the factors involved in maintenance, such as DPPA3, are contributed to the zygote by the oocyte and mediate their protective effect during the very early stages following fertilization, providing an element of maternal control over DNA methylation maintenance. But many others act mostly in the early embryo, some even after implantation and are equally expressed from both the parental genomes (table 1). Certainly after zygotic genome activation, the two parental genomes unite in somatic maintenance of gDMRs, irrespective of their parental origin. While some of the processes of gDMR maintenance depend upon generic mechanisms regulating DNA methylation, there is also the involvement of factors like ZFP57, which themselves are not imprinted, that act selectively at imprinted gDMRs. This suggests the evolution of specific imprinting maintenance factors possibly in concert with the evolution of their target sequences. There may be a point in development, after the phases of greatest epigenetic upheaval, at which maintenance does become the default and specific factors such as ZFP57 become dispensable. Future research into these intriguing issues should benefit from comparisons between different groups of animals, to trace back the evolution of maintenance factors and their involvement in genomic imprinting.
Acknowledgements
We thank all the members of our teams for discussion and helpful comments. R.F. acknowledges grant funding from l'Agence Nationale de la Recherche (ANR), Institut National Contre le Cancer (INCa), the Ligue National Contre le Cancer and the Agency for International Cancer Research (AICR). G.K. acknowledges funding from the UK Biotechnology and Biological Sciences Research Council (BBSRC) and Medical Research Council (MRC). Both authors are affiliated to the FP7 Network of Excellence ‘EpiGeneSys’.
References
- 1.Ferguson-Smith AC. 2011. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 12, 565–575 10.1038/nrg3032 (doi:10.1038/nrg3032) [DOI] [PubMed] [Google Scholar]
- 2.McGrath J, Solter D. 1984. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37, 179–183 10.1016/0092-8674(84)90313-1 (doi:10.1016/0092-8674(84)90313-1) [DOI] [PubMed] [Google Scholar]
- 3.Surani MA, Barton SC, Norris ML. 1984. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308, 548–550 10.1038/308548a0 (doi:10.1038/308548a0) [DOI] [PubMed] [Google Scholar]
- 4.Cattanach BM, Kirk M. 1985. Differential activity of maternal and paternally derived chromosome regions in mice. Nature 315, 496–498 10.1038/315496a0 (doi:10.1038/315496a0) [DOI] [PubMed] [Google Scholar]
- 5.Barlow DP, Stoger R, Herrmann BG, Saito K, Schweifer N. 1991. The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 349, 84–87 10.1038/349084a0 (doi:10.1038/349084a0) [DOI] [PubMed] [Google Scholar]
- 6.DeChiara TM, Robertson EJ, Efstratiadis A. 1991. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849–859 10.1016/0092-8674(91)90513-X (doi:10.1016/0092-8674(91)90513-X) [DOI] [PubMed] [Google Scholar]
- 7.Bartolomei MS, Zemel S, Tilghman SM. 1991. Parental imprinting of the mouse H19 gene. Nature 351, 153–155 10.1038/351153a0 (doi:10.1038/351153a0) [DOI] [PubMed] [Google Scholar]
- 8.Henckel A, Arnaud P. 2010. Genome-wide identification of new imprinted genes. Brief. Funct. Genomics 9, 304–314 10.1093/bfgp/elq016 (doi:10.1093/bfgp/elq016) [DOI] [PubMed] [Google Scholar]
- 9.Williamson CM, Blake A, Thomas S, Beechey CV, Hancock J, Cattanach BM, Peters J. MRC Harwell, Oxfordshire. World Wide Web Site—Mouse Imprinting Data and References. See. 2012. http://www.har.mrc.ac.uk/research/genomic_imprinting/ .
