

Abstract
The pathophysiology of obesity is extremely complex and is associated with extensive gene expression changes in tissues throughout the body. This situation, combined with the fact that all gene expression changes are thought to have associated epigenetic changes, means that the links between obesity and epigenetics will undoubtedly be vast. Much progress in identifying epigenetic changes induced by (or inducing) obesity has already been made, with candidate and genome-wide approaches. These discoveries will aid the clinician through increasing our understanding of the inheritance, development and treatment of obesity. However, they are also of great value for epigenetic researchers, as they have revealed mechanisms of environmental interactions with epigenetics that can produce or perpetuate a disease state. Here, we will review the evidence for four mechanisms through which epigenetics contributes to obesity: as downstream effectors of environmental signals; through abnormal global epigenetic state driving obesogenic expression patterns; through facilitating developmental programming and through transgenerational epigenetic inheritance.
Keywords: obesity, epigenetics, transgenerational epigenetic inheritance, developmental programming
1. Introduction to obesity
Obesity is one of the world's greatest public health challenges, contributing to morbidity and mortality through the increased risk for many chronic diseases, including type 2 diabetes, hypertension, dyslipidemia, coronary artery disease, stroke, osteoarthritis and certain forms of cancer. Obesity is a multifactorial disorder, with key genetic and environmental drivers. Genome-wide association studies have identified more than 50 loci associated with body mass index, although the effect sizes are small [1]. Among the environmental factors, changes in lifestyle, including the increased availability of palatable, energy dense foods, a reduced need for physical activity and a reduction in sleep are thought to contribute [2,3]. Food is also rewarding, in much the same way as drugs of abuse; drug addiction and obesity share neurobiological mechanisms involving dopamine (DA) [4,5].
Predictions of the World Health Organization suggest that by 2015, 75 per cent of the adult population will be overweight, and 41 per cent obese [6]. Thus, the relatively recent rise in obesity appears to be related to gene–environment interactions where our genetic background, coupled with the current obesogenic environment, and the rewarding nature of palatable foods, tends to promote obesity [7]. The other recent changes in the demographic profile in most of the developed world are increased overweight and obesity in pregnant women, which is now considered to be an ‘endemic’ challenge for obstetric care [8]. Maternal obesity is associated with childhood obesity, now considered as epidemic in some areas and on the rise in others [9]. An estimated 22 million children under five are overweight worldwide. The number of overweight children in the USA has doubled, and the number of overweight adolescents has trebled since 1980 [10].
Unravelling the contributors to obesity is complex, given the multiple levels of gut–brain interaction underpinning appetite and feeding regulation, and the redundancy in neurotransmitter systems controlling food intake. In humans, higher centres can override physiological signals, leading to overconsumption. Overall, the rise in obesity suggests an inadequacy of mechanisms regulating body weight to cope with environments that promote overconsumption of energy, and discourage physical activity. While much progress has been made in identifying risk loci for increased body mass index/adiposity, often from a young age, recent data show that epigenetic modification of genes by environmental factors may contribute to the development of obesity, and this issue forms the basis of this review.
2. Introduction to epigenetics
Epigenetics is defined as the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence [11]. Heritability is what makes epigenetics special, as its role in phenotype determination lies in-between DNA sequence that is usually entirely inherited and the more transient activities of transcription factors. Because of this positioning between DNA and transcription factors, epigenetic mechanisms often function to perpetuate a phenotype over several cell divisions, for example in maintaining a cell in a differentiated state (e.g. making the daughter cells of a muscle cell also muscle cells) [12,13]. However, an equally important feature of epigenetic modifications is their ability to be reversed. Owing to this ability, epigenetic mechanisms are used to control processes that are advantageous for the organism when they are stable for several cell divisions but not indefinitely. A classic example of this is inactivation of the X-chromosome in female mammals [14] (box 1). Females silence one copy of the X-chromosome to have equivalent levels of gene products as males, who only have one X. The choice of which X chromosome is silenced in a cell happens during gestation and that choice is maintained for a lifetime, through millions of cell divisions. However, the silenced X-chromosome has to be reactivated between generations so that a female can produce a healthy male child. This reactivation occurs in the cells that are the precursors of the gametes in a process of genome-wide transgenerational epigenetic reprogramming (see below) [13,15]. Other examples of epigenetic regulation in mammals that are part of normal development are described in box 1. Importantly, the reversal of epigenetic processes that are part of normal development or genome function is tightly regulated, occurring only at defined time-points in the life cycle of a mammal. Epigenetic modifications that are directed by the environment are more plastic than those described earlier, yet the degree of plasticity is yet to be determined [11,16]. This is an important question for obesity research, as it is unclear how completely the epigenetic modifications set by a high-fat diet, for example, can be reversed through a reduced calorie diet, or exercise. While some genes that are regulated by normal developmental epigenetic processes have been implicated in obesity (such as those that undergo genomic imprinting), environmental–epigenetic interactions mostly involve other genes.
Box 1. Epigenetic functions in mammals.
Cell differentiation: multiple cell types can be produced from one genome through the expression of different groups of genes that regulate cell function and phenotype. Epigenetic modification of genes helps to coordinate the transition of one cell-type to another through the developmental process of differentiation. Epigenetic modifications also ‘lock-down’ a terminally differentiated cell to ensure that all of its daughter cells are the same cell-type. In particular, developmental gene promoters are regulated by DNA methylation, histone acetylation, and H3K4 and H3K27 methylation.
Dosage compensation: to ensure gene dosage equivalence between male and female mammals, one X-chromosome is inactivated in all somatic cells in females. The silencing is initiated in the early post-implantation embryo when one chromosome is randomly chosen to be inactivated. The inactive chromosome is epigenetically marked with DNA methylation at gene promoters, transcriptionally repressive histone modifications and the silencing requires the ncRNAs Xist and Tsix. High levels of H3K27 trimethylation are present on the inactive X-chromosome.
Genome structure maintenance: telomeres and centromeres have unique epigenetic structures that facilitate chromosome integrity and organization of replication and recombination. Epigenetic modifications also facilitate DNA damage repair.
Genomic/parental imprinting: this is a process whereby the two chromosomal homologues are differently epigenetically marked (imprinted) depending on which parent they are inherited from. This marking leads to genes being differently expressed between the two parental homologues. The imprints are set in the germ line, propagated from the gametes to the adult organism and then erased in primordial germ cells (to be sex-dependently reset in the maturing germ cells to complete the cycle). The molecular regulation of imprinted gene expression varies between imprinted regions but commonly involves DNA methylation, H3K4, H3K27 and H3K9 methylation.
Repetitive element repression: repetitive elements such as retrotransposons, transposons, short interspersed nucleotide elements (SINEs), tandem repeats and microsatellites make up more than half of the genome. They are normally covered with epigenetic modifications that results in a compact DNA/protein structure termed heterochromatin. This compact structure prevents transcription of mobile repetitive elements or unequal recombination between sequences with similar DNA sequence that are located in different parts of the genome. Repression of repetitive elements is achieved through DNA methylation and histone modifications that depend on the class of repeat and the cell-type. For example, in mouse ES cells, intracisternal A particle (IAP) retrotransposons bear high levels of H4K20 trimethylation, whereas DNA transposons and pericentric satellite repeats have high levels of H3K9 trimethylation.
(a). Types of epigenetic modification
At the molecular level, there are three main types of epigenetic modification in mammals, DNA methylation, histone modifications and non-coding RNA (ncRNA; table 1). DNA methylation [12] is the covalent attachment of a methyl group to the base cytosine. The methyl group protrudes into the major groove of the double helix and can inhibit the binding of transcription factors. Therefore, DNA methylation is usually associated with repression of transcription at a locus. DNA methylation in mammals occurs predominantly at CpG dinucleotides (where p indicates the phosphate group of DNA that bridges the nucleotides in the strand). The DNA sequence that base pairs with CpG is also CpG, and this palindromic situation is used for the inheritance of DNA methylation between cellular divisions. During DNA synthesis, maintenance DNA methyltransferase enzymes ‘read’ the methylated CpG on the template strand and then add a methyl group to the paired CpG on the nascent daughter strand. Because of this mechanism, DNA methylation is considered to be a relatively stable epigenetic modification with the ability to be faithfully replicated for an organism's lifetime, if required. Recent discoveries, however, have revealed situations where DNA methylation is more dynamic with replication-independent methylation and demethylation occurring [17]. As yet, it is unclear whether transient DNA methylation is a common feature of gene–environment interactions, including those related to obesity.
Table 1.
Introduction to epigenetic modifications in mammals.
| epigenetic modification | modification enzymes | effect on genome function |
|---|---|---|
| CpG DNA methylation | DNA methyltransferases (DNMT) 1, 3a and 3b. Base excision/DNA repair proteins or simply failure to remethylate at DNA replication are thought to facilitate demethylation | presence at promoters associated with gene silencing. Presence in gene bodies associated with gene activity |
| non-CpG DNA methylation | some evidence points to the known DNA methyltransferases | currently unclear, though its presence at genes is usually associated with gene activity |
| histone acetylation | several histone acetyl transferases (HATs) and histone deacetylases (HDACs) | increases protein access to DNA for transcription or genome-wide reprogramming e.g. sperm protamination |
| histone methylation | several modification-specific histone methyl transferases and histone demethylases | methylation of some amino acid residues associated with transcriptional repression, while others with activation |
| histone variants e.g. H2A.Z, CENP-A, H2AX | various specialized functions including centromere function, DNA repair and gene regulation | |
| small non-coding RNAs, e.g. miRNAs, piRNAs | biogenesis and function requires enzymes such as RNA polymerase II, DICER and ARGONAUTE | various effects such as transcriptional repression and activation, translational repression |
| long non-coding RNAs | biogenesis of most found so far involves RNA polymerase II | known to regulate large-scale transcriptional repression in genomic imprinting. Recent discovery of abundance throughout the genome suggests a high variety of functions |
Histones are the components of the protein complexes (nucleosomes) around which DNA is wound. The termini of histones extend out from the core of the protein like tails and can be modified by the addition of a variety of chemical groups such as methylation, acetylation, phosphorylation and sumoylation [18]. These different modifications can have activating or repressive effects on local transcription. For example, methylation of lysine 9 on histone H3 (H3K9met) is associated with transcriptional repression, whereas H3K4met is associated with transcriptional activation. In addition, a lysine can have multiple (typically up to three) methyl groups attached, each with potentially different effects on regional function. Acetylation is considered to be an activating epigenetic mark as the acetyl group facilitates ‘spacing out’ of nucleosomes, thereby allowing easier access to transcription factors. This expansion or contraction of protein–DNA complexes is at the heart of epigenetic regulation and extends beyond nucleosomes to higher-order chromatin structures—for example, heterochromatin proteins attach to repressive histone modifications, further constricting the chromatin, hiding the DNA from and creating a barrier to transcriptionally activating proteins.
