

Abstract
Transposable elements and their fossil sequences occupy about half of the genome in mammals. While most of these selfish mobile elements have been inactivated by truncations and mutations during evolution, some copies remain competent to transpose and/or amplify, posing an ongoing genetic threat. To control such mutagenic sequences, host genomes have developed multiple layers of defence mechanisms, including epigenetic regulation and RNA silencing. Germ cells, in particular, employ the piwi–small RNA pathway, which plays a central and adaptive role in safeguarding the germline genome from retrotransposons. Recent studies have revealed that a class of developmentally regulated genes, which have long been implicated in germ cell specification and differentiation, such as vasa and tudor family genes, play key roles in the piwi pathway to suppress retrotransposons, indicating that the piwi-mediated genome protection is at the core of germline development. Furthermore, while the piwi system primarily operates post-transcriptionally at the RNA level, it also affects the epigenetics of cognate genome loci, offering an intriguing link between small RNAs and transcriptional control in mammals. In this review, we summarize our current understanding of the piwi pathway in mice, which is emerging as a fundamental component of spermatogenesis that ensures male fertility and genome integrity.
Keywords: piwi, piRNA, DNA methylation, retrotransposon, spermatogenesis, germ cells
1. Introduction
The genome encodes a master blueprint of an organism and species, which however is not unchanging but is constantly subject to various alterations and modifications. Mutation rates of both unicellular and multicellular organisms are estimated at between 1 × 10−9 and 10−10 mutations per base per cell division [1], which corresponds to around 1 × 10−5 mutations per locus per generation in humans. Actual insults to genomic DNA are, however, far more extensive, being constantly generated as consequences of physical and chemical attacks imposed by, for example, oxidative stress, exposure to natural ionizing radiation and uptake of genotoxic reagents, leading to oxidation, hydrolysis and alkylation and so on. Such damages to DNA molecules are estimated to be more than thousands in a cell per day in mammals [2]. To counteract these, organisms have evolved multiple DNA repair systems such as base and nucleotide excision repair, non-homologous and homologous recombinational repair, etc., which constitutively operate to avoid cellular catastrophe.
In addition to physico-chemical attacks on DNA, however, the genome is subject to another severe biological threat, which is encoded by the genome itself, that is, selfish mobile elements or transposons. Transposons are genetic sequences that move or amplify themselves in the genome, thereby altering the genome information and organization [3]. Unlike the structural lesions of DNA molecules as described earlier, transposons do not leave distinct chemical marks of damage that can be recognized by DNA repair proteins. Host genomes instead have to distinguish transposon sequences to control their parasitic activity. A general means of sequence-specific recognition is DNA- or RNA-binding proteins, as exemplified by transcription factors. In transposon regulation, the KAP1/TRIM28 complex recognizes a class of retrotransposon promoters and suppresses their activity in mouse embryonic stem cells [4,5]. However, transposons, especially RNA-mediated retrotransposons, rapidly mutate their sequences when compared with endogenous protein-coding genes and thus can be divergent more than a repertoire of DNA-/RNA-binding proteins encoded in the genome. Another more adaptive way of recognizing specific sequences is RNA interference (RNAi) and its related systems, which use small non-coding RNAs that guide the Argonaute family proteins or associated complexes to cleave or translationally suppress target RNAs or to affect cognate DNA loci [6]. Argonaute proteins loaded with microRNA (miRNA) or small interfering RNA (siRNA) are widespread in eukaryotes (with several notable exceptions, such as Saccharomyces cerevisiae), and they regulate a variety of cellular and developmental processes through controlling the stability or translational activity of endogenous mRNAs or foreign sequences. Such functioning of RNAi likely originates from a key and ancient defensive response to double-stranded RNAs derived from transposons, viruses and repetitive sequences, etc., and there operates another specialized RNAi mechanism, called the piwi pathway, in animals, whose function is to silence retrotransposons in the germline [7]. In this small RNA pathway, Piwi proteins of an Argonaute subfamily are loaded with piwi-interacting RNAs (piRNAs) [8–11] and act to control retrotransposons both transcriptionally and post-transcriptionally. Remarkably, a class of developmentally regulated genes that had long been thought to act in germline specification and differentiation actually play key roles in the piwi pathway [12–15], indicating that transposon control is closely associated with proper germ cell development and that host genomes have invested enormous resources to protect their genetic information in the germline.
In this review, we summarize our current understanding of the piwi–small RNA system in mammals. The whole picture of the piwi pathway remains unclear still, but it is now emerging as a key component of spermatogenesis that ensures male fertility, and we outline the piRNA system focusing on the functional components, biogenesis, retrotransposon control and developmental regulation.
2. Germ cell development in mammals
The germ cell linage is segregated early in development, and is set aside from other somatic lineages in many species. In several model animals, such as Drosophila, C. elegans and Xenopus, primordial germ cells (PGCs) are fate-determined by maternally provided factors transmitted from oocytes, representing a mosaic/determinative development [16–18]. By contrast, in mammals, PGCs are lineage-restricted among a population of pluripotent epiblast cells, depending on inter-cellular induction from surrounding somatic cells, and thus the specification is regulative [19]. In mice, PGCs appear in a posterior region of the extra-embryonic tissue, the allantois, and then the cells proliferate and migrate through the embryo proper to reach gonadal primordia at around mid-gestation. Post migratory PGCs then initiate male or female germ cell differentiation according to the sex of the surrounding somatic cells, with (pro)spermatogonia/gonocytes in the male being arrested at the G1(G0) phase of the mitotic cell cycle, while in the female the prophase of meiosis I starts during embryonic development.
Prospermatogonia then resume mitotic proliferation after birth to become postnatal spermatogonial stem cells, followed by spermatogenic differentiation, which gives rise to meiotic spermatocytes, haploid spermatids and subsequently mature spermatozoa throughout the male life [20]. In the female, by contrast, oocytes start to grow after birth and periodically mature into functional eggs depending on the estrous cycle [21].
During such germline development, dynamic epigenetic reprogramming takes place, including global and progressive CpG demethylation in PGCs [22], followed by de novo establishment of DNA methylation patterns in foetal (pro)spermatogonia/gonocytes in the male and in postnatal growing oocytes in the female [23,24]. This epigenetic rewriting defines how the genetic programme of the germline is read out in the subsequent generation, including genomic imprinting whose parent-of-origin specific marks are reestablished according to the sex of the germ cells. One major drawback of global epigenetic erasure/rewriting is that it also affects selfish transposon sequences. Indeed, transposons lose their silencing marks during global CpG demethylation of PGCs [22], and are actually activated to detectable levels in subsequent prospermatogonia/gonocytes and in growing oocytes [25]. Such transposon activity is an outcome of the balance between the activation programme and host silencing mechanisms. If host defence systems fail, the consequence is catastrophic for the genome information, which will either be inherited by the next generation or will trigger gross germ cell death, leading to sterility. The piRNA machinery acts to establish retrotransposon silencing in foetal prospermatogonia/gonocytes, through piwi-pathway-mediated DNA methylation, to ensure proper postnatal spermatogenesis (see relevant later text).
3. Transposable elements
Transposons are widespread in the three branches of life: prokaryote, eukaryote and archaea. These selfish mobile elements tend to increase their number during evolution, with higher organisms having an increasingly higher proportion of transposon copies in their genomes. In mammals, transposons and their fossil sequences occupy about half of the genome (at least approx. 45% in humans and 37% in mice), which contrasts with 1–2% of protein-coding exonic sequences [26,27]. Although most of these transposon elements have been inactivated by mutations and truncations, some sequences remain active and are still expanding, causing new transposon copies to appear in the genome.