- 10.Fowden AL, Coan PM, Angiolini E, Burton GJ, Constancia M. 2011. Imprinted genes and the epigenetic regulation of placental phenotype. Prog. Biophys. Mol. Biol. 106, 281–288 10.1016/j.pbiomolbio.2010.11.005 (doi:10.1016/j.pbiomolbio.2010.11.005) [DOI] [PubMed] [Google Scholar]
- 11.Charalambous M, da Rocha ST, Ferguson-Smith AC. 2007. Genomic imprinting, growth control and the allocation of nutritional resources: consequences for postnatal life. Curr. Opin. Endocrinol. Diabetes Obes. 14, 3–12 10.1097/MED.0b013e328013daa2 (doi:10.1097/MED.0b013e328013daa2) [DOI] [PubMed] [Google Scholar]
- 12.Frontera M, Dickins B, Plagge A, Kelsey G. 2008. Imprinted genes, postnatal adaptations and enduring effects on energy homeostasis. Adv. Exp. Med. Biol. 626, 41–61 10.1007/978-0-387-77576-0_4 (doi:10.1007/978-0-387-77576-0_4) [DOI] [PubMed] [Google Scholar]
- 13.Wilkinson LS, Davies W, Isles AR. 2007. Genomic imprinting effects on brain development and function. Nat. Rev. Neurosci. 8, 832–843 10.1038/nrn2235 (doi:10.1038/nrn2235) [DOI] [PubMed] [Google Scholar]
- 14.Wan LB, Bartolomei MS. 2008. Regulation of imprinting in clusters: noncoding RNAs versus insulators. Adv. Genet. 61, 207–223 10.1016/S0065-2660(07)00007-7 (doi:10.1016/S0065-2660(07)00007-7) [DOI] [PubMed] [Google Scholar]
- 15.Spahn L, Barlow DP. 2003. An ICE pattern crystallizes. Nat. Genet. 35, 11–12 10.1038/ng0903-11 (doi:10.1038/ng0903-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomizawa S, Sasaki H. 2012. Genomic imprinting and its relevance to congenital disease, infertility, molar pregnancy and induced pluripotent stem cell. J. Hum. Genet. 57, 84–91 10.1038/jhg.2011.151 (doi:10.1038/jhg.2011.151) [DOI] [PubMed] [Google Scholar]
- 17.Stöger R, Kubicka P, Liu CG, Kafri T, Razin A, Cedar H, Barlow DP. 1993. Maternal-specific methylation of the imprinted mouse Igf2r locus identifies the expressed locus as carrying the imprinting signal. Cell 73, 61–71 10.1016/0092-8674(93)90160-R (doi:10.1016/0092-8674(93)90160-R) [DOI] [PubMed] [Google Scholar]
- 18.Wutz A, Smrzka OW, Schweifer N, Schellander K, Wagner EF, Barlow DP. 1997. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 389, 745–749 10.1038/39631 (doi:10.1038/39631) [DOI] [PubMed] [Google Scholar]
- 19.Tomizawa S, Kobayashi H, Watanabe T, Andrews S, Hata K, Kelsey G, Sasaki H. 2011. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development 138, 811–820 10.1242/dev.061416 (doi:10.1242/dev.061416) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li E, Beard C, Jaenisch R. 1993. Role for DNA methylation in genomic imprinting. Nature 366, 362–365 10.1038/366362a0 (doi:10.1038/366362a0) [DOI] [PubMed] [Google Scholar]
- 21.Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. 2001. Dnmt3L and the establishment of maternal genomic imprints. Science 294, 2536–2539 10.1126/science.1065848 (doi:10.1126/science.1065848) [DOI] [PubMed] [Google Scholar]
- 22.Hata K, Okano M, Lei H, Li E. 2002. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129, 1983–1993 [DOI] [PubMed] [Google Scholar]
- 23.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. 2004. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429, 900–903 10.1038/nature02633 (doi:10.1038/nature02633) [DOI] [PubMed] [Google Scholar]
- 24.Neumann B, Kubicka P, Barlow DP. 1995. Characteristics of imprinted genes. Nat. Genet. 9, 12–13 10.1038/ng0195-12 (doi:10.1038/ng0195-12) [DOI] [PubMed] [Google Scholar]
- 25.Shemer R, Hershko AY, Perk J, Mostoslavsky R, Tsuberi B, Cedar H, Buiting K, Razin A. 2000. The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat. Genet. 26, 440–443 10.1038/82571 (doi:10.1038/82571) [DOI] [PubMed] [Google Scholar]
- 26.Kantor B, Makedonski K, Green-Finberg Y, Shemer R, Razin A. 2004. Control elements within the PWS/AS imprinting box and their function in the imprinting process. Hum. Mol. Genet. 13, 751–762 10.1093/hmg/ddh085 (doi:10.1093/hmg/ddh085) [DOI] [PubMed] [Google Scholar]
- 27.Ruf N, Bähring S, Galetzka D, Pliushch G, Luft FC, Nürnberg P, Haaf T, Kelsey G, Zechner U. 2007. Sequence-based bioinformatic prediction and QUASEP identify genomic imprinting of the KCNK9 potassium channel gene in mouse and human. Hum. Mol. Genet. 16, 2591–2599 10.1093/hmg/ddm216 (doi:10.1093/hmg/ddm216) [DOI] [PubMed] [Google Scholar]