A huge variety of ncRNA are transcribed from the genome, and several classes have been shown to perpetuate a phenotype in an epigenetic manner [19]. In particular, they can recruit transcriptional activating or repressing complexes site-specifically through base-complementation. This highlights the multi-level nature of epigenetic regulation as ncRNA, DNA methylation enzymes, histone modifiers and larger-scale chromatin remodellers usually coexist in large protein complexes where one epigenetic modification stimulates or represses another type. This crosstalk between different types of modification can reprogramme, or synergistically repress or activate a locus. ncRNA is an exciting new area of epigenetics [19,20] and its importance for adipogenesis has been shown [21]. However, the study of its possible involvement in obesity is relatively new; so here we will focus on the larger body of work from the longer-established areas of DNA methylation and histone modifications in obesity.
(b). Global epigenetic reprogramming: windows of environmental sensitivity
The mammalian life cycle has two major epigenetic reprogramming events. These events are needed to erase epigenetic errors (epimutations) that may have arisen through the lifetime of the parent and to provide the correct epigenetic patterns throughout the genome to ensure the proper initiation of embryonic gene expression [13,15].
The first genome-wide reprogramming event occurs in the cells which will go on to make the gametes, the primordial germ cells (PGCs). In mice, these cells start to be reprogrammed in the developing embryo at about mid-way through gestation. In females and males the timing of events differs slightly, reflecting the different end products of the cell differentiation pathway, oocytes and sperm. However, PGCs in both germ lines undergo extensive genome-wide (also known as ‘global’) DNA demethylation. Normally stable DNA methylation on the silent X-chromosome and genomic imprints is erased. However, some classes of retrotransposon and subtelomeric regions retain methylation [22–24]. The former is presumably due to retrotransposon transcriptional activation, through the loss of DNA-methylation-mediated repression, being extremely undesirable for the genome. Retrotransposons insert into new loci, and recombination occurs between unmethylated repeats from different areas of the genome. The subtelomeric resistance to demethylation may be due to its importance for maintaining chromosome end stability [23]. DNA methylation levels return to those of somatic cells a few days after the demethylation began in males, but not until after birth in females [25]. Genomic imprints at these points are set according to the sex of the individual.
A rapid reprogramming of histone modifications throughout the genome also occurs in PGCs [26]. The histone reprogramming is more transient than the DNA methylation erasure probably as a result of the requirement of histone modifications for coordinating the gene expression programmes of germ cell development. Another genome-wide histone reprogramming event occurs in adult males when nearly all histones are replaced with protamines that can be more tightly packed, thus enabling the DNA to be condensed in the sperm [27].
The second genome-wide reprogramming timepoint occurs immediately after fertilization and continues until the blastocyst stage. The sperm, upon entry into the oocyte, is stripped of protamines, which are replaced with histones from maternal stores, and some repetitive elements in the sperm DNA are actively demethylated [28]. The chromosomes in the oocyte are less methylated, yet during development to the blastocyst, a further reduction in methylation occurs on both sets of chromosomes, presumably through exclusion of DNA methylation enzymes from the nucleus. The demethylation process in the preimplantation embryo is less extreme than in the germ line, as some types of retrotransposon and genomic imprints are not demethylated [29], and a recent study has also detected significant levels of global methylation throughout the preimplantation period [30]. The reprogramming events involving histone modifications are more complex than DNA methylation. It appears that apart from the extensive deprotamination and histone-replacement across the sperm DNA, most chromatin changes reflect gene regulatory changes that coordinate this highly dynamic period of development [31].
These two reprogramming events are the times of the life cycle that have the most extensive changes to epigenetic state. The epigenetic state that is set at the end of either of the events, at some loci, persists for the lifetime of that individual. Therefore, any epigenetic modifications that are triggered by unusual environmental conditions, or are just plain errors, can affect long-term phenotype. Furthermore, unlike epigenetic changes in adult tissues, a change in epigenetic modifications in the gamete or early embryo can affect the whole body, as all organs stem from these initial cells [32]. For these reasons, researchers that are trying to explain the mechanisms of inheritance and individual risk of developing diseases such as obesity are interested in the genome-wide reprogramming events.
(c). Cautionary points for those investigating epigenetics in disease
Firstly, cause and effect of epigenetic modifications are notoriously hard to distinguish [33]. Researchers should always keep in mind that the modification may not actually determine the expression at a locus; it may be the other way around. Secondly, the importance of DNA sequence in determining epigenetic state at a locus should not be forgotten. Almost always, the presence or absence of an epigenetic modification is dependent on an underlying (or even distal) DNA sequence [34,35]. Finally, an epigenetic change that is detected in a tissue may sometimes not be due to the reprogramming of the locus, but rather could reflect the change in the relative proportion of cell-types that have differing epigenetic states [36].
(d). Environment–epigenetic interactions
The recent expansion in examples of epigenetic modifications being set by environmental triggers has been exciting for epigenetics researchers [11,16]. This is because previously most work in the field focused on epigenetic changes that happened to all individuals as part of normal development, for example in Hox clusters and genomic/parental imprinting. The other large area of epigenetic study at the time, in mammals was disease, predominantly cancer. Environmentally induced epigenetic change was new in that it linked the epigenetic mechanisms to reactive systems that were not always used. This non-obligatory use of epigenetics also provided a new mechanism for the development of organismal individuality. Previously, the only involvement of epigenetics in phenotype variation was due to the stochastic nature of some epigenetic processes [16].
Epigenetic research into obesity has illuminated new molecular mechanisms. These mechanisms facilitate the environmental triggering of epigenetic state, molecular aetiology of disease and disease inheritance. Here, we highlight four aspects of obesity that have progressed our understanding of epigenetic mechanisms.
3. Epigenetics as a downstream effector of environmental signals
At the heart of cellular interactions with the environment, and the coordination of different cell types in metazoa is the cell surface receptor. They facilitate the uptake of nutrients, communication between connected and distally located cells and ultimately, through signalling cascades, direct an appropriate gene expression change to modify the cell's function. The gene expression changes are usually associated with epigenetic modifications at single or multiple loci. All tissues in the body are at least indirectly affected by obesity through this general mechanism as all import nutrients such as glucose and lipids, and an increase in circulating levels of these, especially during development, alters cell state. However, obesogenic processes such as appetite and reward pathways in the brain, adipogenesis and fat deposition, inflammation, insulin signalling and glucose and lipid metabolism have unique as well as overlapping cell receptors [37–39]. For some of these processes, the route from external stimulus, through receptor and signalling pathways to epigenetic changes has been described (see figure 1). Here, we will give two examples of how this general mechanism can drive obesity.
Figure 1.
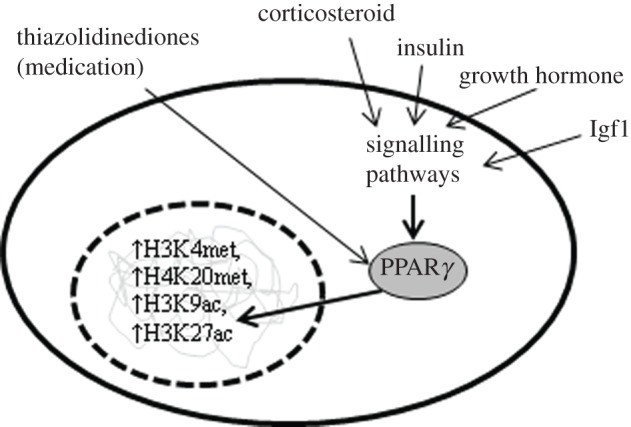
Multiple extracellular signals stimulate adipogenesis through activation of PPARγ, which translocates to the nucleus and directs epigenetic remodelling of target genes.
(a). Adipogenesis
The cellular differentiation process whereby preadipocytes become adipocytes (adipogenesis) is a major obesogenic process. The mechanisms of this process have been extensively researched, partly due to its importance in human health and partly due to the availability of cell culture models [40]. The pathways that facilitate or inhibit adipogenesis have both been shown to be epigenetically regulated (figure 1). In vivo, there are many external signals that bind cell surface or cytosolic receptors to promote or inhibit adipogenesis, including hormones, fatty acids, growth factors, interferons [40] and the peroxisome proliferator-activated receptor gamma (PPARγ) activating drugs thiazolidinediones [41]. The differentiation process has multiple stages with a cascade of sequentially expressed adipogenic transcription factors interacting with each other and extracellular signals [42]. Two key transcription factors are PPARγ and Ccaat-enhancer-binding proteins alpha (C/EBPα), which act together to activate hundreds of genes to produce a mature adipocyte [43,44]. The H3, lysine 4 methyltransferases (H3K4MTs) mixed-lineage leukaemia (MLL) proteins MLL3 and MLL4, and their respective complexes have been shown in several studies to be critical for adipogenesis through their activation of PPARγ and C/EBPα [45–47]. Furthermore, PPARγ has been shown to then increase transcription of the histone H4 Lysine 20 (H4K20) monomethyltransferase PR-Set7/Setd8, which then, in a feedback loop, activates PPARγ and the many targets of PPARγ to promote adipogenesis [48]. DNA methylation too appears to be part of this mechanism, as the PPARγ promoter is demethylated during adipogenesis, and adipose tissue from obese mice was found to have less methylation than normal-sized mice [49]. The repression of adipogenesis is facilitated by transcription factors of the Wnt families and β-catenin, which are themselves silenced in preadipocytes through the repressive histone mark H3K27 methylation [50]. Deletion of the histone methyltransferase that is responsible for this mark, Ezh2, derepresses Wnt genes, thus activating the Wnt/β-catenin signalling, which in turn inhibits adipogenesis by preventing the transcription of PPARγ and C/EBPα [51].