Transposons are classified into two classes (DNA transpososns and RNA-mediated retrotransposons) according to how they move and/or amplify their sequences. DNA transposons, which move from one genomic site to another, use a transposase together with host repair proteins to ‘cut and paste’ themselves into the genome [28]. They occupy about 2–3% of mammalian genomes, but all copies are inactive because of accumulated mutations, with a possible exception of vespertilionid bat (Myotis lucifugus) piggyBac-like elements [29]. Currently, only artificially resurrected DNA transposons, derived from a variety of species, are being used in mutagenesis studies, etc.
The other class of transposons, retrotransposons, amplify themselves from RNA intermediates transcribed from one locus, which are then reverse-transcribed back to DNA and transpose into new genome positions (copy and paste). Retrotransposons are further divided into three main subclasses, depending on their structures: (i) long terminal repeat (LTR) retrotransposons, (ii) non-LTR autonomous long interspersed elements (LINEs), and (iii) non-LTR non-autonomous short interspersed elements (SINEs) [30].
LTR retrotransposons are similar to retroviruses, and encode gag (group-specific antigen) and pol (reverse transcriptase, etc.) proteins but not the env protein, which is necessary to make the envelope of a retrovirus particle (or only a fragment of env remains in several classes of LTR retrotransposons). LTR retrotransposons occupy about 8–10% of mammalian genomes but are mostly inactive in humans, while the mouse genome contains active copies as exemplified by intracisternal A particles (IAPs) [31].
Another class of retrotransposons, non-LTR LINEs, are different from LTR retrotransposons in their terminal sequences as well as coding proteins. LINEs possess a 5′UTR that contains an internal promoter(s) and a 3′UTR with a poly A signal(s), and two open reading frames (ORF1 and ORF2) are encoded in a bicistronic mRNA. ORF1 is an RNA-binding protein and ORF2 has endonuclease and reverse transcriptase domains. LINE retrotransposition occurs by a mechanism termed target-site-primed reverse transcription in the nucleus, in contrast to LTR type retrotransposition, which occurs in the cytoplasm. LINEs are the most abundant retrotransposons in mammalian genomes: L1, the most common type of LINEs, is present in more than 500 000 copies, comprising approximately 20 per cent of the human and mouse genomes. Among these, there are about 100 copies of full-length active elements in the human genome and about 3000 copies in the mouse genome [3].
The third class of retrotransposons, SINEs, are short (a few hundred bp when compared with 5–10 kb of LTR retrotransposons and LINEs) and do not encode functional proteins, being unable to transpose by their own machinery. Instead, to expand in the genome, these non-autonomous sequences use LINE proteins, mainly ORF2, that associate with the 3′ end sequence of SINEs, which share a significant homology with that of LINEs [32]. Alu sequences are the most abundant SINEs in humans, comprising about 10 per cent of the genome, while B1 and B2 elements are the most prevalent elements in mice, each occupying 2–3% of the genome. The number of active copies of SINEs is not well estimated, but a significant proportion of them remains active and can transpose depending on the LINEs’ activity in trans.
Among the three classes of retrotransposons, LINEs and SINEs are most actively expanding in mammalian genomes. In humans, at least one in every 50 individuals has a new copy of L1, while one in every 30 individuals contains a novel Alu transposition. In mice, the estimates are even higher, and novel transposon insertions are the major source of spontaneous phenotypic variations among inbred mice [3,31]. Naturally, transposons expand themselves in a population through their activity in the germline, which includes early pluripotent cells and fate-determined germ cells. Indeed, retrotransposons are expressed in both early embryos and germ cells [25], in addition to several somatic cell lineages and some cancerous cells. How retrotransposons are actively transcribed, especially in the germline, is not well understood, but they co-opt cellular transcription machinery such as RNA polymerase II and transcription factors, including YY1, SOX2, SOX11 and RUNX3, for LINE1 promoter regulation [33].
To fight against such transposon activity, host genomes use multiple layers of molecular defence systems. In mammals, one key pathway is genome DNA methylation. A maintenance DNA methyl transferase, DNMT1, acts to suppress transposons and prevent embryonic lethality [34,35], whereas DNMT3L has a more specific role in regulating retrotransposons in germ cells [36]. Histone modifications also have critical functions in epigenetic silencing of transposons, as was shown for Setdb1/Eset and H3K9 in mouse embryonic stem cells [4,5]. In addition to epigenetic regulation, retrotransposon transcripts after their expression are further targeted by adaptive RNAi, mainly by recognition of the presence of both sense and anti-sense strands of RNA. In particular, the piwi system specifically operates in the germline and is essential to protect the genome stability of the germline and reproductive fitness.
4. Piwi proteins and piRNAs
RNA interference (RNAi) and related systems represent a posttranscriptional gene-silencing mechanism mediated by small non-coding RNAs that guide Argonaute family proteins to cleave or translationally suppress complementary target RNAs [37,38]. Argonaute family proteins encode PIWI (endonuclease), PAZ (single-stranded RNA binding) and MID (5′ nucleotide binding) domains, and are further classified into Ago and Piwi clades. Argonaute clade members are rather ubiquitously expressed in multicellular organisms and bind to approximately 21–24 nucleotide small RNAs, such as miRNAs derived from hairpin precursors and siRNAs processed from sense and anti-sense hybrids [6]. The other Argonaute clade comprises Piwi proteins, which are only found in animals and are specifically expressed in germ cells (and certain gonadal somatic cells of several species, including Drosophila) [7]. Piwi proteins bind to piwi-interacting RNAs (piRNAs), which are approximately 24–30 nucleotide single-stranded RNAs (8–11). In mice, the genome encodes three piwi proteins: PIWIL1/MIWI, PIWIL2/MILI and PIWIL4/MIWI2 [39–42]. These three PIWI proteins are primarily expressed in male germ cells (MILI is also detectable in oocytes [43–45]) and show sequential and overlapping expression patterns during spermatogenic differentiation. MILI is first expressed in male foetal germ cells at around the time of sex differentiation and then continues its expression in prospermatogonia, postnatal spermatogonia, pachytene speramtocytes and early round spermatids (MILI is not detectable in leptotene-zygotene spermatocytes; S. Chuma 2012, unpublished data). MIWI2 is expressed in prospermatogonia but shows a narrow expression window and is diminished a few days after birth. In contrast to MILI and MIWI2, MIWI expression starts postnatally and is expressed in pachytene spermatocytes and later including round to elongating spermatids. How such differential expression of the piwi family members is regulated is currently unknown.
In mammals, piRNAs are classified mainly into three categories (foetal/prenatal, postnatal prepachytene and pachytene piRNAs), according to the developmental stages of their expression. In prospermatogonia, MILI and MIWI2 are loaded with foetal piRNAs, whose biogenesis is closely linked to epigenetic control over retrotransposons (see below), while MILI alone is expressed in postnatal spermatogonia, wherein piRNAs (postnatal prepachytene piRNAs) are less abundant and less well characterized. Then during meiosis, the amount of piRNAs highly increases at the pachytene stage of spermatocytes and also in post-meiotic round spermatids [8–11,44,46,47]. Deep sequencing studies revealed that piRNAs, which are very complex in their sequence profiles when compared with miRNAs (millions of piRNAs versus a few hundreds or thousands of miRNAs), drastically change their sequences during development and the three piRNA groups are quite distinct. While pachytene piRNAs are enriched with inter-genic, non-annotated sequences with relatively low abundance of transposon-derived sequences (up to 20%), pre-pachytene piRNAs expressed in postnatal spermatogonia are more abundant with transposon-derived sequences (−40%) as well as genic sequences (−20%). Foetal piRNAs also contain 40–50% of transposon sequences but with different composition compared with those in postnatal spermatogonia and with less exonic sequences (−3%). Genome mapping of such piRNA sequences revealed that piRNAs mostly originate from distinct genome clusters, termed piRNA clusters [8–11,44,46,47], which are a few to hundreds of kb in length, with piRNA clusters of each developmental stage showing little overlap and being derived from different chromosomal locations. How such piRNA clusters are defined is currently unclear, but given piRNA sequences per se are not well conserved among different species and, rather, the cluster synteny is surprisingly conserved, some chromatin context(s) is most probably involved.