- 28.Wood AJ, Roberts RG, Monk D, Moore GE, Schulz R, Oakey RJ. 2007. A screen for retrotransposed imprinted genes reveals an association between X chromosome homology and maternal germ-line methylation. PLoS Genet. 3, e20. 10.1371/journal.pgen.0030020 (doi:10.1371/journal.pgen.0030020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ooi SK, et al. 2007. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717 10.1038/nature05987 (doi:10.1038/nature05987) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, James K, Dean W, Kelsey G. 2009. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 23, 105–117 10.1101/gad.495809 (doi:10.1101/gad.495809) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henckel A, Chebli K, Kota SK, Arnaud P, Feil R. 2011. Transcription and histone methylation changes correlate with imprint acquisition in male germ cells. EMBO J. 31, 606–615 10.1038/emboj.2011.425 (doi:10.1038/emboj.2011.425) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe T, et al. 2011. Role of piRNA and non-coding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 332, 848–852 10.1126/science.1203919 (doi:10.1126/science.1203919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smallwood SA, et al. 2011. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 43, 811–814 10.1038/ng.864 (doi:10.1038/ng.864) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi H, et al. 2012. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 8, e1002440. 10.1371/journal.pgen.1002440 (doi:10.1371/journal.pgen.1002440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484, 339–344 10.1038/nature10960 (doi:10.1038/nature10960) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kota SK, Feil R. 2010. Epigenetic transitions in germ cell development and meiosis. Dev. Cell 19, 675–686 10.1016/j.devcel.2010.10.009 (doi:10.1016/j.devcel.2010.10.009) [DOI] [PubMed] [Google Scholar]
- 37.Arnaud P. 2010. Genomic imprinting in germ cells: imprints are under control. Reproduction 140, 411–423 10.1530/REP-10-0173 (doi:10.1530/REP-10-0173) [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi H, Suda C, Abe T, Kohara Y, Ikemura T, Sasaki H. 2006. Bisulfite sequencing and dinucleotide content analysis of 15 imprinted mouse differentially methylated regions (DMRs): paternally methylated DMRs contain less CpGs than maternally methylated DMRs. Cytogenet. Genome Res. 113, 130–137 10.1159/000090824 (doi:10.1159/000090824) [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi H, Sakurai T, Sato S, Nakabayashi K, Hata K, Kono T. 2012. Imprinted DNA methylation reprogramming during early mouse embryogenesis at the Gpr1-Zdbf2 locus is linked to long cis-intergenic transcription. FEBS Lett. 586, 827–833 10.1016/j.febslet.2012.01.059 (doi:10.1016/j.febslet.2012.01.059) [DOI] [PubMed] [Google Scholar]
- 40.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. 2010. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463, 1101–1105 10.1038/nature08829 (doi:10.1038/nature08829) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guibert S, Forné T, Weber M. 2012. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 22, 633–641 10.1101/gr.130997.111 (doi:10.1101/gr.130997.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. 2002. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 117, 15–23 10.1016/S0925-4773(02)00181-8 (doi:10.1016/S0925-4773(02)00181-8) [DOI] [PubMed] [Google Scholar]
- 43.Lee J, Inoue K, Ono R, Ogonuki N, Kohda T, Kaneko-Ishino T, Ogura A, Ishino F. 2002. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development 129, 1807–1817 [DOI] [PubMed] [Google Scholar]
- 44.Lucifero D, Mann MR, Bartolomei MS, Trasler JM. 2004. Gene-specific timing and epigenetic memory in oocyte imprinting. Hum. Mol. Genet. 13, 839–849 10.1093/hmg/ddh104 (doi:10.1093/hmg/ddh104) [DOI] [PubMed] [Google Scholar]
- 45.Hiura H, Obata Y, Komiyama J, Shirai M, Kono T. 2006. Oocyte growth-dependent progression of maternal imprinting in mice. Genes Cells 11, 353–361 10.1111/j.1365-2443.2006.00943.x (doi:10.1111/j.1365-2443.2006.00943.x) [DOI] [PubMed] [Google Scholar]