(b). Neural reward pathways
The appetite and reward pathways in the brain are important for the development of obesity through their regulation of food intake. For example, the neurotransmitter dopamine (DA) is released upon feeding to induce consummatory pleasure [52]. Chronic high-fat diet reduces the dopaminergic pathways in the brain, which means that an individual needs more stimulation (in the form of feeding) in order to receive a sufficient feeling of reward, a situation that can induce obesity [53]. The upstream signal for the dopaminergic pathway reduction is not yet known though evidence points to the hormone leptin [54,55]. However, more is known about the downstream epigenetic effects of DA. Vucetic et al. [53] showed that the decreased reward circuitry in rats fed with a high-fat diet after weaning involved DNA-methylation-associated repression at the promoters of tyrosine hydroxylase (TH), the rate-limiting enzyme in DA synthesis, and the dopamine transporter (DAT) in the area of the brain that governs hedonically driven feeding [53]. In another paper, using the same experimental model, the same group described high-fat-diet-induced changes in the expression of another gene with importance in the reward pathways, the transmembrane opioid receptor (μOR). In this work, they not only correlated increased DNA methylation at the gene promoter with decreased expression, but also identified other repressive epigenetic features, namely increased H3K9 methylation, decreased H3 acetylation and methyl CpG binding protein 2 (MeCP2), a transcriptional-repressor binding [56]. The epigenetic complexes that target TH, DAT and μOR have not been characterized, but considering the variety of epigenetic change at the μOR promoter, multiple reprogramming enzymes will be involved.
In conclusion, changes to epigenetic state at the promoters of genes is now accepted as a feature of obesity owing to the extensive examples obtained from research in rodents and humans cell culture models (reviewed in [11,37–39], and new experimental data [57–63]). As the activity of many of those genes is known to be influenced by external signals, the mechanism of epigenetic changes at disease genes being caused by upstream signalling pathways is likely to be important for obesity.
4. Could abnormal global epigenetic state drive obesogenic expression patterns?
In contrast to the established ‘top down’ mechanisms of a signal from outside the cell stimulating a disease epigenetic state, we propose here the possibility for a ‘bottom-up’ mechanism (figure 2). In this currently unproven model, an individual has a genotype or dietary deficiency that either alters or ablates the function of a gene that is involved in epigenetic reprogramming. The faulty function, expression or absence of the modifier initiates a gene expression program in one or several cell-types that ultimately induces obesity. As elaborated below, there is some evidence for this mechanism from mouse mutants, human disease and nutritional studies.
Figure 2.

Two hypothetical mechanisms of genome-wide epigenetic change that could increase the risk of obesity. (a) normally an EM is transcribed, translated and the protein enters the nucleus to regulate genomic epigenetic state (black dots). (b) mutation (or loss) of an EM causes abnormal regulation of one or many genes. (c) in another scenario, normal levels of methyl-donors (grey triangle) ensure appropriate epigenetic modification of the genome. (d) if the availability of methyl-donors is reduced, abnormal epigenetic modification of the genome prevents normal gene regulation.
(a). Mouse models of epigenetic-modifier-driven obesity
No mice with genetically increased or decreased levels of DNA methyltransferases have been reported to be obese. The de novo methyltransferase Dnmt3a (but not Dnmt1 or Dnmt3b) was found to be more than doubled in white adipose tissue of obese mice [64]. However, importantly, in the same study, a transgenic mouse that had threefold elevated Dnmt3a mRNA levels in adipose tissue did not manifest weight gain or increased adiposity. These data argue against DNA methyltransferase-driven obesity.
More potential for this mechanism of epigenetically driven obesity may lie with other modifiers, in particular those that have more specialized functions and tissue-specific expression compared with the ubiquitous DNA methyltransferases. In mouse, different histone deacetylases are upregulated in the hypothalamus in response to fasting (Hdac3 and -4), feeding on a normal diet (Hdhac10 and -11) or a high-fat diet (Hdac5 and -8) [63]. The epigenetic reprogramming of the genome caused by these proteins may be extensive, as the changes they caused to histone acetylation were detectable with immunohistochemistry. Specific histone methyltransferases have also been found to be important for adipogenesis (see above). Mice with mutations in the histone H3, lysine 4 methyltransferase (H3K4MT), MLL3 have reduced adipogenesis leading to considerably less white adipose tissue when fed with a high-fat diet [45]. Also, mice that have loss of function of the H3K9-specific demethylase JmjC domain-containing histone demethylase 2A (Jhdm2a) have obesity and hyperlipidemia [65,66]. The histone demethylase activity of Jhdm2a activates key metabolic regulators of the conversion of fat to heat in brown adipose tissue, and β-oxidation and glycerol release from skeletal muscle. Finally, mice that are heterozygous for a mutation in Trim28 (also known as KAP1 or TIF1-β), the central component of an epigenetic-modifier complex that directs repressive chromatin state, develop liver steatosis, adipocyte hypertrophy and impaired glucose tolerance [67,68]. Therefore, there is precedence for an epigenetic-modifier mutation being the primary cause of, or protective against, obesity (figure 2a,b).
(b). Evidence in humans for obesity associated with epigenetic-modifier dysfunction
There are few human conditions known to be caused by mutations in epigenetic-modifier genes. This is likely due to heterozygosity for mutations usually resulting in a normal phenotype, and null mutations being embryonic lethal (as evidenced by mouse models). However, one epigenetic-modifier disease does have obesity as one of its many downstream consequences. Rett syndrome is a developmental disorder affecting approximately one in 10 000 females [69]. Around 95 per cent of cases have a mutation in MECP2 gene, which encodes a protein that binds throughout the genome at regions of methylated DNA and complexes with chromatin remodelling proteins to regulate transcription [70]. Patients with milder forms of Rett syndrome are often obese [71,72], and mouse models typically get fat when older [73]. This obesity appears to be caused by hypothalamic regulation of appetite, as a conditional deletion in neurons in the hypothalamus of mice made them anxious, hyperphagic and obese [73]. The neurotransmitter neuropeptide Y (NPY) was increased, suggesting that the hyperphagia may be partly explained by altered hypothalamic feeding regulators.
Linkage analyses of cases of familial obesity, and genome-wide association studies with body mass index (BMI) in humans have not uncovered many loci containing epigenetic modifiers (EMS) [1]. This may indicate the rarity of cases of epigenetic-modifier-driven obesity, or the extremely polygenic nature of BMI determinants [74]. The former explanation would require the rare cases to have large effects on disease risk [75], which is possible, considering the extensive phenotype changes caused by MECP2 mutation, and in another epigenetic-modifier disease, immunodeficiency centromeric instability and facial anomalies syndrome (ICF syndrome) [76]. It would be interesting to test this theory by looking for mutations in known epigenetic-modifier genes in case–control studies.
(c). Evidence for nutrition or chemicals that cause epigenetic changes to induce obesity in adults
Several chemicals in the form of environmental toxins or medicines are known to induce obesity [2], and the range of metabolic processes and genes affected involve alteration to epigenetic patterns [11,77,78]. Most of these act through mechanisms such as those described in the first section of this review—the epigenetic changes are downstream consequences of an environmental trigger. There are a few examples, however, that primarily alter epigenetic patterns and these can cause obesity (figure 2c,d).
An example of this is the histone deacetylase valproic acid, which has been successfully used to treat epilepsy and bipolar disorder for over 40 years. Apart from the brain, histone acetylation changes occur in a variety of tissues, and one of the many side effects of the treatment is weight gain and insulin resistance [79]. The mechanism of valproic-acid-induced obesity is unclear but thought to be partly due to a reduction in the expression of adiponectin, a hormone that regulates energy homeostasis, insulin sensitivity and mitochondrial biogenesis [80,81]. An alternative possibility is upregulation of NPY in the central nervous system [82].
Methylation of DNA and histones occurs through the transfer of a methyl group from S-adenosyl-l-methionine (SAM). Many dietary factors (such as folate, methionine, choline, betaine and vitamins B2, B6 and B12) are methyl-donors that contribute to the production of SAM through one-carbon metabolism. Dietary decreases in methyl donors have been shown to have harmful effects, especially in the development of fatty-liver disease [83,84]. Adult mice that were fed with a methyl-deficient diet had extensive epigenetic changes in liver, including reduced global DNA methylation, reactivation of normally silenced repetitive elements and altered global levels of histone modifications (H3K9, H4K20 and H3K27 trimethylation) [84]. It is unclear whether dietary deficiency of methyl-donors provides a novel route to obesity, as no mention was made of other metabolic abnormalities in the mouse model. Fatty-liver disease is not only a feature of obesity (where it is caused by an oversupply of lipids or insulin resistance), but is also a feature of other conditions such as alcoholism or hepatitis. Indeed, fatty-liver disease associated with global DNA hypomethylation and global histone changes has been described with rats exposed to various hepatocarcinogens; so genome-wide epigenetic change may be a common reaction in the liver to environmental stress [85]. Furthermore, the brains of rats fed with a methyl-donor-deficient diet actually had a slight increase in global methylation [86]. Nonetheless, the extensive changes to lipid metabolism (including fatty acid uptake, de novo triglyceride synthesis and fatty acid β-oxidation) caused by methyl-deficiency may promote obesity [87]. Increased dietary methyl donors in adults can also alter organism phenotype [88–90]. However, no animal or human studies have so far reported an induction of obesity from the treatment of adults. In fact, methyl-supplementation may even be protective against obesity [91]. Indeed, the best evidence for methyl-donor availability inducing obesity comes from the developmental programming field (see below).