Another characteristic feature of piRNA clusters is strand asymmetry; that is, most piRNAs map to only one genome strand of each cluster or a segment of it, suggesting that long single-stranded RNAs transcribed from the piRNA clusters are precursors of piRNAs. Such precursor transcripts are then probably processed by (an) unknown nuclease(s) to produce 5′ ends with a phosphate group, which are subsequently loaded onto PIWI proteins. Then, 3′ sequences are cleaved again by (an) unidentified nuclease producing an OH end (and 2′-O-methylation) with the size of piRNAs being determined by the footprint of each PIWI protein (average 26 nucleotides for MILI, 28 nucleotides for MIWI2 and 29–30 nucleotides for MIWI) [7,48]. This mechanism of piRNA biogenesis is called the primary processing of piRNAs (figure 1). Whether the primary piRNA biogenesis has any selectivity to preferentially recognize retrotransposon transcripts is not well understood. In addition to the primary pathway, there operates another, secondary mechanism to amplify piRNAs, which is important for retrotransposon control. When piRNA populations are examined for their sequence complementarity, a significant proportion of them clearly show a 5′ overlap of precisely 10 nucleotides, with one strand enriched for uridine at the 5′ end and the other complementary strand enriched for adenine at position 10 [63]. This characteristic signature is explained by a proposed secondary biogenesis pathway of piRNAs, termed the ping-pong or feed-forward amplification cycle. In this pathway, PIWI proteins loaded with primary piRNAs first recognize complementary target RNAs, and then using the slicer activity of the PIWI domain, the target RNAs are cleaved at the nucleotide position complementary to the 10th nucleotide from the 5′ end of the primary piRNA, producing secondary piRNA precursors. The 3′ end of secondary piRNA precursors is then processed probably by the same mechanism as that for primary piRNAs. Such sequence-complementarity-dependent recognition and cleavage form a cycle that amplifies piRNAs derived from transcripts having complementary counterparts in the cellular transcriptome, and thus this mechanism preferentially targets genome-repetitive sequences, including retrotransposons [7,63] (figure 1).
Figure 1.
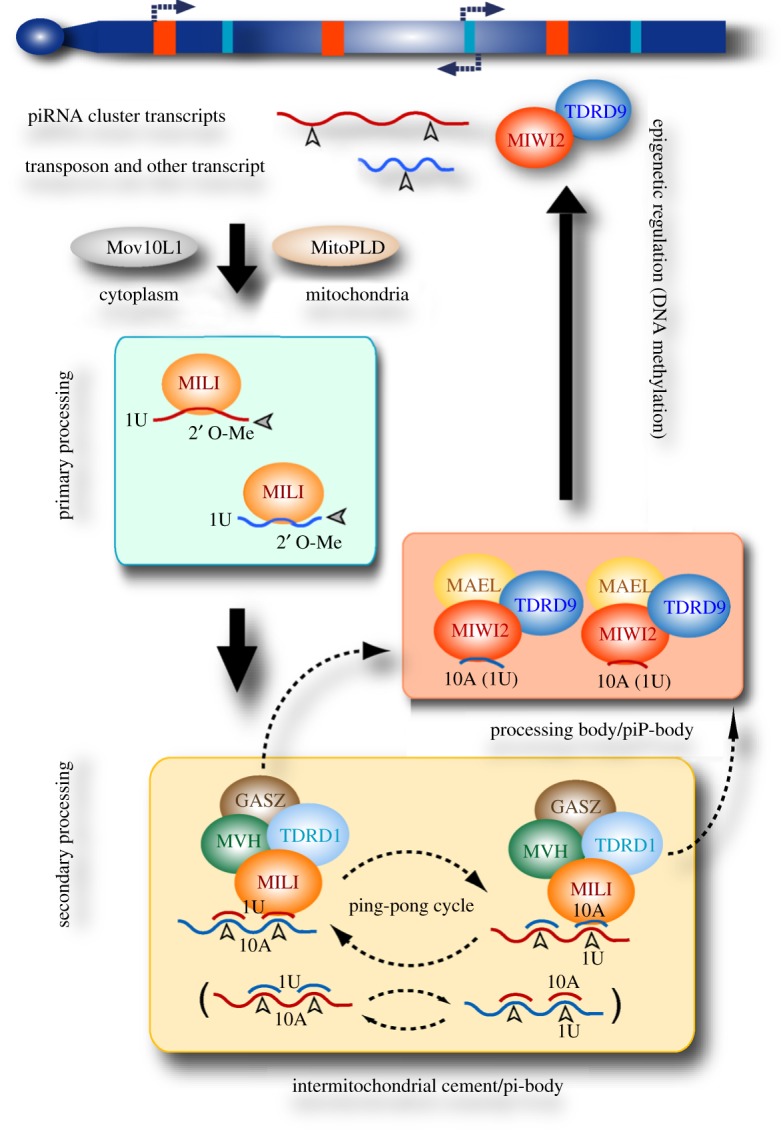
A schematic showing piRNA biogenesis in foetal prospermatogonia in mice. piRNA precursors transcribed from piRNA clusters (red) and from transposon and other (genic, etc.) loci (blue) are 5′ processed by an unknown nuclease with a preference for uridine at the 5′ end (1U) and loaded onto MILI. The 3′ end is trimmed by again an unknown nuclease and modified by HEN1 to produce 2′-O-methylation. The primary piRNAs guide MILI to their complementary target RNAs and the slicer activity of MILI cleaves the secondary piRNA precursors with a 5′ overlap of 10 nucleotides (the 1U bias of primary piRNAs leads to a preference for adenine at position 10 (10A) of secondary piRNAs). MIWI2, on the other hand, does not have a slicer activity (as suggested by a genetic study, see [49]), but is loaded with secondary piRNAs (as well as primary piRNAs) and most likely guides DNA methylation of retrotransposon loci. For details, see the main text and table 1.
Such a piRNA amplification loop should effectively contribute to reducing the transcript level of retrotransposon RNAs. However, the piwi pathway not only operates at the RNA level post-transcriptionally, but also exerts an intriguing control over epigenetic regulation. In the absence of piwi genes in mice (Mili2 and Miwi2), foetal piRNA biogenesis and/or sequence profiles are severely disrupted, and de novo genome CpG methylation of cognate retrotranspon loci in (pro)spermatogonia is not properly established [42,44,46,47]. This hypomethylation is thought to cause an epigenetic situation that later triggers a transcriptional activation of retrotransposons in postnatal spermatocytes, wherein gross cell death occurs. These phenotypes, retrotransposon demethylation and meiotic catastrophe, are reminiscent of those seen in Dnmt3L mutants [36], suggesting that DNA demethylation alone could be the major cause of Mili2 and Miwi2 mutant phenotypes. A possible molecular link between the piwi pathway and de novo DNA methylation is not yet understood. However, because DNA hypomethylation in the piwi mutants is selective for retrotransposons, there should be some mechanism(s) of target discrimination, most likely via piRNAs recognizing homologous genome DNA sequences or nascent RNA transcripts, as is suggested for RNA-induced transcriptional silencing in plants and yeasts [64].