- 46.Obata Y, Kono T. 2002. Maternal primary imprinting is established at a specific time for each gene throughout oocyte growth. J. Biol. Chem. 277, 5285–5289 10.1074/jbc.M108586200 (doi:10.1074/jbc.M108586200) [DOI] [PubMed] [Google Scholar]
- 47.Bourc'his D, Proudhon C. 2008. Sexual dimorphism in parental imprint ontogeny and contribution to embryonic development. Mol. Cell. Endocrinol. 282, 87–94 10.1016/j.mce.2007.11.025 (doi:10.1016/j.mce.2007.11.025) [DOI] [PubMed] [Google Scholar]
- 48.Davis TL, Yang GJ, McCarrey JR, Bartolomei MS. 2000. The H19 methylation imprint is erased and re-established differentially on the parental alleles during male germ cell development . Hum. Mol. Genet. 9, 2885–2894 10.1093/hmg/9.19.2885 (doi:10.1093/hmg/9.19.2885) [DOI] [PubMed] [Google Scholar]
- 49.Bourc'his D, Bestor TH. 2004. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431, 96–99 10.1038/nature02886 (doi:10.1038/nature02886) [DOI] [PubMed] [Google Scholar]
- 50.Kato Y, et al. 2007. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum. Mol. Genet. 16, 2272–2280 10.1093/hmg/ddm179 (doi:10.1093/hmg/ddm179) [DOI] [PubMed] [Google Scholar]
- 51.Hutter B, Helms V, Paulsen M. 2006. Tandem repeats in the CpG islands of imprinted genes. Genomics 88, 323–332 10.1016/j.ygeno.2006.03.019 (doi:10.1016/j.ygeno.2006.03.019) [DOI] [PubMed] [Google Scholar]
- 52.Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. 2006. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 20, 1268–1282 10.1101/gad.1416906 (doi:10.1101/gad.1416906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. 2007. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449, 248–251 10.1038/nature06146 (doi:10.1038/nature06146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD. 2011. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146, 1029–1041 10.1016/j.cell.2011.08.016 (doi:10.1016/j.cell.2011.08.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borgel J, Guibert S, Li Y, Chiba H, Schübeler D, Sasaki H, Forné T, Weber M. 2010. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 42, 1093–1100 10.1038/ng.708 (doi:10.1038/ng.708) [DOI] [PubMed] [Google Scholar]
- 56.Mackay DJ, et al. 2008. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 40, 949–951 10.1038/ng.187 (doi:10.1038/ng.187) [DOI] [PubMed] [Google Scholar]
- 57.Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. 2008. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev. Cell 15, 547–557 10.1016/j.devcel.2008.08.014 (doi:10.1016/j.devcel.2008.08.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quenneville S, et al. 2011. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell 44, 361–372 10.1016/j.molcel.2011.08.032 (doi:10.1016/j.molcel.2011.08.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, et al. 2010. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 38, 4246–4253 10.1093/nar/gkq147 (doi:10.1093/nar/gkq147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bernstein BE, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 10.1016/j.cell.2006.02.041 (doi:10.1016/j.cell.2006.02.041) [DOI] [PubMed] [Google Scholar]
- 61.Mikkelsen TS, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 10.1038/nature06008 (doi:10.1038/nature06008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao XD, et al. 2007. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 1, 286–298 10.1016/j.stem.2007.08.004 (doi:10.1016/j.stem.2007.08.004) [DOI] [PubMed] [Google Scholar]
- 63.Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T. 2009. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461, 415–418 10.1038/nature08315 (doi:10.1038/nature08315) [DOI] [PubMed] [Google Scholar]
- 64.Delaval K, Govin J, Cerqueira F, Rousseaux S, Khochbin S, Feil R. 2007. Differential histone modifications mark mouse imprinting control regions during spermatogenesis. EMBO J. 26, 720–729 10.1038/sj.emboj.7601513 (doi:10.1038/sj.emboj.7601513) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. 2009. Distinctive chromatin in human sperm packages genes for embryo development. Nature 460, 473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brykczynska U, et al. 2010. Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat. Struct. Mol. Biol. 17, 679–687 10.1038/nsmb.1821 (doi:10.1038/nsmb.1821) [DOI] [PubMed] [Google Scholar]