In conclusion, current evidence supports the feasibility of the mechanism of epigenetic-modifiers-driven obesity, though not it being a common driver of obesity. Further, animal models with tissue-specific under/overexpression of EMs are needed to resolve this. Also, studies where epigenetic-modifier mutants are exposed to a high-fat diet would be informative.
5. Developmental programming of adult obesity
The greatest area of research on the involvement of epigenetic mechanisms in obesity is on the developmental origins of adult health and disease (DOHaD), otherwise known as gestational/foetal/developmental programming. This is when an early-life experience of an organism increases its risk of developing disease as an adult. The types of early-life experience that have been shown to cause this include changes to nutrition (excess and starvation), chemical exposures and stress [92–94]. The resulting diseases are often metabolic although sometimes cancer, behavioural or reproductive disease. There are two reasons why epigenetic processes are important for developmental programming. Firstly, epigenetic modifications, through their stability over cell divisions, serve to maintain the molecular effects of an early-life experience until adulthood [11]. Secondly, the large-scale epigenetic changes that naturally occur throughout development create ‘windows of opportunity’ for environmental modulation of epigenetic state [16]. This contrasts with the relative stability of epigenetic state in the adult.
Early-life exposures to stress [95], under or overnutrition during gestation or lactation [7,96] and chemicals such as endocrine disruptors [78] have all been associated with increased risk of obesity in adulthood.
Obesity is part of a range of programmed disorders that are collectively known as ‘metabolic syndrome’, the others being hyperglycaemia, insulin resistance, hyperlipidemia, hyperinsulinemia and hypertension. The cause of the syndrome is thought to be due to a mismatch in the environment experienced by the organism during development compared with later life [97,98]. For example, if an organism experiences reduced nutrients during development, it can be ‘programmed’ to anticipate the same environment in later life. This would involve hyperphagia, increased fat storage and a preference for a high-fat diet. Such programming may have been advantageous in ancient human populations, as these ‘predictive adaptive responses’ [99] would increase the chances of the individual surviving in a resource-limited environment. However, if an individual is programmed in this way owing to an abnormal shortage of food (such as famine) or exposure to a chemical that mimics the famine programming, signals continue in a time of abundant food supply, and the mismatch could lead to obesity. As well as altered systemic metabolism, affecting glucose and lipid homeostasis, the programming of appetite and reward pathways in the brain [7], adipogenesis [100–102] and inflammation [103] are thought to be important programmed processes relevant to adult obesity.
DNA methylation or histone modification changes associated with the developmental programming of obesity are numerous and have been extensively reviewed recently [37,38,104]. For example, epigenetic changes have been detected at genes that regulate growth factors [105], adipogenesis [106], brain appetite and reward pathways [107,108] and glucose homeostasis [58,109]. The precise mechanisms of developmental programming are not understood but are expected to be varied, as several initial triggers of programming are known. Programmed epigenetic changes that are triggered by hormones, endocrine disruptors, circulating levels of different nutrients or social stress will likely be downstream consequences of environmental signals (figure 1). Alternatively, some evidence also supports alteration of epigenetic state during development as the underlying cause adult phenotype (figure 2). Methyl-donor deficiency or supplementation during gestation can cause DNA methylation changes that persist throughout the life of the offspring. Deficiency during gestation can cause genome-wide hypomethylation [110], while in utero supplementation has been shown to alter the methylation of specific genes [111,112].
The presumed large variety of programming mechanisms is also related to the timepoint of the environmental exposure, as different developmental processes occur at different times. Epigenetic abnormalities can be caused by extremely early developmental changes or postnatal ones. For example, many studies have shown epigenetic changes in offspring that are generated with assisted reproductive technologies where the preimplantation embryo was exposed only briefly to an unnatural environment [113]. Nutritional alterations in the periconceptional period also in natural matings can have effects on offspring obesity, with associated epigenetic changes [114]. Conversely, exposure to only high fat during lactation can programme offspring adiposity and neural stress responses [115]. Therefore, it seems that there are different windows of opportunity for the programming of epigenetically labile genes [16].
Importantly, there is now evidence for developmental programming to affect multiple generations. This is based on human epidemiological studies where nutritional availability in the grandparental (F0) generation has been shown to be associated with cardiovascular and metabolic health of the third generation (F2) [116–118]. These and other observations have created the possibility that, as obese mothers are more likely to have obese children, human populations may be facing an intergenerational cycle of obesity [114,119]. Mouse models have attempted to test this and have confirmed transgenerational amplification of obesity and hepatic steatosis [91,120]. Interestingly, however, a diet enriched in methyl-donors prevented the transgenerational amplification of obesity, which may suggest that epigenetic mechanisms could play a role not only in the inheritance, but also in the treatment of the cycle.
As well as transgenerational effects owing to maternal health, our group showed that the offspring of obese male rats had impaired insulin secretion and glucose tolerance [121]. Another group also reported altered lipid metabolism in the livers of offspring from fathers fed with a low protein diet [122]. As, in these cases, the father only contributed to the offspring at conception, an epigenetic factor in the sperm seems the likely mechanism [123].
As well as continuing to characterize the mechanisms of DOHaD, current research is focusing on identifying epigenetic modifications that are markers of developmental abnormalities with a view to reducing the risk of later disease by chemical (e.g. nutrient supplementation) or lifestyle (diet or exercise) interventions.
6. Transgenerational epigenetic inheritance and obesity
The situation in DOHaD of an environmental exposure (e.g. high-fat diet) during pregnancy influencing offspring phenotype can be considered a transgenerational effect; however, the offspring phenotypes cannot truly be considered ‘inherited’ as the offspring (filial 1, or F1 generation) actually directly experienced the high-fat diet themselves while in utero. In fact, as the cells within the F1 animal that will go on to produce the F2 generation (the germ cells) have already been specified by mid-gestation; therefore the grandchildren (F2) of the pregnant animal (F0) can also be considered to have directly experienced the diet/toxin. When a male experiences an environmental exposure, he (the F0) and the next generation (F1) in the form of his germ cells will also have directly been exposed [124,125].
As mentioned in §2, extensive epigenetic reprogramming occurs between generations to prevent epigenetic modifications that are associated with a disease state from being inherited. In spite of these processes, evidence is emerging that epigenetic determinants of disease are inherited. To be sure that a phenotype seen in successive generations is due to the resistance of an epigenetic mark to reprogramming rather than caused by the direct exposure of both generations, effects need to be seen in the F3 generation of a pregnant female or F2 generation down the male line [125]. These stringent criteria for transgenerational effects being truly due to epigenetic inheritance have limited the number of documented cases in mammals [126–129]. However, research in the field of under and overnutrition is starting to generate some of the most compelling evidence for transgenerational epigenetic inheritance in mammals [124,130,131].
Down the female line, one study found that glucose metabolism was altered in F3 rats when the pregnant (F0) females were fed with a protein-restricted diet [132]. Down the male lineage, reproductive [133] and glucose homeostasis abnormalities [134,135] have been detected in the F2 generation, and body-size changes in the F3 of F0 obese male mice. Postnatal overfeeding of F0 mice also was found to cause changes to F2 glucose homeostasis through the male line [136]. Finally, an allele that causes reduced food intake in mice also caused reduced feeding in two subsequent generations that did not inherit the mutation, only down the male line [137]. In some of the above studies [133,135], the same phenotypes were inherited down the male and female lines. This is surprising considering the large differences between male and female gametogenesis [138]. Continued efforts in characterizing epigenetic molecules in the gametes may explain this.
The transgenerational transmission of effects caused by ancestral obesity are likely to involve regions of DNA methylation or histone modifications that are resistant to reprogramming, or the inheritance of RNA. The germ line and preimplantation reprogramming events provide multiple challenges for the transmission of an epigenetic mark between generations. However, there are increasing numbers of reports of sequences that avoid one or both reprogramming events. In the last few years, it has also been shown that some histones survive protamination during late spermatogenesis [139–141]. These histone-bound regions are common in genes that are active in early development, which supports the theory that the histones ‘prime’ the genes for transcription in the next generation. An alteration to this process may be able to transmit diseases down the male line. Alternatively, abnormal deposits of RNA in sperm could facilitate disease inheritance, in a mechanism similar to those discovered by Rassoulzadegan and co-workers (reviewed in [129]). Experimental testing of these hypotheses is underway in several laboratories around the world.
Despite the less extreme reprogramming in the female germ line compared with the male—there is no protamination or great reduction of RNA—paradoxically, less is known about maternal inheritance of these molecules. This is because genome-wide analyses such as chromatin immunoprecipitation require millions of cells, and are thus feasible on sperm, and also because it is hard to separate offspring phenotypes that are caused by oocyte epigenetic molecules from gestational interactions between the foetus and the mother. Therefore, further research into epigenetic inheritance down the female line awaits technological improvements and technically challenging embryo transfer strategies.
The best-characterized, inherited epigenetic modification is DNA methylation. Certain classes of retrotransposons retain DNA methylation through both reprogramming events [23,29]. Subtelomeric regions also are resistant to at least germ-cell demethylation [23], and single copy loci have been found that resist one or both events [23,142]. Therefore, DNA methylation at one of these regions or a nearby gene could maintain a disease state between generations. Alternatively, transgenerational epigenetic mechanisms could involve a modulation of the gene network that undergoes genomic imprinting [134]. These genes have a long history in obesity research as many of them regulate adipogenesis, appetite and metabolism and there are numerous examples of their abnormal expression causing obesity [143]. Changes to DNA methylation at loci that regulate imprinting (differentially methylated regions, DMRs) can also be altered by gestational or postnatal diet [89,144]. Furthermore, superovulation of mice or the treatment of pregnant mice with endocrine disrupting chemicals can induce small methylation defects at DMRs that appear to survive transgenerational reprogramming down the male line [145–147]. These studies could therefore explain transgenerational effects caused by ancestral nutritional state. Three cautionary notes for this theory are that the imprint defects caused by endocrine disruptors and superovulation were rarely found in tissues other than sperm. That argues against their being a source of systemic gene dysregulation. Also in the male-line transmitted effects of a protein restricted diet no imprint defects in sperm were detected [122]. Third, a recent study found that imprinted genes were not more likely than non-imprinted genes to have altered expression in an intergenerational model of developmental programming [148].