5. Other piwi pathway components
Piwi proteins and piRNAs are the core effector components of the piwi–small RNA machinery, but they do not act alone. Instead, they form larger ribonucleoprotein (RNP) complexes with other functional components (table 1). Tudor family proteins were the first to be identified to interact with Piwi proteins in mice [13–15,51,52,54,65–68]. There are about 30 genes that encode a tudor domain(s) in mammalian genomes, and a class of tudor domain containing (Tdrd) proteins, including TDRD1 and TDRD9, act in the piwi pathway. Tudor domains in general recognize arginine dimethylation of target proteins [69], and recent studies identified that the N termini of mouse Piwi proteins are arginine dimethylated and are indeed recognized by the tudor domains of the TDRD proteins [15]. More specifically, MILI binds to TDRD1, MIWI2 makes a complex with TDRD9, MIWI complexes with TDRD6 and so on. Among these, Tdrd1 and Tdrd9 mutations exhibit LINE1 activation during spermatogenesis with piRNA profiles being significantly altered. IAP activation is not detectable unlike Mili and Miwi2 mutations, the reason for which is currently unknown. In Tdrd1 and Tdrd9 mutants, de novo DNA methylation in (pro)spermatogonia is clearly reduced at LINE1 promoters, indicating that the two tudor domain proteins, which associate with the piwi proteins, are essential for LINE1 regulation at both the transcriptional and post-transcriptional level. Other Tdrd members, including Tdrd5 and Tdrd7, also show retrotransposon desilencing with Tdrd5 implicated in the piwi pathway, while Tdrd7 may act differently [53,70–72]. At the molecular level, TDRD proteins most likely function as scaffolds to assemble macromolecular complexes via their tudor domains, as well as other domains in each member. Evolutionarily, tudor family genes in other species including Drosophila tudor (the founding member of the tudor family), spn-E/homeless (a homologue of Tdrd9) and others (Tejas, Yb and Krimper, etc.), also act in the piwi pathway [73,74]. Tudor family genes are now emerging as key conserved components of the piwi–small RNA system that ensures the germline integrity of diverse animals.
Table1.
piRNA pathway components in mice.
| gene symbol | synonym | protein domain | protein localization | retroposon activation in mutant | piRNA biogenesis in mutanta | spermatogenesis phenotype in mutant | references |
|---|---|---|---|---|---|---|---|
| Piwil1 | Miwi | Piwi, Paz | chromatoid body | LINE1 | pachytene piRNA loss | spermatid | [40,50] |
| Piwil2 | Mili | Piwi, Paz | intermitochondrial cement, chromatoid body | LINE1, IAP | foetal piRNA loss | spermatocyte | [41,44,46,47,49] |
| Piwil4 | Miwi2 | Piwi, Paz | processing body, nucleus | LINE1, IAP | foetal piRNA affected | spermatocyte | [42,44,47,49] |
| Tdrd1 | Mtr-1 | Tudor, Mynd | intermitochondrial cement, chromatoid body | LINE1 | foetal piRNA affected | spermatocyte, spermatid | [13–15,51,52] |
| Tdrd5 | — | Tudor, Lotus | intermitochondrial cement, chromatoid body | LINE1 | NA | spermatid | [53] |
| Tdrd9 | — | Tudor, helicase | processing body, chromatoid body, nucleus | LINE1 | foetal piRNA affected | spermatocyte | [54] |
| Ddx4 | Mvh | helicase | intermitochondrial cement, chromatoid body | LINE1, IAP | foetal piRNA affected | spermatocyte | [12,55] |
| Mov10L1 | Champ, Csm | helicase | cytoplasmic diffuse | LINE1, IAP | perinatal piRNA loss | spermatocyte | [56,57] |
| Mael | — | high mobility group | processing body, chromatoid body | LINE1, IAP | transient foetal piRNA loss | spermatocyte | [58,59] |
| Asz1 | Gasz | ankyrin repeats, sterile alpha motif | intermitochondrial cement | LINE1, IAP | postnatal prepachytene piRNA affected | spermatocyte | [60] |
| Pld6 | Zuc/mitoPLD | phospholipase D | mitochondria | LINE1 | foetal piRNA affected | spermatocyte | [61,62] |
apiRNA phenotypes are roughly classified into ‘loss’ (severe reduction or absence) and ‘affected’ (reduced in amount and/or sequence profile altered). For Mov10L1 and Asz1 mutants, piRNA sequence data were obtained from postnatal day 0 and 7 testes, respectively. For details, see references. NA, data not available.
Another intriguing and conserved component of the piwi pathway is vasa (mouse vasa homologue (Mvh)/Ddx4 in mice [55,75]). Vasa genes are evolutionarily conserved and have long been used as a specific marker of germ cells of a wide variety of animals. Vasa proteins have an RNA helicase domain and have been implicated in RNA metabolism, but the detailed molecular function remained unclear. Recently, studies on vasa mutants of mice and Drosophila revealed that vasa primarily acts in the piwi pathway [12,76]. In mice, Mvh/Ddx4 mutation leads to clear upregulation of LINE1 and IAP expression with spermatogenesis being blocked during meiosis of spermatocytes. In the mutant, foetal piRNAs loaded onto MIWI2 are abolished and cognate DNA methylation is impaired, which clearly identified vasa as an essential factor of the piwi pathway. In the developmental biology field, vasa and tudor have long been considered as key developmental genes essential for the germline specification (Drosophila) and differentiation (mice) [51,55,77–79]. However, it now has turned out that these genes, as well as a group of other genes (including piwi) that have been implicated in the germline development, actually participate in transposon control through the piwi–small RNA pathway, illustrating the importance and intimate integration of transposon control in germ cell development.
Mov10L1 (a homologue of Drosophila Armitage) is another RNA helicase required for the primary biogenesis and/or loading of piRNAs and for suppression of LINE1 and IAP in spermatogenesis [56,57]. Together with Mvh/Ddx4 and Tdrd9 (which also has a helicase domain) [12,54], these RNA helicases are evolutionarily conserved and are essential for the operation of the piwi pathway.
Other components of the piwi pathway identified so far include Maelstrom [58,80] and GASZ/ASZ1 [60]. Maelstrom has a HMG-box and is necessary for LINE1 and IAP suppression, with its loss of function causing a defect in the piRNA profile and transient hypomethylation of LINE1 in (pro)spermatogonia, of which the underlying mechanism is unclear. Gasz/Asz1 encodes Ankyrin repeats and a SAM domain, and acts to suppress LINE1 and IAP, possibly through the stabilization of MILI and other piwi pathway factors.
Another recently identified, intriguing component of the piwi pathway is Mitopld/Zucchini/PLD6, a member of the phospholipase D family, which hydrolyses phospholipids and mediates lipid signalling [61,62]. Mitopld/Zucchini has also been assumed to be a putative nuclease responsible for piRNA processing, but its nuclease activity has not been established, while a lipase activity was biochemically demonstrated [81]. Mitopld/Zucchini mutations result in LINE1 derepression with cognate DNA hypomethylation and a severe defect in primary piRNA biogenesis. Interestingly, the Mitopld/Zucchini protein is located at the outer membrane of mitochondria, and its overexpression (in somatic cells) facilitates mitochondrial fusion through lipid signalling [81], while its loss-of-function disrupts the distribution of mitochondria and delocalizes piwi pathway components such as MILI and TDRD1. Mitopld/Zucchini provides an important clue that links mitochondrial membrane regulation and/or lipid signalling to the piwi pathway.
6. Spermatogenesis and the piwi pathway in mice
The piwi pathway primarily functions in the male germline in mice, and all loss of function mutations of the piwi pathway genes reported to date lead to spermatogenesis defects. The phenotypes are mainly manifested at two distinct stages of spermatogenic differentiation, during meiosis of spermatocytes (Mili/Piwil2, Miwi2/Piwil4, Tdrd1, Tdrd9, Mvh/Ddx4, Mov10L1, Maelstrom, Gasz/Asz1, Zucchini/Mitopld/Pld6) [12–15,41,42,46,47,49–52,54,56–58,60–62,80] and during later spermiogenesis of haploid spermatids (Miwi/Piwil1, Tdrd1, Tdrd5; Tdrd1 mutants show both spermatocyte and spermatid defects) [40,51,53].