- 67.Tamaru H, Zhang X, McMillen D, Singh PB, Nakayama J, Grewal SI, Allis CD, Cheng X, Selker EU. 2003. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat. Genet. 34, 75–79 10.1038/ng1143 (doi:10.1038/ng1143) [DOI] [PubMed] [Google Scholar]
- 68.Zhao Q, et al. 2009. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 16, 304–311 10.1038/nsmb.1568 (doi:10.1038/nsmb.1568) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jelinic P, Stehle JC, Shaw P. 2006. The testis-specific factor CTCFL cooperates with the protein methyltransferase PRMT7 in H19 imprinting control region methylation. PLoS Biol. 4, e355. 10.1371/journal.pbio.0040355 (doi:10.1371/journal.pbio.0040355) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Illingworth RS, et al. 2010. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 6, e1001134. 10.1371/journal.pgen.1001134 (doi:10.1371/journal.pgen.1001134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maunakea AK, et al. 2010. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257 10.1038/nature09165 (doi:10.1038/nature09165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kanber D, Berulava T, Ammerpohl O, Mitter D, Richter J, Siebert R, Horsthemke B, Lohmann D, Buiting K. 2009. The human retinoblastoma gene is imprinted. PLoS Genet. 5, e1000790. 10.1371/journal.pgen.1000790 (doi:10.1371/journal.pgen.1000790) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Horsthemke B, Wagstaff J. 2008. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A. 146A, 2041–2052 10.1002/ajmg.a.32364 (doi:10.1002/ajmg.a.32364) [DOI] [PubMed] [Google Scholar]
- 74.Dittrich B, et al. 1996. Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat. Genet. 14, 163–170 10.1038/ng1096-163 (doi:10.1038/ng1096-163) [DOI] [PubMed] [Google Scholar]
- 75.Mapendano CK, Kishino T, Miyazaki K, Kondo S, Yoshiura K, Hishikawa Y, Koji T, Niikawa N, Ohta T. 2006. Expression of the Snurf-Snrpn IC transcript in the oocyte and its putative role in the imprinting establishment of the mouse 7C imprinting domain. J. Hum. Genet. 51, 236–243 10.1007/s10038-005-0351-8 (doi:10.1007/s10038-005-0351-8) [DOI] [PubMed] [Google Scholar]
- 76.Bastepe M, Fröhlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, Jüppner H. 2005. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat. Genet. 37, 25–27 10.1038/ng1560 (doi:10.1038/ng1560) [DOI] [PubMed] [Google Scholar]
- 77.Bastepe M, et al. 2003. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J. Clin. Invest. 112, 1255–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fröhlich LF, Mrakovcic M, Steinborn R, Chung UI, Bastepe M, Jüppner H. 2010. Targeted deletion of the Nesp55 DMR defines another Gnas imprinting control region and provides a mouse model of autosomal dominant PHP-Ib. Proc. Natl Acad. Sci. USA 107, 9275–9280 10.1073/pnas.0910224107 (doi:10.1073/pnas.0910224107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Williamson CM, et al. 2011. Uncoupling antisense-mediated silencing and DNA methylation in the imprinted Gnas cluster. PLoS Genet. 7, e1001347. 10.1371/journal.pgen.1001347 (doi:10.1371/journal.pgen.1001347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith EY, Futtner CR, Chamberlain SJ, Johnstone KA, Resnick JL. 2011. Transcription is required to establish maternal imprinting at the Prader–Willi syndrome and Angelman syndrome locus. PLoS Genet. 7, e1002422. 10.1371/journal.pgen.1002422 (doi:10.1371/journal.pgen.1002422) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smallwood SA, Kelsey G. 2012. De novo DNA methylation: a germ cell perspective. Trends Genet. 28, 33–42 10.1016/j.tig.2011.09.004 (doi:10.1016/j.tig.2011.09.004) [DOI] [PubMed] [Google Scholar]
- 82.O'Doherty AM, Rutledge CE, Sato S, Thakur A, Lees-Murdock DJ, Hata K, Walsh CP. 2011. DNA methylation plays an important role in promoter choice and protein production at the mouse Dnmt3L locus. Dev. Biol. 356, 411–420 10.1016/j.ydbio.2011.05.665 (doi:10.1016/j.ydbio.2011.05.665) [DOI] [PubMed] [Google Scholar]