The potential for epigenetic mechanisms explaining ‘missing heritability’, i.e. when genetic inheritance cannot fully account for the inherited component of disease, or in providing new mechanisms of evolutionary adaptation have made it an area of much current interest and debate. Human epidemiological studies of metabolic disease and animal models of abnormal nutrition will be at the forefront of determining the feasibility and impact of these mechanisms in the coming decade.
7. Conclusions
Work performed with the ultimate aim of reducing the frequency of obesity, or its associated health problems, has uncovered new examples of environmental modulation of epigenetic state. Work in the field has also started to reveal how epigenetic alterations in early-life and even in previous generations can influence the risk of an individual becoming obese. It will be interesting to see whether the same genes, tissues and epigenetic modifications are responsible for early-life, transgenerational and adult-lifestyle-induced obesity or whether several different molecular mechanisms feed into the same phenotypes. A future evaluation of the importance of epigenetics compared with other determinants of obesity [2] would also help to reveal the relative potential of epigenetic therapies.
Acknowledgements
We thank Enda Byrne and Lucia Daxinger for helpful comments in the preparation of this manuscript. Also, we wish to apologize to the authors that we did not have space to cite.
References
- 1.Day FR, Loos RJ. 2011. Developments in obesity genetics in the era of genome-wide association studies. J. Nutrigenet. Nutrigenomics 4, 222–238 10.1159/000332158 (doi:10.1159/000332158) [DOI] [PubMed] [Google Scholar]
- 2.McAllister EJ, et al. 2009. Ten putative contributors to the obesity epidemic. Crit. Rev. Food Sci. Nutr. 49, 868–913 10.1080/10408390903372599 (doi:10.1080/10408390903372599) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mavanji V, Billington CJ, Kotz CM, Teske JA. 2012. Sleep and obesity: a focus on animal models. Neurosci. Biobehav. Rev. 36, 1015–1029 10.1016/j.neubiorev.2012.01.001 (doi:10.1016/j.neubiorev.2012.01.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volkow ND, Wang GJ, Baler RD. 2011. Reward, dopamine and the control of food intake: implications for obesity. Trends Cogn. Sci. 15, 37–46 10.1016/j.tics.2010.11.001 (doi:10.1016/j.tics.2010.11.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gearhardt AN, Grilo CM, DiLeone RJ, Brownell KD, Potenza MN. 2011. Can food be addictive? Public health and policy implications. Addiction 106, 1208–1212 10.1111/j.1360-0443.2010.03301.x (doi:10.1111/j.1360-0443.2010.03301.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.WHO 2007. Global database on body mass index. See http://www.who.int/bmi/index.jsp [Google Scholar]
- 7.Morris MJ. 2009. Early life influences on obesity risk: maternal overnutrition and programming of obesity. Expert Rev. Endocronol. Metab. 4, 625–637 10.1586/eem.09.45 (doi:10.1586/eem.09.45) [DOI] [PubMed] [Google Scholar]
- 8.McIntyre HD, Gibbons KS, Flenady VJ, Callaway LK. 2012. Overweight and obesity in Australian mothers: epidemic or endemic? Med. J. Austr. 196, 184–188 10.5694/mja11.11120 (doi:10.5694/mja11.11120) [DOI] [PubMed] [Google Scholar]
- 9.CDC. Centers for Disease Control and Prevention; 2010. Overweight and obesity: childhood overweight and obesity: data and statistics. [Google Scholar]
- 10.Watkins M, Clark K, Foster C, Welch K, Kasa-Vubu J. 2007. Relationships among body mass index, parental perceptions, birthweight and parental weight after referral to a weight clinic. J. Natl Med. Assoc. 99, 908–913 [PMC free article] [PubMed] [Google Scholar]
- 11.Feil R, Fraga MF. 2011. Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13, 97–109 [DOI] [PubMed] [Google Scholar]
- 12.Law JA, Jacobsen SE. 2010. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 10.1038/nrg2719 (doi:10.1038/nrg2719) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reik W. 2007. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432 10.1038/nature05918 (doi:10.1038/nature05918) [DOI] [PubMed] [Google Scholar]
- 14.Payer B, Lee JT, Namekawa SH. 2011. X-inactivation and X-reactivation: epigenetic hallmarks of mammalian reproduction and pluripotent stem cells. Hum. Genet. 130, 265–280 10.1007/s00439-011-1024-7 (doi:10.1007/s00439-011-1024-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lange UC, Schneider R. 2010. What an epigenome remembers. BioEssays 32, 659–668 10.1002/bies.201000030 (doi:10.1002/bies.201000030) [DOI] [PubMed] [Google Scholar]
- 16.Faulk C, Dolinoy DC. 2011. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics 6, 791–797 10.4161/epi.6.7.16209 (doi:10.4161/epi.6.7.16209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamagata Y, Szabo P, Szuts D, Bacquet C, Aranyi T, Paldi A. 2012. Rapid turnover of DNA methylation in human cells. Epigenetics 7, 141–145 10.4161/epi.7.2.18906 (doi:10.4161/epi.7.2.18906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou VW, Goren A, Bernstein BE. 2011. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12, 7–18 10.1038/nrg2905 (doi:10.1038/nrg2905) [DOI] [PubMed] [Google Scholar]
- 19.Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. 2010. Non-coding RNAs: regulators of disease. J. Pathol. 220, 126–139 10.1002/path.2638 (doi:10.1002/path.2638) [DOI] [PubMed] [Google Scholar]
- 20.Heneghan HM, Miller N, Kerin MJ. 2010. Role of microRNAs in obesity and the metabolic syndrome. Obes. Rev. 11, 354–361 10.1111/j.1467-789X.2009.00659.x (doi:10.1111/j.1467-789X.2009.00659.x) [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Xie H, Mori MA, Alexander R, Yuan B, Hattangadi SM, Liu Q, Kahn CR, Lodish HF. 2011. Mir193b-365 is essential for brown fat differentiation. Nat. Cell Biol. 13, 958–965 10.1038/ncb2286 (doi:10.1038/ncb2286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. 2002. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 117, 15–23 10.1016/S0925-4773(02)00181-8 (doi:10.1016/S0925-4773(02)00181-8) [DOI] [PubMed] [Google Scholar]
- 23.Guibert S, Forne T, Weber M. 2012. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 22, 633–641 10.1101/gr.130997.111 (doi:10.1101/gr.130997.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. 2010. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463, 1101–1105 10.1038/nature08829 (doi:10.1038/nature08829) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sasaki H, Matsui Y. 2008. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat. Rev. Genet. 9, 129–140 10.1038/nrg2295 (doi:10.1038/nrg2295) [DOI] [PubMed] [Google Scholar]
- 26.Hajkova P, et al. 2008. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452, 877–881 10.1038/nature06714 (doi:10.1038/nature06714) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward WS, Coffey DS. 1991. DNA packaging and organization in mammalian spermatozoa: comparison with somatic cells. Biol. Reprod. 44, 569–574 10.1095/biolreprod44.4.569 (doi:10.1095/biolreprod44.4.569) [DOI] [PubMed] [Google Scholar]
- 28.Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484, 339–344 10.1038/nature10960 (doi:10.1038/nature10960) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, Reik W. 2003. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis 35, 88–93 10.1002/gene.10168 (doi:10.1002/gene.10168) [DOI] [PubMed] [Google Scholar]
- 30.Li Y, O'Neill C. 2012. Persistence of cytosine methylation of DNA following fertilisation in the mouse. PLoS ONE 7, e30687. 10.1371/journal.pone.0030687 (doi:10.1371/journal.pone.0030687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burton A, Torres-Padilla ME. 2010. Epigenetic reprogramming and development: a unique heterochromatin organization in the preimplantation mouse embryo. Brief. Funct. Genomics 9, 444–454 10.1093/bfgp/elq027 (doi:10.1093/bfgp/elq027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hitchins MP. 2010. Inheritance of epigenetic aberrations (constitutional epimutations) in cancer susceptibility. Adv. Genet. 70, 201–243 10.1016/B978-0-12-380866-0.60008-3 (doi:10.1016/B978-0-12-380866-0.60008-3) [DOI] [PubMed] [Google Scholar]
- 33.Martin DI, Cropley JE, Suter CM. 2011. Epigenetics in disease: leader or follower? Epigenetics 6, 843–848 10.4161/epi.6.7.16498 (doi:10.4161/epi.6.7.16498) [DOI] [PubMed] [Google Scholar]
- 34.Hesson LB, Hitchins MP, Ward RL. 2010. Epimutations and cancer predisposition: importance and mechanisms. Curr. Opin. Genet. Dev. 20, 290–298 10.1016/j.gde.2010.02.005 (doi:10.1016/j.gde.2010.02.005) [DOI] [PubMed] [Google Scholar]
- 35.Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schubeler D. 2011. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 43, 1091–1097 10.1038/ng.946 (doi:10.1038/ng.946) [DOI] [PubMed] [Google Scholar]
- 36.Zierath JR, Barres RE. 2011. Nutritional status affects the epigenomic profile of peripheral blood cells. Epigenomics 3, 259–260 10.2217/epi.11.24 (doi:10.2217/epi.11.24) [DOI] [PubMed] [Google Scholar]