In the former group of mutants, spermatocytes are arrested at around the zygotene stage, showing a synapsis failure of homologous chromosomes with increased DNA double-strand breaks (DSBs), of which the simplest explanation would be that elevated activities of retrotransposons cause genome-wide DNA damage. Such DSBs are likely to be independent of the endogenous meiotic recombination programme, as was exemplified by the persistent DSBs observed in Maelstrom mutant spermatocytes in the absence of Spo11, a meiosis specific topoisomerase [58]. Remarkably, while the developmental defect of these mutants is evident during meiosis of spermatocytes, distinct molecular changes are seen at much earlier stages in foetal (pro)spermatogonia during embryonic development. In mutant (pro)spermatogonia, in which no detectable defect is observed at the cellular level, the biogenesis of piRNAs per se or their sequence profiles are clearly impaired with the expression of retrotransposons being upregulated. Further, de novo CpG methylation of retrotransposon loci, which is normally established in (pro)spermatogonia after the genome-wide demethylation in PGCs [23], is also defective in the piwi pathway mutants that show postnatal meiotic catastrophe. This epigenetic status in (pro)spermatogonia probably explains the delayed cellular phenotype. CpG hypomethylation at retrotranspson loci should be transmitted to postnatal spermatogonial stem cells and then to meiotic spermatocytes, wherein retrotransposons, especially LINE1, are transcriptionally activated to a moderate permissive level in the wild-type, while in the piwi pathway mutants, which lack repressive methylation marks, much higher activation occurs to a detrimental level leading to gross cell death. Another compelling possibility is that an unusual chromatin conformation caused by hypomethylation of retrotransposon loci leads to non-allelic homologous recombination or aberrant chromosome condensation, which should then trigger checkpoint activation.
Compared with the ‘early’ meiotic phenotype, the later spermatid defect of piwi pathway mutants (Miwi, Tdrd1, Tdrd5) is less well characterized [40,51,53]. Because pachytene piRNAs expressed in pachytene spermatocytes and later spermatids are not enriched with transposon derived sequences, the piwi system has not been thought to function in transposon control post-meiotically. However, a recent study revealed that LINE1 retrotransposon is actually activated in Miwi mutants, and that Miwi slicer activity cleaves LINE1 transcripts to reduce their abundance and to produce repeat derived piRNAs [50]. This action of MIWI however does not affect DNA methylation of cognate LINE1 loci and thus is likely post-transcriptional. Another post-meiotic defect is seen in Tdrd5 mutants. However, although the Tdrd5 loss-of-function mutant shows a phenotype in round spermatids reminiscent of the Miwi mutation, it also brings about earlier molecular changes in foetal (pro)spermatogonia, with LINE1 expression being upregulated, genome DNA hypomethylated at LINE1 loci and MIWI2 and TDRD9, which act in the secondary pathway, delocalized. One explanation for such a long lag period between the molecular and cellular phenotypes is that the extent of retrotransposon desilencing in this mutant may be relatively moderate or that Tdrd5 may have independent functions in foetal (pro)spermatogonia and postnatal spermatids.
Together, genetic studies have unveiled that the piwi pathway is essential for male fertility in mice with each component acting at several distinct stages of spermatogenesis and having non-redundant functions. In the female, by contrast, although several piwi pathway components, such as Mili, Tdrd1 and Tdrd9, as well as piRNAs are expressed in oocytes [43–45,51,54,82], there has been no report of female sterility or oocyte degeneration in such piwi pathway mutants. This may be because endogenous siRNAs and the canonical RNAi pathway act to suppress retrotransposons in oocytes [45,82]. Alternatively, oocytes of the piwi pathway mutants are indeed affected, but the effect may be below the threshold of fatal oocyte damage. Supporting the latter notion, Mili mutant oocytes show a moderate increase in retrotransposon expression [45], but they are still functional and fertilizable. Whether the increased retrotransposon expression causes novel insertions in oocytes and in subsequent offspring awaits future investigations.
7. Experimental systems
Our current understanding of the piwi pathway has mainly come from a combination of genetic studies of mutant animals and deep sequencing profiling of piRNAs therein. Such studies unambiguously uncovered a novel paradigm of transposon regulation by this small RNA system in the germline. However, to further elucidate the detailed molecular processes, cell culture models, if available, would greatly assist easier experimental manipulation. In several insect species, gonadal somatic cells express a Piwi protein(s) and produce piRNAs, and several cell lines established from insect gonads have been successfully used to analyse the piwi pathway. For example, a Drosophila ovarian somatic cell line, OSC, was established from follicle cells and expresses the Piwi protein (but not Aub and Ago3) [83]. In this cell line, piRNAs are mainly derived from primary loading onto the Piwi protein and the secondary ping-pong signature is negligible. Such a cell line is useful for characterizing piRNAs from a pure cell population and allows for easy access to RNAi and mutagenesis experiments. Another insect cell line, BmN4, a Bombyx mori (silkworm) ovary derived cell line, expresses two Piwi proteins, Siwi and BmAgo3, which as expected act in the ping-pong amplification of piRNAs [84]. Cellular extracts from this cell line have been effectively used to load the Piwi proteins with synthetic RNA substrates to examine 5′ nucleotide preference and 3′ end processing, etc [85].
In mammals, germline stem (GS) cells are a good candidate cell line for piRNA study. GS cells are established from spermatogonial stem cells and can expand in culture in the presence of growth factors such as glial cell line-derived neurotrophic factor and basic fibroblast growth factor [86]. Remarkably, GS cells produce functional sperm when transplanted back into testes (seminiferous tubules) of recipient mice and normal live offspring can be derived. In this sense, GS cells authentically function as GS cells. In GS cells, Mili is expressed and primary biogenesis of piRNAs operates (T. Nakano 2012, unpublished data), which corresponds to the piwi system in postnatal spermatogonia in vivo, in which MIWI2 has been turned off and before the onset of MIWI expression. This cell line should provide a good resource to study the primary processing of mammalian piRNAs, the identification of novel piwi pathway components and their biochemistry. Whether GS cells can be reconstituted with the foetal piRNA pathway by expressing Miwi2, etc. will be worthy of further investigation.
8. Perspective
The battle between host genomes and transposable elements over long evolutionary history is still ongoing. While piRNAs should have been recognized for years (for instance, pachytene piRNAs in mammalian testes are very abundant), it was not until recently that we became aware that such small RNA species are at the core of the genome defence system against transposons in the germline. Still, much remains to be learned about this ancient small RNA pathway. One key issue is how or whether retrotransposon transcripts are preferentially incorporated into the piwi pathway. The feed-forward/ping-pong amplification mechanism is beautiful to capture those transcripts that have both sense and anti-sense complementary copies, but the selection mechanism of the primary loading of the cellular transcriptome, if any, onto the piwi machinery is not well explained. In addition, how genome piRNA clusters are defined, such as their transcriptional regulation, developmental control and syntenic conservation, is not understood. Another unsolved issue is sub-cellular RNP assembly of piwi pathway components. The piwi pathway seems to be closely associated with germinal granules/nuage, a cytoplasmic RNP compartment characteristically observed in the germline [87]. Germinal granules/nuage have long been implicated in the germline specification in early embryos of several model animals, such as Drosophila and C. elegans [16–18], but in mammals the structure is only assembled during later differentiation stages of germ cells [87,88]. Recent studies found that many piwi pathway proteins are enriched at germinal granules/nuage or at another relevant RNP structure, the processing body [54,59,71,89,90]. It remains elusive how such subcellular localization contributes to the piwi pathway function and whether there is any crosstalk between piwi factors and other components of germinal granules/nuage.