- 83.Kaneda M, Hirasawa R, Chiba H, Okano M, Li E, Sasaki H. 2010. Genetic evidence for Dnmt3a-dependent imprinting during oocyte growth obtained by conditional knockout with Zp3-Cre and complete exclusion of Dnmt3b by chimera formation. Genes Cells 15, 169–179 10.1111/j.1365-2443.2009.01374.x (doi:10.1111/j.1365-2443.2009.01374.x) [DOI] [PubMed] [Google Scholar]
- 84.Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. 2010. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 285, 26114–26120 10.1074/jbc.M109.089433 (doi:10.1074/jbc.M109.089433) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wagner EJ, Carpenter PB. 2012. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell. Biol. 13, 115–126 10.1038/nrm3274 (doi:10.1038/nrm3274) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yoh SM, Lucas JS, Jones KA. 2008. The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 22, 3422–3434 10.1101/gad.1720008 (doi:10.1101/gad.1720008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xie L, Pelz C, Wang W, Bashar A, Varlamova O, Shadle S, Impey S. 2011. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 30, 1473–1484 10.1038/emboj.2011.91 (doi:10.1038/emboj.2011.91) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fang R, et al. 2010. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 39, 222–233 10.1016/j.molcel.2010.07.008 (doi:10.1016/j.molcel.2010.07.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ. 2010. CpG islands recruit a histone H3 lysine 36 demethylase. Mol. Cell 38, 179–190 10.1016/j.molcel.2010.04.009 (doi:10.1016/j.molcel.2010.04.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoon BJ, Herman H, Sikora A, Smith LT, Plass C, Soloway PD. 2002. Regulation of DNA methylation of Rasgrf1. Nat. Genet. 30, 92–96 10.1038/ng795 (doi:10.1038/ng795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aravin AA, Bourc'his D. 2008. Small RNA guides for de novo DNA methylation in mammalian germ cells. Genes Dev. 22, 970–975 10.1101/gad.1669408 (doi:10.1101/gad.1669408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Augui S, Nora EP, Heard E. 2011. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet. 12, 429–442 10.1038/nrg2987 (doi:10.1038/nrg2987) [DOI] [PubMed] [Google Scholar]
- 93.Mak W, Nesterova TB, de Napoles M, Appanah R, Yamanaka S, Otte AP, Brockdorff N. 2004. Reactivation of the paternal X chromosome in early mouse embryos. Science 303, 666–669 10.1126/science.1092674 (doi:10.1126/science.1092674) [DOI] [PubMed] [Google Scholar]
- 94.Patrat C, Okamoto I, Diabangouaya P, Vialon V, Le Baccon P, Chow J, Heard E. 2009. Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proc. Natl Acad. Sci. USA 106, 5198–5203 10.1073/pnas.0810683106 (doi:10.1073/pnas.0810683106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goto Y, Takagi N. 2000. Maternally inherited X chromosome is not inactivated in mouse blastocysts due to parental imprinting. Chromosome Res. 8, 101–109 10.1023/A:1009234217981 (doi:10.1023/A:1009234217981) [DOI] [PubMed] [Google Scholar]
- 96.Chiba H, Hirasawa R, Kaneda M, Amakawa Y, Li E, Sado T, Sasaki H. 2008. De novo DNA methylation independent establishment of maternal imprint on X chromosome in mouse oocytes. Genesis 46, 768–774 10.1002/dvg.20438 (doi:10.1002/dvg.20438) [DOI] [PubMed] [Google Scholar]
- 97.Okae H, et al. 2012. Re-investigation and RNA sequencing-based identification of genes with placenta-specific imprinted expression. Hum. Mol. Genet. 21, 548–558 10.1093/hmg/ddr488 (doi:10.1093/hmg/ddr488) [DOI] [PubMed] [Google Scholar]
- 98.Wang Q, et al. 2011. Recent acquisition of imprinting at the rodent Sfmbt2 locus correlates with insertion of a large block of miRNAs. BMC Genomics 12, 204. 10.1186/1471-2164-12-204 (doi:10.1186/1471-2164-12-204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arnaud P, Hata K, Kaneda M, Li E, Sasaki H, Feil R, Kelsey G. 2006. Stochastic imprinting in the progeny of Dnmt3L–/– females. Hum. Mol. Genet. 15, 589–598 10.1093/hmg/ddi475 (doi:10.1093/hmg/ddi475) [DOI] [PubMed] [Google Scholar]