- 37.Lavebratt C, Almgren M, Ekstrom TJ. 2011. Epigenetic regulation in obesity. Int. J. Obes. (Lond). 36, 757–765 10.1038/ijo.2011.178 (doi:10.1038/ijo.2011.178) [DOI] [PubMed] [Google Scholar]
- 38.Lillycrop KA, Burdge GC. 2011. Epigenetic changes in early life and future risk of obesity. Int. J. Obes. (Lond.) 35, 72–83 10.1038/ijo.2010.122 (doi:10.1038/ijo.2010.122) [DOI] [PubMed] [Google Scholar]
- 39.Campion J, Milagro F, Martinez JA. 2010. Epigenetics and obesity. Prog. Mol. Biol. Transl. Sci. 94, 291–347 10.1016/B978-0-12-375003-7.00011-X (doi:10.1016/B978-0-12-375003-7.00011-X) [DOI] [PubMed] [Google Scholar]
- 40.Gregoire FM, Smas CM, Sul HS. 1998. Understanding adipocyte differentiation. Physiol. Rev. 78, 783–809 [DOI] [PubMed] [Google Scholar]
- 41.Berger J, Bailey P, Biswas C, Cullinan CA, Doebber TW, Hayes NS, Saperstein R, Smith RG, Leibowitz MD. 1996. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology 137, 4189–4195 10.1210/en.137.10.4189 (doi:10.1210/en.137.10.4189) [DOI] [PubMed] [Google Scholar]
- 42.Ge K. 2012. Epigenetic regulation of adipogenesis by histone methylation. Biochim. Biophys. Acta 1819, 727–732 10.1016/j.bbagrm.2011.12.008 (doi:10.1016/j.bbagrm.2011.12.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen R, et al. 2008. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 22, 2953–2967 10.1101/gad.501108 (doi:10.1101/gad.501108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lefterova MI, et al. 2008. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 22, 2941–2952 10.1101/gad.1709008 (doi:10.1101/gad.1709008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J, et al. 2008. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc. Natl Acad. Sci. USA 105, 19 229–19 234 10.1073/pnas.0810100105 (doi:10.1073/pnas.0810100105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho YW, Hong S, Jin Q, Wang L, Lee JE, Gavrilova O, Ge K. 2009. Histone methylation regulator PTIP is required for PPARgamma and C/EBPalpha expression and adipogenesis. Cell Metab. 10, 27–39 10.1016/j.cmet.2009.05.010 (doi:10.1016/j.cmet.2009.05.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Y, Kan L, Qi C, Kanwar YS, Yeldandi AV, Rao MS, Reddy JK. 2000. Isolation and characterization of peroxisome proliferator-activated receptor (PPAR) interacting protein (PRIP) as a coactivator for PPAR. J. Biol. Chem. 275, 13 510–13 516 10.1074/jbc.275.18.13510 (doi:10.1074/jbc.275.18.13510) [DOI] [PubMed] [Google Scholar]
- 48.Wakabayashi K, et al. 2009. The peroxisome proliferator-activated receptor gamma/retinoid X receptor alpha heterodimer targets the histone modification enzyme PR-Set7/Setd8 gene and regulates adipogenesis through a positive feedback loop. Mol. Cell Biol. 29, 3544–3555 10.1128/MCB.01856-08 (doi:10.1128/MCB.01856-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujiki K, Kano F, Shiota K, Murata M. 2009. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 7, 38. 10.1186/1741-7007-7-38 (doi:10.1186/1741-7007-7-38) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, Rosen ED. 2010. Comparative epigenomic analysis of murine and human adipogenesis. Cell 143, 156–169 10.1016/j.cell.2010.09.006 (doi:10.1016/j.cell.2010.09.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Jin Q, Lee JE, Su IH, Ge K. 2010. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl Acad. Sci. USA 107, 7317–7322 10.1073/pnas.1000031107 (doi:10.1073/pnas.1000031107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rada P, Bocarsly ME, Barson JR, Hoebel BG, Leibowitz SF. 2010. Reduced accumbens dopamine in Sprague-Dawley rats prone to overeating a fat-rich diet. Physiol. Behav. 101, 394–400 10.1016/j.physbeh.2010.07.005 (doi:10.1016/j.physbeh.2010.07.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vucetic Z, Carlin JL, Totoki K, Reyes TM. 2012. Epigenetic dysregulation of the dopamine system in diet-induced obesity. J. Neurochem. 120, 891–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krugel U, Schraft T, Kittner H, Kiess W, Illes P. 2003. Basal and feeding-evoked dopamine release in the rat nucleus accumbens is depressed by leptin. Eur. J. Pharmacol. 482, 185–187 10.1016/j.ejphar.2003.09.047 (doi:10.1016/j.ejphar.2003.09.047) [DOI] [PubMed] [Google Scholar]
- 55.Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. 2006. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51, 801–810 10.1016/j.neuron.2006.08.023 (doi:10.1016/j.neuron.2006.08.023) [DOI] [PubMed] [Google Scholar]
- 56.Vucetic Z, Kimmel J, Reyes TM. 2011. Chronic high-fat diet drives postnatal epigenetic regulation of mu-opioid receptor in the brain. Neuropsychopharmacology 36, 1199–1206 10.1038/npp.2011.4 (doi:10.1038/npp.2011.4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wheatley KE, Nogueira LM, Perkins SN, Hurstig SD. 2011. Differential effects of calorie restriction and exercise on the adipose transcriptome in diet-induced obese mice. J. Obes. 2011, 265417. 10.1155/2011/265417 (doi:10.1155/2011/265417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nikoshkov A, Sunkari V, Savu O, Forsberg E, Catrina SB, Brismar K. 2011. Epigenetic DNA methylation in the promoters of the Igf1 receptor and insulin receptor genes in db/db mice. Epigenetics 6, 405–409 10.4161/epi.6.4.14791 (doi:10.4161/epi.6.4.14791) [DOI] [PubMed] [Google Scholar]
- 59.Almen MS, Jacobsson JA, Moschonis G, Benedict C, Chrousos GP, Fredriksson R, Schioth HB. 2012. Genome wide analysis reveals association of a FTO gene variant with epigenetic changes. Genomics 99, 132–137 10.1016/j.ygeno.2011.12.007 (doi:10.1016/j.ygeno.2011.12.007) [DOI] [PubMed] [Google Scholar]
- 60.Wang X, et al. 2010. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 8, 87. 10.1186/1741-7015-8-87 (doi:10.1186/1741-7015-8-87) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milagro FI, Campion J, Cordero P, Goyenechea E, Gomez-Uriz AM, Abete I, Zulet MA, Martinez JA. 2011. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB J. 25, 1378–1389 10.1096/fj.10-170365 (doi:10.1096/fj.10-170365) [DOI] [PubMed] [Google Scholar]
- 62.Lee J, Mi Seok S, Yu P, Kim K, Smith Z, Rivas-Astroza M, Zhong S, Kim Kemper J. 2012. Genomic analysis of hepatic farnesoid X receptor (FXR) binding sites reveals altered binding in obesity and direct gene repression by FXR. Hepatology 56, 108–117 10.1002/hep.25609 (doi:10.1002/hep.25609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Funato H, Oda S, Yokofujita J, Igarashi H, Kuroda M. 2011. Fasting and high-fat diet alter histone deacetylase expression in the medial hypothalamus. PLoS ONE 6, e18950. 10.1371/journal.pone.0018950 (doi:10.1371/journal.pone.0018950) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kamei Y, et al. 2010. Increased expression of DNA methyltransferase 3a in obese adipose tissue: studies with transgenic mice. Obesity (Silver Spring, MD) 18, 314–321 10.1038/oby.2009.246 (doi:10.1038/oby.2009.246) [DOI] [PubMed] [Google Scholar]
- 65.Tateishi K, Okada Y, Kallin EM, Zhang Y. 2009. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458, 757–761 10.1038/nature07777 (doi:10.1038/nature07777) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inagaki T, et al. 2009. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 14, 991–1001 10.1111/j.1365-2443.2009.01326.x (doi:10.1111/j.1365-2443.2009.01326.x) [DOI] [PubMed] [Google Scholar]
- 67.Iyengar S, Farnham PJ. 2011. KAP1 protein: an enigmatic master regulator of the genome. J. Biol. Chem. 286, 26 267–26 276 10.1074/jbc.R111.252569 (doi:10.1074/jbc.R111.252569) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whitelaw NC, Chong S, Morgan DK, Nestor C, Bruxner TJ, Ashe A, Lambley E, Meehan R, Whitelaw E. 2010. Reduced levels of two modifiers of epigenetic gene silencing, Dnmt3a and Trim28, cause increased phenotypic noise. Genome Biol. 11, R111. 10.1186/gb-2010-11-11-r111 (doi:10.1186/gb-2010-11-11-r111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moretti P, Zoghbi HY. 2006. MeCP2 dysfunction in Rett syndrome and related disorders. Curr. Opin. Genet. Dev. 16, 276–281 10.1016/j.gde.2006.04.009 (doi:10.1016/j.gde.2006.04.009) [DOI] [PubMed] [Google Scholar]
- 70.Guy J, Cheval H, Selfridge J, Bird A. 2011. The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652 10.1146/annurev-cellbio-092910-154121 (doi:10.1146/annurev-cellbio-092910-154121) [DOI] [PubMed] [Google Scholar]
- 71.Kleefstra T, Yntema HG, Oudakker AR, Romein T, Sistermans E, Nillessen W, van Bokhoven H, de Vries BB, Hamel BC. 2002. De novo MECP2 frameshift mutation in a boy with moderate mental retardation, obesity and gynaecomastia. Clin. Genet. 61, 359–362 10.1034/j.1399-0004.2002.610507.x (doi:10.1034/j.1399-0004.2002.610507.x) [DOI] [PubMed] [Google Scholar]
- 72.Couvert P, et al. 2001. MECP2 is highly mutated in X-linked mental retardation. Hum. Mol. Genet. 10, 941–946 10.1093/hmg/10.9.941 (doi:10.1093/hmg/10.9.941) [DOI] [PubMed] [Google Scholar]