Last, the epigenetic link between the piwi pathway and cognate genome CpG methylation is quite important, given that very little is known about the physiological function of endogenous small RNAs in epigenetic regulation in mammals, unlike plants and yeasts [64]. The piwi pathway in foetal (pro)spermatogonia provides a precious experimental model to study the mechanism by which small RNAs guide epigenetic control over specific genome loci. A recent study reported that DNA methylation of a paternally imprinted gene, Rasgrf1, is regulated under the control of the piwi pathway via a transcript of a neighbouring repeat element and piRNAs generated from a remote locus [91]. Retrotransposons are quite abundant in the genome and epigenetic control could exert a long distance effect in cis; therefore, the piwi pathway may have more widespread effects beyond retrotransposon silencing in the genome as well as in the cellular transcriptome.
Acknowledgements
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank all the members of our laboratories for their helpful discussions and advice. S.C. is also grateful to Norio Nakatsuji for his support and encouragement.
References
- 1.Gundry M, Vijg J. 2012. Direct mutation analysis by high-throughput sequencing: from germline to low-abundant, somatic variants. Mutat. Res. 729, 1–15 10.1016/j.mrfmmm.2011.10.001 (doi:10.1016/j.mrfmmm.2011.10.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2005. DNA repair and mutagenesis, 2nd edn. Washington, DC: ASM Press [Google Scholar]
- 3.Kazazian HH, Jr, 2004. Mobile elements: drivers of genome evolution. Science 303, 1626–1632 10.1126/science.1089670 (doi:10.1126/science.1089670) [DOI] [PubMed] [Google Scholar]
- 4.Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y. 2010. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464, 927–931 10.1038/nature08858 (doi:10.1038/nature08858) [DOI] [PubMed] [Google Scholar]
- 5.Rowe HM, et al. 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240 10.1038/nature08674 (doi:10.1038/nature08674) [DOI] [PubMed] [Google Scholar]
- 6.Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136, 642–655 10.1016/j.cell.2009.01.035 (doi:10.1016/j.cell.2009.01.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aravin AA, Hannon GJ, Brennecke J. 2007. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 318, 761–764 10.1126/science.1146484 (doi:10.1126/science.1146484) [DOI] [PubMed] [Google Scholar]
- 8.Aravin A, et al. 2006. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203–207 10.1038/nature04916 (doi:10.1038/nature04916) [DOI] [PubMed] [Google Scholar]
- 9.Girard A, Sachidanandam R, Hannon GJ, Carmell MA. 2006. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442, 199–202 10.1038/nature04917 (doi:10.1038/nature04917) [DOI] [PubMed] [Google Scholar]
- 10.Grivna ST, Beyret E, Wang Z, Lin H. 2006. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 20, 1709–1714 10.1101/gad.1434406 (doi:10.1101/gad.1434406) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, Minami N, Imai H. 2006. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev. 20, 1732–1743 10.1101/gad.1425706 (doi:10.1101/gad.1425706) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuramochi-Miyagawa S, et al. 2010. MVH in piRNA processing and gene silencing of retrotransposons. Genes Dev. 24, 887–892 10.1101/gad.1902110 (doi:10.1101/gad.1902110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Saxe JP, Tanaka T, Chuma S, Lin H. 2009. Mili interacts with tudor domain-containing protein 1 in regulating spermatogenesis. Curr. Biol. 19, 640–644 10.1016/j.cub.2009.02.061 (doi:10.1016/j.cub.2009.02.061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reuter M, Chuma S, Tanaka T, Franz T, Stark A, Pillai RS. 2009. Loss of the Mili-interacting Tudor domain-containing protein-1 activates transposons and alters the Mili-associated small RNA profile. Nat. Struct. Mol. Biol. 16, 639–646 10.1038/nsmb.1615 (doi:10.1038/nsmb.1615) [DOI] [PubMed] [Google Scholar]
- 15.Vagin VV, et al. 2009. Proteomic analysis of murine Piwi proteins reveals a role for arginine methylation in specifying interaction with Tudor family members. Genes Dev. 23, 1749–1762 10.1101/gad.1814809 (doi:10.1101/gad.1814809) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saffman EE, Lasko P. 1999. Germline development in vertebrates and invertebrates. Cell Mol. Life Sci. 55, 1141–1163 10.1007/s000180050363 (doi:10.1007/s000180050363) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seydoux G, Braun RE. 2006. Pathway to totipotency: lessons from germ cells. Cell 127, 891–904 10.1016/j.cell.2006.11.016 (doi:10.1016/j.cell.2006.11.016) [DOI] [PubMed] [Google Scholar]
- 18.Strome S, Lehmann R. 2007. Germ versus soma decisions: lessons from flies and worms. Science 316, 392–393 10.1126/science.1140846 (doi:10.1126/science.1140846) [DOI] [PubMed] [Google Scholar]
- 19.McLaren A. 2003. Primordial germ cells in the mouse. Dev. Biol. 262, 1–15 10.1016/S0012-1606(03)00214-8 (doi:10.1016/S0012-1606(03)00214-8) [DOI] [PubMed] [Google Scholar]
- 20.Yoshida S. 2010. Stem cells in mammalian spermatogenesis. Dev. Growth Diff. 52, 311–317 10.1111/j.1440-169X.2010.01174.x (doi:10.1111/j.1440-169X.2010.01174.x) [DOI] [PubMed] [Google Scholar]
- 21.Handel MA, Eppig JJ. 1998. Sexual dimorphism in the regulation of mammalian meiosis. Curr. Top. Dev. Biol. 37, 333–358 10.1016/S0070-2153(08)60179-9 (doi:10.1016/S0070-2153(08)60179-9) [DOI] [PubMed] [Google Scholar]
- 22.Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. 2002. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 117, 15–23 10.1016/S0925-4773(02)00181-8 (doi:10.1016/S0925-4773(02)00181-8) [DOI] [PubMed] [Google Scholar]
- 23.Schaefer CB, Ooi SK, Bestor TH, Bourc'his D. 2007. Epigenetic decisions in mammalian germ cells. Science 316, 398–399 10.1126/science.1137544 (doi:10.1126/science.1137544) [DOI] [PubMed] [Google Scholar]
- 24.Sasaki H, Matsui Y. 2008. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat. Rev. Genet. 9, 129–140 10.1038/nrg2295 (doi:10.1038/nrg2295) [DOI] [PubMed] [Google Scholar]
- 25.Trelogan SA, Martin SL. 1995. Tightly regulated, developmentally specific expression of the first open reading frame from LINE-1 during mouse embryogenesis. Proc. Natl Acad. Sci. USA 92, 1520–1524 10.1073/pnas.92.5.1520 (doi:10.1073/pnas.92.5.1520) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lander ES, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409, 860–921 10.1038/35057062 (doi:10.1038/35057062) [DOI] [PubMed] [Google Scholar]
- 27.Waterston RH, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 10.1038/nature01262 (doi:10.1038/nature01262) [DOI] [PubMed] [Google Scholar]
- 28.Feschotte C, Pritham EJ. 2007. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 41, 331–368 10.1146/annurev.genet.40.110405.090448 (doi:10.1146/annurev.genet.40.110405.090448) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray DA, Feschotte C, Pagan HJ, Smith JD, Pritham EJ, Arensburger P, Atkinson PW, Craig NL. 2008. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res. 18, 717–728 10.1101/gr.071886.107 (doi:10.1101/gr.071886.107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jurka J, Kapitonov VV, Kohany O, Jurka MV. 2007. Repetitive sequences in complex genomes: structure and evolution. Annu. Rev. Genomics Hum. Genet. 8, 241–259 10.1146/annurev.genom.8.080706.092416 (doi:10.1146/annurev.genom.8.080706.092416) [DOI] [PubMed] [Google Scholar]