- 100.Henckel A, Nakabayashi K, Sanz LA, Feil R, Hata K, Arnaud P. 2009. Histone methylation is mechanistically linked to DNA methylation at imprinting control regions in mammals. Hum. Mol. Genet. 18, 3375–3383 10.1093/hmg/ddp277 (doi:10.1093/hmg/ddp277) [DOI] [PubMed] [Google Scholar]
- 101.Tanimoto K, Shimotsuma M, Matsuzaki H, Omori A, Bungert J, Engel JD, Fukamizu A. 2005. Genomic imprinting recapitulated in the human beta-globin locus. Proc. Natl Acad. Sci. USA 102, 10 250–10 255 10.1073/pnas.0409541102 (doi:10.1073/pnas.0409541102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsuzaki H, Okamura E, Shimotsuma M, Fukamizu A, Tanimoto K. 2009. A randomly integrated transgenic H19 imprinting control region acquires methylation imprinting independently of its establishment in germ cells. Mol. Cell. Biol. 29, 4595–4603 10.1128/MCB.00275-09 (doi:10.1128/MCB.00275-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shoji M, et al. 2009. The TDRD9-MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev. Cell 17, 775–787 10.1016/j.devcel.2009.10.012 (doi:10.1016/j.devcel.2009.10.012) [DOI] [PubMed] [Google Scholar]
- 104.Murdoch S, et al. 2006. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat. Genet. 38, 300–302 10.1038/ng1740 (doi:10.1038/ng1740) [DOI] [PubMed] [Google Scholar]
- 105.Meyer E, Lim D, Pasha S, Tee LJ, Rahman F, Yates JR, Woods CG, Reik W, Maher ER. 2009. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith–Wiedemann Syndrome). PLoS Genet. 5, e1000423. 10.1371/journal.pgen.1000423 (doi:10.1371/journal.pgen.1000423) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Parry DA, et al. 2011. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am. J. Hum. Genet. 89, 451–458 10.1016/j.ajhg.2011.08.002 (doi:10.1016/j.ajhg.2011.08.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nakamura T, et al. 2007. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat. Cell Biol. 9, 64–71 10.1038/ncb1519 (doi:10.1038/ncb1519) [DOI] [PubMed] [Google Scholar]
- 108.Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB. 2012. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 335, 1499–1502 10.1126/science.1216154 (doi:10.1126/science.1216154) [DOI] [PubMed] [Google Scholar]
- 109.Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, Sasaki H. 2008. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 22, 1607–1616 10.1101/gad.1667008 (doi:10.1101/gad.1667008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, Chaillet JR. 2001. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 104, 829–838 10.1016/S0092-8674(01)00280-X (doi:10.1016/S0092-8674(01)00280-X) [DOI] [PubMed] [Google Scholar]
- 111.Sharif J, et al. 2007. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912 10.1038/nature06397 (doi:10.1038/nature06397) [DOI] [PubMed] [Google Scholar]
- 112.Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, Stein A, Woodcock CL, Skoultchi AI. 2005. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212 10.1016/j.cell.2005.10.028 (doi:10.1016/j.cell.2005.10.028) [DOI] [PubMed] [Google Scholar]
- 113.Reese KJ, Lin S, Verona RI, Schultz RM, Bartolomei MS. 2007. Maintenance of paternal methylation and repression of the imprinted H19 gene requires MBD3. PLoS Genet. 3, e137. 10.1371/journal.pgen.0030137 (doi:10.1371/journal.pgen.0030137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ma P, Lin S, Bartolomei MS, Schultz RM. 2010. Metastasis tumor antigen 2 (MTA2) is involved in proper imprinted expression of H19 and Peg3 during mouse preimplantation development. Biol. Reprod. 83, 1027–1035 10.1095/biolreprod.110.086397 (doi:10.1095/biolreprod.110.086397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu MY, Tsai TF, Beaudet AL. 2006. Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the PWS/AS domain. Genes Dev. 20, 2859–2870 10.1101/gad.1452206 (doi:10.1101/gad.1452206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schoenherr CJ, Levorse JM, Tilghman SM. 2003. CTCF maintains differential methylation at the Igf2/H19 locus. Nat. Genet. 33, 66–69 10.1038/ng1057 (doi:10.1038/ng1057) [DOI] [PubMed] [Google Scholar]