- 73.Fyffe SL, et al. 2008. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 59, 947–958 10.1016/j.neuron.2008.07.030 (doi:10.1016/j.neuron.2008.07.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang J, et al. 2011. Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 43, 519–525 10.1038/ng.823 (doi:10.1038/ng.823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bogardus C. 2009. Missing heritability and GWAS utility. Obesity (Silver Spring, MD) 17, 209–210 10.1038/oby.2008.613 (doi:10.1038/oby.2008.613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matarazzo MR, De Bonis ML, Vacca M, Della Ragione F, D'Esposito M. 2009. Lessons from two human chromatin diseases, ICF syndrome and Rett syndrome. Int. J. Biochem. Cell Biol. 41, 117–126 10.1016/j.biocel.2008.07.026 (doi:10.1016/j.biocel.2008.07.026) [DOI] [PubMed] [Google Scholar]
- 77.Csoka AB, Szyf M. 2009. Epigenetic side-effects of common pharmaceuticals: a potential new field in medicine and pharmacology. Med. Hypotheses 73, 770–780 10.1016/j.mehy.2008.10.039 (doi:10.1016/j.mehy.2008.10.039) [DOI] [PubMed] [Google Scholar]
- 78.Newbold RR. 2010. Impact of environmental endocrine disrupting chemicals on the development of obesity. Hormones (Athens) 9, 206–217 [DOI] [PubMed] [Google Scholar]
- 79.Verrotti A, Basciani F, De Simone M, Trotta D, Morgese G, Chiarelli F. 2002. Insulin resistance in epileptic girls who gain weight after therapy with valproic acid. J. Child Neurol. 17, 265–268 10.1177/088307380201700405 (doi:10.1177/088307380201700405) [DOI] [PubMed] [Google Scholar]
- 80.Qiao L, Kinney B, Yoo HS, Lee B, Schaack J, Shao J. 2012. Adiponectin increases skeletal muscle mitochondrial biogenesis by suppressing mitogen-activated protein kinase phosphatase-1. Diabetes 61, 1463–1470 10.2337/db11-1475 (doi:10.2337/db11-1475) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qiao L, Schaack J, Shao J. 2006. Suppression of adiponectin gene expression by histone deacetylase inhibitor valproic acid. Endocrinology 147, 865–874 10.1210/en.2005-1030 (doi:10.1210/en.2005-1030) [DOI] [PubMed] [Google Scholar]
- 82.Brill J, Lee M, Zhao S, Fernald RD, Huguenard JR. 2006. Chronic valproic acid treatment triggers increased neuropeptide y expression and signaling in rat nucleus reticularis thalami. J. Neurosci. 26, 6813–6822 10.1523/JNEUROSCI.5320-05.2006 (doi:10.1523/JNEUROSCI.5320-05.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pogribny IP, Shpyleva SI, Muskhelishvili L, Bagnyukova TV, James SJ, Beland FA. 2009. Role of DNA damage and alterations in cytosine DNA methylation in rat liver carcinogenesis induced by a methyl-deficient diet. Mutat. Res. 669, 56–62 10.1016/j.mrfmmm.2009.05.003 (doi:10.1016/j.mrfmmm.2009.05.003) [DOI] [PubMed] [Google Scholar]
- 84.Pogribny IP, Tryndyak VP, Bagnyukova TV, Melnyk S, Montgomery B, Ross SA, Latendresse JR, Rusyn I, Beland FA. 2009. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol. 51, 176–186 10.1016/j.jhep.2009.03.021 (doi:10.1016/j.jhep.2009.03.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tryndyak VP, Muskhelishvili L, Kovalchuk O, Rodriguez-Juarez R, Montgomery B, Churchwell MI, Ross SA, Beland FA, Pogribny IP. 2006. Effect of long-term tamoxifen exposure on genotoxic and epigenetic changes in rat liver: implications for tamoxifen-induced hepatocarcinogenesis. Carcinogenesis 27, 1713–1720 10.1093/carcin/bgl050 (doi:10.1093/carcin/bgl050) [DOI] [PubMed] [Google Scholar]
- 86.Pogribny IP, Karpf AR, James SR, Melnyk S, Han T, Tryndyak VP. 2008. Epigenetic alterations in the brains of Fisher 344 rats induced by long-term administration of folate/methyl-deficient diet. Brain Res. 1237, 25–34 10.1016/j.brainres.2008.07.077 (doi:10.1016/j.brainres.2008.07.077) [DOI] [PubMed] [Google Scholar]
- 87.Pogribny IP, James SJ, Beland FA. 2012. Molecular alterations in hepatocarcinogenesis induced by dietary methyl deficiency. Mol. Nutr. Food Res. 56, 116–125 10.1002/mnfr.201100524 (doi:10.1002/mnfr.201100524) [DOI] [PubMed] [Google Scholar]
- 88.Waterland RA. 2006. Assessing the effects of high methionine intake on DNA methylation. J. Nutr. 136(Suppl. 6), S1706–S1710 [DOI] [PubMed] [Google Scholar]
- 89.Waterland RA, Lin JR, Smith CA, Jirtle RL. 2006. Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum. Mol. Genet. 15, 705–716 10.1093/hmg/ddi484 (doi:10.1093/hmg/ddi484) [DOI] [PubMed] [Google Scholar]
- 90.Tian W, Zhao M, Li M, Song T, Zhang M, Quan L, Li S, Sun ZS. 2012. Reversal of cocaine-conditioned place preference through methyl supplementation in mice: altering global DNA methylation in the prefrontal cortex. PLoS ONE 7, e33435. 10.1371/journal.pone.0033435 (doi:10.1371/journal.pone.0033435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S. 2008. Methyl donor supplementation prevents transgenerational amplification of obesity. Int. J. Obes. (Lond.) 32, 1373–1379 10.1038/ijo.2008.100 (doi:10.1038/ijo.2008.100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. 2004. Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854 10.1038/nn1276 (doi:10.1038/nn1276) [DOI] [PubMed] [Google Scholar]
- 93.Szyf M, Weaver I, Meaney M. 2007. Maternal care, the epigenome and phenotypic differences in behavior. Reprod. Toxicol. 24, 9–19 10.1016/j.reprotox.2007.05.001 (doi:10.1016/j.reprotox.2007.05.001) [DOI] [PubMed] [Google Scholar]
- 94.Hanson M, Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD. 2011. Developmental plasticity and developmental origins of non-communicable disease: theoretical considerations and epigenetic mechanisms. Prog. Biophys. Mol. Biol. 106, 272–280 10.1016/j.pbiomolbio.2010.12.008 (doi:10.1016/j.pbiomolbio.2010.12.008) [DOI] [PubMed] [Google Scholar]
- 95.Pervanidou P, Chrousos GP. 2011. Metabolic consequences of stress during childhood and adolescence. Metabolism 61, 611–619 10.1016/j.metabol.2011.10.005 (doi:10.1016/j.metabol.2011.10.005) [DOI] [PubMed] [Google Scholar]
- 96.Bruce KD, Cagampang FR. 2011. Epigenetic priming of the metabolic syndrome. Toxicol. Mech. Methods 21, 353–361 10.3109/15376516.2011.559370 (doi:10.3109/15376516.2011.559370) [DOI] [PubMed] [Google Scholar]
- 97.Bateson P. 2001. Fetal experience and good adult design. Int. J. Epidemiol. 30, 928–934 10.1093/ije/30.5.928 (doi:10.1093/ije/30.5.928) [DOI] [PubMed] [Google Scholar]
- 98.Gluckman PD, Hanson MA. 2004. The developmental origins of the metabolic syndrome. Trends Endocrinol. Metab. 15, 183–187 10.1016/j.tem.2004.03.002 (doi:10.1016/j.tem.2004.03.002) [DOI] [PubMed] [Google Scholar]
- 99.Gluckman PD, Hanson MA, Spencer HG. 2005. Predictive adaptive responses and human evolution. Trends Ecol. Evol. 20, 527–533 10.1016/j.tree.2005.08.001 (doi:10.1016/j.tree.2005.08.001) [DOI] [PubMed] [Google Scholar]
- 100.Budge H, Gnanalingham MG, Gardner DS, Mostyn A, Stephenson T, Symonds ME. 2005. Maternal nutritional programming of fetal adipose tissue development: long-term consequences for later obesity. Birth Defects Res. C 75, 193–199 10.1002/bdrc.20044 (doi:10.1002/bdrc.20044) [DOI] [PubMed] [Google Scholar]
- 101.Taylor PD, Poston L. 2007. Developmental programming of obesity in mammals. Exp. Physiol. 92, 287–298 10.1113/expphysiol.2005.032854 (doi:10.1113/expphysiol.2005.032854) [DOI] [PubMed] [Google Scholar]
- 102.Samuelsson AM, et al. 2008. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 51, 383–392 10.1161/HYPERTENSIONAHA.107.101477 (doi:10.1161/HYPERTENSIONAHA.107.101477) [DOI] [PubMed] [Google Scholar]
- 103.Martinez JA, Cordero P, Campion J, Milagro FI. 2012. Interplay of early-life nutritional programming on obesity, inflammation and epigenetic outcomes. Proc. Nutr. Soc. 71, 276–283 10.1017/S0029665112000055 (doi:10.1017/S0029665112000055) [DOI] [PubMed] [Google Scholar]
- 104.Wang J, Wu Z, Li D, Li N, Dindot SV, Satterfield MC, Bazer FW, Wu G. 2012. Nutrition, epigenetics, and metabolic syndrome. Antioxid. Redox Signal. 17, 282–301 10.1089/ars.2011.4381 (doi:10.1089/ars.2011.4381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. 2008. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl Acad. Sci. USA 105, 17 046–17 049 10.1073/pnas.0806560105 (doi:10.1073/pnas.0806560105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Godfrey KM, et al. 2011. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes 60, 1528–1534 10.2337/db10-0979 (doi:10.2337/db10-0979) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vucetic Z, Kimmel J, Totoki K, Hollenbeck E, Reyes TM. 2010. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology 151, 4756–4764 10.1210/en.2010-0505 (doi:10.1210/en.2010-0505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stevens A, Begum G, Cook A, Connor K, Rumball C, Oliver M, Challis J, Bloomfield F, White A. 2010. Epigenetic changes in the hypothalamic proopiomelanocortin and glucocorticoid receptor genes in the ovine fetus after periconceptional undernutrition. Endocrinology 151, 3652–3664 10.1210/en.2010-0094 (doi:10.1210/en.2010-0094) [DOI] [PubMed] [Google Scholar]