- 31.Deininger PL, Moran JV, Batzer MA, Kazazian HH., Jr 2003. Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 13, 651–658 10.1016/j.gde.2003.10.013 (doi:10.1016/j.gde.2003.10.013) [DOI] [PubMed] [Google Scholar]
- 32.Kramerov DA, Vassetzky NS. 2005. Short retroposons in eukaryotic genomes. Int. Rev. Cytol. 247, 165–221 10.1016/S0074-7696(05)47004-7 (doi:10.1016/S0074-7696(05)47004-7) [DOI] [PubMed] [Google Scholar]
- 33.Farkash EA, Luning Prak ET. 2006. DNA damage and L1 retrotransposition. J. Biomed. Biotechnol. 2006, 37285. 10.1155/JBB/2006/37285 (doi:10.1155/JBB/2006/37285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li E, Bestor TH, Jaenisch R. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 10.1016/0092-8674(92)90611-F (doi:10.1016/0092-8674(92)90611-F) [DOI] [PubMed] [Google Scholar]
- 35.Walsh CP, Chaillet JR, Bestor TH. 1998. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20, 116–117 10.1038/2413 (doi:10.1038/2413) [DOI] [PubMed] [Google Scholar]
- 36.Bourc'his D, Bestor TH. 2004. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431, 96–99 10.1038/nature02886 (doi:10.1038/nature02886) [DOI] [PubMed] [Google Scholar]
- 37.Hutvagner G, Simard MJ. 2008. Argonaute proteins: key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 9, 22–32 10.1038/nrm2321 (doi:10.1038/nrm2321) [DOI] [PubMed] [Google Scholar]
- 38.Tolia NH, Joshua-Tor L. 2007. Slicer and the argonautes. Nat. Chem. Biol. 3, 36–43 10.1038/nchembio848 (doi:10.1038/nchembio848) [DOI] [PubMed] [Google Scholar]
- 39.Cox DN, Chao A, Baker J, Chang L, Qiao D, Lin H. 1998. A novel class of evolutionarily conserved genes defined by piwi are essential for stem cell self-renewal. Genes Dev. 12, 3715–3727 10.1101/gad.12.23.3715 (doi:10.1101/gad.12.23.3715) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng W, Lin H. 2002. miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev. Cell 2, 819–830 10.1016/S1534-5807(02)00165-X (doi:10.1016/S1534-5807(02)00165-X) [DOI] [PubMed] [Google Scholar]
- 41.Kuramochi-Miyagawa S, et al. 2004. Mili, a mammalian member of piwi family gene, is essential for spermatogenesis. Development 131, 839–849 10.1242/dev.00973 (doi:10.1242/dev.00973) [DOI] [PubMed] [Google Scholar]
- 42.Carmell MA, Girard A, van de Kant HJ, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ. 2007. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev. Cell 12, 503–514 10.1016/j.devcel.2007.03.001 (doi:10.1016/j.devcel.2007.03.001) [DOI] [PubMed] [Google Scholar]
- 43.Kuramochi-Miyagawa S, Kimura T, Yomogida K, Kuroiwa A, Tadokoro Y, Fujita Y, Sato M, Matsuda Y, Nakano T. 2001. Two mouse piwi-related genes: miwi and mili. Mech. Dev. 108, 121–133 10.1016/S0925-4773(01)00499-3 (doi:10.1016/S0925-4773(01)00499-3) [DOI] [PubMed] [Google Scholar]
- 44.Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. 2008. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 31, 785–799 10.1016/j.molcel.2008.09.003 (doi:10.1016/j.molcel.2008.09.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe T, et al. 2008. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453, 539–543 10.1038/nature06908 (doi:10.1038/nature06908) [DOI] [PubMed] [Google Scholar]
- 46.Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. 2007. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316, 744–747 10.1126/science.1142612 (doi:10.1126/science.1142612) [DOI] [PubMed] [Google Scholar]
- 47.Kuramochi-Miyagawa S, et al. 2008. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 22, 908–917 10.1101/gad.1640708 (doi:10.1101/gad.1640708) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siomi MC, Sato K, Pezic D, Aravin AA. 2011. PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 12, 246–258 10.1038/nrm3089 (doi:10.1038/nrm3089) [DOI] [PubMed] [Google Scholar]
- 49.De Fazio S, et al. 2011. The endonuclease activity of Mili fuels piRNA amplification that silences LINE1 elements. Nature 480, 259–263 10.1038/nature10547 (doi:10.1038/nature10547) [DOI] [PubMed] [Google Scholar]
- 50.Reuter M, Berninger P, Chuma S, Shah H, Hosokawa M, Funaya C, Antony C, Sachidanandam R, Pillai RS. 2011. Miwi catalysis is required for piRNA amplification-independent LINE1 transposon silencing. Nature 480, 264–267 10.1038/nature10672 (doi:10.1038/nature10672) [DOI] [PubMed] [Google Scholar]
- 51.Chuma S, Hosokawa M, Kitamura K, Kasai S, Fujioka M, Hiyoshi M, Takamune K, Noce T, Nakatsuji N. 2006. Tdrd1/Mtr-1, a tudor-related gene, is essential for male germ-cell differentiation and nuage/germinal granule formation in mice. Proc. Natl Acad. Sci. USA 103, 15 894–15 899 10.1073/pnas.0601878103 (doi:10.1073/pnas.0601878103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kojima K, Kuramochi-Miyagawa S, Chuma S, Tanaka T, Nakatsuji N, Kimura T, Nakano T. 2009. Associations between PIWI proteins and TDRD1/MTR-1 are critical for integrated subcellular localization in murine male germ cells. Genes Cells 14, 1155–1165 10.1111/j.1365-2443.2009.01342.x (doi:10.1111/j.1365-2443.2009.01342.x) [DOI] [PubMed] [Google Scholar]
- 53.Yabuta Y, Ohta H, Abe T, Kurimoto K, Chuma S, Saitou M. 2011. TDRD5 is required for retrotransposon silencing, chromatoid body assembly and spermiogenesis in mice. J. Cell Biol. 192, 781–795 10.1083/jcb.201009043 (doi:10.1083/jcb.201009043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shoji M, et al. 2009. The TDRD9-MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev. Cell 17, 775–787 10.1016/j.devcel.2009.10.012 (doi:10.1016/j.devcel.2009.10.012) [DOI] [PubMed] [Google Scholar]
- 55.Tanaka SS, Toyooka Y, Akasu R, Katoh-Fukui Y, Nakahara Y, Suzuki R, Yokoyama M, Noce T. 2000. The mouse homolog of Drosophila Vasa is required for the development of male germ cells. Genes Dev. 14, 841–853 [PMC free article] [PubMed] [Google Scholar]