- 117.Kim JD, Kim H, Ekram MB, Yu S, Faulk C, Kim J. 2011. Rex1/Zfp42 as an epigenetic regulator for genomic imprinting. Hum. Mol. Genet. 20, 1353–1362 10.1093/hmg/ddr017 (doi:10.1093/hmg/ddr017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, Ren B. 2012. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148, 816–831 10.1016/j.cell.2011.12.035 (doi:10.1016/j.cell.2011.12.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reik W. 2007. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432 10.1038/nature05918 (doi:10.1038/nature05918) [DOI] [PubMed] [Google Scholar]
- 120.Liu YJ, Nakamura T, Nakano T. 2011. Essential role of DPPA3 for chromatin condensation in mouse oocytogenesis. Biol. Reprod. 86, 40. 10.1095/biolreprod.111.095018 (doi:10.1095/biolreprod.111.095018) [DOI] [PubMed] [Google Scholar]
- 121.Wossidlo M, et al. 2011. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2, 241. 10.1038/ncomms1240 (doi:10.1038/ncomms1240) [DOI] [PubMed] [Google Scholar]
- 122.Yuan P, et al. 2009. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev. 23, 2507–2520 10.1101/gad.1831909 (doi:10.1101/gad.1831909) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Engel N, Thorvaldsen JL, Bartolomei MS. 2006. CTCF binding sites promote transcription initiation and prevent DNA methylation on the maternal allele at the imprinted H19/Igf2 locus. Hum. Mol. Genet. 15, 2945–2954 10.1093/hmg/ddl237 (doi:10.1093/hmg/ddl237) [DOI] [PubMed] [Google Scholar]
- 124.Stadler MB, et al. 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495 10.1038/nature10716 (doi:10.1038/nature10716) [DOI] [PubMed] [Google Scholar]
- 125.Branco MR, Ficz G, Reik W. 2011. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 13, 7–13 10.1038/nrn3125 (doi:10.1038/nrn3125) [DOI] [PubMed] [Google Scholar]
- 126.Tahiliani M, et al. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 10.1126/science.1170116 (doi:10.1126/science.1170116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gu TP, et al. 2011. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610 10.1038/nature10443 (doi:10.1038/nature10443) [DOI] [PubMed] [Google Scholar]
- 128.Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. 2010. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329, 643–648 10.1126/science.1190830 (doi:10.1126/science.1190830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.DeVeale B, van der Kooy D, Babak T. 2012. Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet. 8, e1002600. 10.1371/journal.pgen.1002600 (doi:10.1371/journal.pgen.1002600) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fauque P, Jouannet P, Lesaffre C, Ripoche MA, Dandolo L, Vaiman D, Jammes H. 2007. Assisted Reproductive Technology affects developmental kinetics, H19 Imprinting Control Region methylation and H19 gene expression in individual mouse embryos. BMC Dev. Biol. 7, 116. 10.1186/1471-213X-7-116 (doi:10.1186/1471-213X-7-116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Market-Velker BA, Zhang L, Magri LS, Bonvissuto AC, Mann MR. 2010. Dual effects of superovulation: loss of maternal and paternal imprinted methylation in a dose-dependent manner. Hum. Mol. Genet. 19, 36–51 10.1093/hmg/ddp465 (doi:10.1093/hmg/ddp465) [DOI] [PubMed] [Google Scholar]
- 132.Kim JM, Ogura A. 2009. Changes in allele-specific association of histone modifications at the imprinting control regions during mouse preimplantation development. Genesis 47, 611–616 10.1002/dvg.20541 (doi:10.1002/dvg.20541) [DOI] [PubMed] [Google Scholar]
- 133.Choo JH, Kim JD, Chung JH, Stubbs L, Kim J. 2006. Allele-specific deposition of macroH2A1 in imprinting control regions. Hum. Mol. Genet. 15, 717–724 10.1093/hmg/ddi485 (doi:10.1093/hmg/ddi485) [DOI] [PubMed] [Google Scholar]
- 134.Mann MR, Lee SS, Doherty AS, Verona RI, Nolen LD, Schultz RM, Bartolomei MS. 2004. Selective loss of imprinting in the placenta following preimplantation development in culture. Development 131, 3727–3735 10.1242/dev.01241 (doi:10.1242/dev.01241) [DOI] [PubMed] [Google Scholar]
- 135.Moore T, Haig D. 1991. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 7, 45–49 [DOI] [PubMed] [Google Scholar]