- 109.Raychaudhuri N, Raychaudhuri S, Thamotharan M, Devaskar SU. 2008. Histone code modifications repress glucose transporter 4 expression in the intrauterine growth-restricted offspring. J. Biol. Chem. 283, 13 611–13 626 10.1074/jbc.M800128200 (doi:10.1074/jbc.M800128200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McKay JA, Waltham KJ, Williams EA, Mathers JC. 2011. Folate depletion during pregnancy and lactation reduces genomic DNA methylation in murine adult offspring. Genes Nutr. 6, 189–196 10.1007/s12263-010-0199-1 (doi:10.1007/s12263-010-0199-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hollingsworth JW, et al. 2008. In utero supplementation with methyl donors enhances allergic airway disease in mice. J. Clin. Invest. 118, 3462–3469 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 112.Waterland RA, Jirtle RL. 2003. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 23, 5293–5300 10.1128/MCB.23.15.5293-5300.2003 (doi:10.1128/MCB.23.15.5293-5300.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Montfoort AP, Hanssen LL, de Sutter P, Viville S, Geraedts JP, de Boer P. 2012. Assisted reproduction treatment and epigenetic inheritance. Hum. Reprod. Update 18, 171–197 10.1093/humupd/dmr047 (doi:10.1093/humupd/dmr047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang S, Rattanatray L, McMillen IC, Suter CM, Morrison JL. 2011. Periconceptional nutrition and the early programming of a life of obesity or adversity. Prog. Biophys. Mol. Biol. 106, 307–314 10.1016/j.pbiomolbio.2010.12.004 (doi:10.1016/j.pbiomolbio.2010.12.004) [DOI] [PubMed] [Google Scholar]
- 115.Trottier G, Koski KG, Brun T, Toufexis DJ, Richard D, Walker CD. 1998. Increased fat intake during lactation modifies hypothalamic-pituitary-adrenal responsiveness in developing rat pups: a possible role for leptin. Endocrinology 139, 3704–3711 10.1210/en.139.9.3704 (doi:10.1210/en.139.9.3704) [DOI] [PubMed] [Google Scholar]
- 116.Stein AD, Lumey LH. 2000. The relationship between maternal and offspring birth weights after maternal prenatal famine exposure: the Dutch Famine Birth Cohort Study. Hum. Biol. 72, 641–654 [PubMed] [Google Scholar]
- 117.Kaati G, Bygren LO, Edvinsson S. 2002. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur. J. Hum. Genet. 10, 682–688 10.1038/sj.ejhg.5200859 (doi:10.1038/sj.ejhg.5200859) [DOI] [PubMed] [Google Scholar]
- 118.Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjostrom M, Golding J. 2006. Sex-specific, male-line transgenerational responses in humans. Eur. J. Hum. Genet. 14, 159–166 10.1038/sj.ejhg.5201538 (doi:10.1038/sj.ejhg.5201538) [DOI] [PubMed] [Google Scholar]
- 119.Whitaker RC. 2004. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics 114, e29–e36 10.1542/peds.114.1.e29 (doi:10.1542/peds.114.1.e29) [DOI] [PubMed] [Google Scholar]
- 120.Li M, Sloboda M, Vickers MH. 2011. Maternal obesity and developmental programming of metabolic disorders in offspring: evidence from animal models. Exp. Diabetes Res. 2011, 1–9 10.1155/2011/592408 (doi:10.1155/2011/592408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. 2010. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 467, 963–966 10.1038/nature09491 (doi:10.1038/nature09491) [DOI] [PubMed] [Google Scholar]
- 122.Carone BR, et al. 2010. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143, 1084–1096 10.1016/j.cell.2010.12.008 (doi:10.1016/j.cell.2010.12.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Youngson NA, Whitelaw E. 2011. The effects of acquired paternal obesity on the next generation. Asian J. Androl. 13, 195–196 10.1038/aja.2010.163 (doi:10.1038/aja.2010.163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Youngson NA, Whitelaw E. 2008. Transgenerational epigenetic effects. Annu. Rev. Genomics Hum. Genet. 9, 233–257 10.1146/annurev.genom.9.081307.164445 (doi:10.1146/annurev.genom.9.081307.164445) [DOI] [PubMed] [Google Scholar]
- 125.Skinner MK. 2008. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod. Toxicol. 25, 2–6 10.1016/j.reprotox.2007.09.001 (doi:10.1016/j.reprotox.2007.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Anway MD, Cupp AS, Uzumcu M, Skinner MK. 2005. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308, 1466–1469 10.1126/science.1108190 (doi:10.1126/science.1108190) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. 1999. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 23, 314–318 10.1038/15490 (doi:10.1038/15490) [DOI] [PubMed] [Google Scholar]
- 128.Rakyan VK, Chong S, Champ ME, Cuthbert PC, Morgan HD, Luu KV, Whitelaw E. 2003. Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc. Natl Acad. Sci. USA 100, 2538–2543 10.1073/pnas.0436776100 (doi:10.1073/pnas.0436776100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cuzin F, Rassoulzadegan M. 2010. Non-Mendelian epigenetic heredity: gametic RNAs as epigenetic regulators and transgenerational signals. Essays Biochem. 48, 101–106 10.1042/bse0480101 (doi:10.1042/bse0480101) [DOI] [PubMed] [Google Scholar]
- 130.Daxinger L, Whitelaw E. 2012. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 13, 153–162 10.1038/nrm3288 (doi:10.1038/nrm3288) [DOI] [PubMed] [Google Scholar]
- 131.Daxinger L, Whitelaw E. 2010. Transgenerational epigenetic inheritance: more questions than answers. Genome Res. 20, 1623–1628 10.1101/gr.106138.110 (doi:10.1101/gr.106138.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Benyshek DC, Johnston CS, Martin JF. 2006. Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia 49, 1117–1119 10.1007/s00125-006-0196-5 (doi:10.1007/s00125-006-0196-5) [DOI] [PubMed] [Google Scholar]
- 133.Fullston T, Palmer NO, Owens JA, Mitchell M, Bakos HW, Lane M. 2012. Diet-induced paternal obesity in the absence of diabetes diminishes the reproductive health of two subsequent generations of mice. Hum. Reprod. 27, 1391–1400 10.1093/humrep/des030 (doi:10.1093/humrep/des030) [DOI] [PubMed] [Google Scholar]
- 134.Dunn GA, Bale TL. 2011. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 152, 2228–2236 10.1210/en.2010-1461 (doi:10.1210/en.2010-1461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dunn GA, Bale TL. 2009. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology 150, 4999–5009 10.1210/en.2009-0500 (doi:10.1210/en.2009-0500) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pentinat T, Ramon-Krauel M, Cebria J, Diaz R, Jimenez-Chillaron JC. 2010. Transgenerational inheritance of glucose intolerance in a mouse model of neonatal overnutrition. Endocrinology 151, 5617–5623 10.1210/en.2010-0684 (doi:10.1210/en.2010-0684) [DOI] [PubMed] [Google Scholar]
- 137.Yazbek SN, Spiezio SH, Nadeau JH, Buchner DA. 2010. Ancestral paternal genotype controls body weight and food intake for multiple generations. Hum. Mol. Genet. 19, 4134–4144 10.1093/hmg/ddq332 (doi:10.1093/hmg/ddq332) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kobayashi H, et al. 2012. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 8, e1002440. 10.1371/journal.pgen.1002440 (doi:10.1371/journal.pgen.1002440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arpanahi A, et al. 2009. Endonuclease-sensitive regions of human spermatozoal chromatin are highly enriched in promoter and CTCF binding sequences. Genome Res. 19, 1338–1349 10.1101/gr.094953.109 (doi:10.1101/gr.094953.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. 2009. Distinctive chromatin in human sperm packages genes for embryo development. Nature 460, 473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brykczynska U, et al. 2010. Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat. Struct. Mol. Biol. 17, 679–687 10.1038/nsmb.1821 (doi:10.1038/nsmb.1821) [DOI] [PubMed] [Google Scholar]
- 142.Borgel J, Guibert S, Li Y, Chiba H, Schubeler D, Sasaki H, Forne T, Weber M. 2010. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 42, 1093–1100 10.1038/ng.708 (doi:10.1038/ng.708) [DOI] [PubMed] [Google Scholar]
- 143.Weinstein LS, Xie T, Qasem A, Wang J, Chen M. 2010. The role of GNAS and other imprinted genes in the development of obesity. Int. J. Obes. (Lond.) 34, 6–17 10.1038/ijo.2009.222 (doi:10.1038/ijo.2009.222) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gallou-Kabani C, et al. 2010. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PLoS ONE 5, e14398. 10.1371/journal.pone.0014398 (doi:10.1371/journal.pone.0014398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stouder C, Paoloni-Giacobino A. 2011. Specific transgenerational imprinting effects of the endocrine disruptor methoxychlor on male gametes. Reproduction 141, 207–216 10.1530/REP-10-0400 (doi:10.1530/REP-10-0400) [DOI] [PubMed] [Google Scholar]
- 146.Stouder C, Paoloni-Giacobino A. 2010. Transgenerational effects of the endocrine disruptor vinclozolin on the methylation pattern of imprinted genes in the mouse sperm. Reproduction 139, 373–379 10.1530/REP-09-0340 (doi:10.1530/REP-09-0340) [DOI] [PubMed] [Google Scholar]
- 147.Stouder C, Deutsch S, Paoloni-Giacobino A. 2009. Superovulation in mice alters the methylation pattern of imprinted genes in the sperm of the offspring. Reprod. Toxicol. 28, 536–541 10.1016/j.reprotox.2009.06.009 (doi:10.1016/j.reprotox.2009.06.009) [DOI] [PubMed] [Google Scholar]
- 148.Radford EJ, et al. 2012. An unbiased assessment of the role of imprinted genes in an intergenerational model of developmental programming. PLoS genetics 8, e1002605. 10.1371/journal.pgen.1002605 (doi:10.1371/journal.pgen.1002605) [DOI] [PMC free article] [PubMed] [Google Scholar]