- 56.Frost RJ, Hamra FK, Richardson JA, Qi X, Bassel-Duby R, Olson EN. 2010. MOV10L1 is necessary for protection of spermatocytes against retrotransposons by Piwi-interacting RNAs. Proc. Natl Acad. Sci. USA 107, 11 847–11 852 10.1073/pnas.1007158107 (doi:10.1073/pnas.1007158107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng K, et al. 2010. Mouse MOV10L1 associates with Piwi proteins and is an essential component of the Piwi-interacting RNA (piRNA) pathway. Proc. Natl Acad. Sci. USA 107, 11 841–11 846 10.1073/pnas.1003953107 (doi:10.1073/pnas.1003953107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soper SF, van der Heijden GW, Hardiman TC, Goodheart M, Martin SL, de Boer P, Bortvin A. 2008. Mouse maelstrom, a component of nuage, is essential for spermatogenesis and transposon repression in meiosis. Dev. Cell 15, 285–297 10.1016/j.devcel.2008.05.015 (doi:10.1016/j.devcel.2008.05.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aravin AA, van der Heijden GW, Castaneda J, Vagin VV, Hannon GJ, Bortvin A. 2009. Cytoplasmic compartmentalization of the fetal piRNA pathway in mice. PLoS Genet. 5, e1000764. 10.1371/journal.pgen.1000764 (doi:10.1371/journal.pgen.1000764) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma L, et al. 2009. GASZ is essential for male meiosis and suppression of retrotransposon expression in the male germline. PLoS Genet. 5, e1000635. 10.1371/journal.pgen.1000635 (doi:10.1371/journal.pgen.1000635) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, Morris AJ, Frohman MA. 2011. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev. Cell 20, 376–387 10.1016/j.devcel.2011.01.004 (doi:10.1016/j.devcel.2011.01.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Watanabe T, et al. 2011. MITOPLD is a mitochondrial protein essential for nuage formation and piRNA biogenesis in the mouse germline. Dev. Cell 20, 364–375 10.1016/j.devcel.2011.01.005 (doi:10.1016/j.devcel.2011.01.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128, 1089–1103 10.1016/j.cell.2007.01.043 (doi:10.1016/j.cell.2007.01.043) [DOI] [PubMed] [Google Scholar]
- 64.Zhang X, Rossi JJ. 2011. Phylogenetic comparison of small RNA-triggered transcriptional gene silencing. J. Biol. Chem. 286, 29 443–29 448 10.1074/jbc.R111.276378 (doi:10.1074/jbc.R111.276378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chuma S, Hiyoshi M, Yamamoto A, Hosokawa M, Takamune K, Nakatsuji N. 2003. Mouse Tudor Repeat-1 (MTR-1) is a novel component of chromatoid bodies/nuages in male germ cells and forms a complex with snRNPs. Mech. Dev. 120, 979–990 10.1016/S0925-4773(03)00181-3 (doi:10.1016/S0925-4773(03)00181-3) [DOI] [PubMed] [Google Scholar]
- 66.Hosokawa M, Shoji M, Kitamura K, Tanaka T, Noce T, Chuma S, Nakatsuji N. 2007. Tudor-related proteins TDRD1/MTR-1, TDRD6 and TDRD7/TRAP: domain composition, intracellular localization and function in male germ cells in mice. Dev. Biol. 301, 38–52 10.1016/j.ydbio.2006.10.046 (doi:10.1016/j.ydbio.2006.10.046) [DOI] [PubMed] [Google Scholar]
- 67.Vasileva A, Tiedau D, Firooznia A, Muller-Reichert T, Jessberger R. 2009. Tdrd6 is required for spermiogenesis, chromatoid body architecture and regulation of miRNA expression. Curr. Biol. 19, 630–639 10.1016/j.cub.2009.02.047 (doi:10.1016/j.cub.2009.02.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen C, et al. 2009. Mouse Piwi interactome identifies binding mechanism of Tdrkh Tudor domain to arginine methylated Miwi. Proc. Natl Acad. Sci. USA 106, 20 336–20 341 10.1073/pnas.0911640106 (doi:10.1073/pnas.0911640106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen C, Nott TJ, Jin J, Pawson T. 2011. Deciphering arginine methylation: Tudor tells the tale. Nat. Rev. Mol. Cell Biol. 12, 629–642 10.1038/nrm3185 (doi:10.1038/nrm3185) [DOI] [PubMed] [Google Scholar]
- 70.Smith JM, Bowles J, Wilson M, Teasdale RD, Koopman P. 2004. Expression of the tudor-related gene Tdrd5 during development of the male germline in mice. Gene Expr. Patterns 4, 701–705 10.1016/j.modgep.2004.04.002 (doi:10.1016/j.modgep.2004.04.002) [DOI] [PubMed] [Google Scholar]
- 71.Tanaka T, et al. 2011. Tudor domain containing 7 (Tdrd7) is essential for dynamic ribonucleoprotein (RNP) remodeling of chromatoid bodies during spermatogenesis. Proc. Natl Acad. Sci. USA 108, 10 579–10 584 10.1073/pnas.1015447108 (doi:10.1073/pnas.1015447108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lachke SA, et al. 2011. Mutations in the RNA granule component TDRD7 cause cataract and glaucoma. Science 331, 1571–1576 10.1126/science.1195970 (doi:10.1126/science.1195970) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Handler D, Olivieri D, Novatchkova M, Gruber FS, Meixner K, Mechtler K, Stark A, Sachidanandam R, Brennecke J. 2011. A systematic analysis of Drosophila TUDOR domain-containing proteins identifies Vreteno and the Tdrd12 family as essential primary piRNA pathway factors. Embo J. 30, 3977–3993 10.1038/emboj.2011.308 (doi:10.1038/emboj.2011.308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siomi MC, Mannen T, Siomi H. 2010. How does the royal family of Tudor rule the PIWI-interacting RNA pathway? Genes Dev. 24, 636–646 10.1101/gad.1899210 (doi:10.1101/gad.1899210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fujiwara Y, Komiya T, Kawabata H, Sato M, Fujimoto H, Furusawa M, Noce T. 1994. Isolation of a DEAD-family protein gene that encodes a murine homolog of Drosophila vasa and its specific expression in germ cell lineage. Proc. Natl Acad. Sci. USA 91, 12 258–12 262 10.1073/pnas.91.25.12258 (doi:10.1073/pnas.91.25.12258) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ. 2009. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137, 522–535 10.1016/j.cell.2009.03.040 (doi:10.1016/j.cell.2009.03.040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boswell RE, Mahowald AP. 1985. tudor, a gene required for assembly of the germ plasm in Drosophila melanogaster. Cell 43, 97–104 10.1016/0092-8674(85)90015-7 (doi:10.1016/0092-8674(85)90015-7) [DOI] [PubMed] [Google Scholar]
- 78.Schupbach T, Wieschaus E. 1986. Germline autonomy of maternal-effect mutations altering the embryonic body pattern of Drosophila. Dev. Biol. 113, 443–448 10.1016/0012-1606(86)90179-X (doi:10.1016/0012-1606(86)90179-X) [DOI] [PubMed] [Google Scholar]
- 79.Ephrussi A, Lehmann R. 1992. Induction of germ cell formation by oskar. Nature 358, 387–392 10.1038/358387a0 (doi:10.1038/358387a0) [DOI] [PubMed] [Google Scholar]
- 80.Zhang D, Xiong H, Shan J, Xia X, Trudeau VL. 2008. Functional insight into Maelstrom in the germline piRNA pathway: a unique domain homologous to the DnaQ-H 3′-5′ exonuclease, its lineage-specific expansion/loss and evolutionarily active site switch. Biol. Direct. 3, 48. 10.1186/1745-6150-3-48 (doi:10.1186/1745-6150-3-48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. 2006. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 8, 1255–1262 10.1038/ncb1487 (doi:10.1038/ncb1487) [DOI] [PubMed] [Google Scholar]
- 82.Tam OH, et al. 2008. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 453, 534–538 10.1038/nature06904 (doi:10.1038/nature06904) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saito K, et al. 2009. A regulatory circuit for piwi by the large Maf gene traffic jam in Drosophila. Nature 461, 1296–1299 10.1038/nature08501 (doi:10.1038/nature08501) [DOI] [PubMed] [Google Scholar]
- 84.Kawaoka S, Hayashi N, Suzuki Y, Abe H, Sugano S, Tomari Y, Shimada T, Katsuma S. 2009. The Bombyx ovary-derived cell line endogenously expresses PIWI/PIWI-interacting RNA complexes. RNA 15, 1258–1264 10.1261/rna.1452209 (doi:10.1261/rna.1452209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kawaoka S, Izumi N, Katsuma S, Tomari Y. 2011. 3′ end formation of PIWI-interacting RNAs in vitro. Mol. Cell 43, 1015–1022 10.1016/j.molcel.2011.07.029 (doi:10.1016/j.molcel.2011.07.029) [DOI] [PubMed] [Google Scholar]
- 86.Kanatsu-Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S, Shinohara T. 2003. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol. Reprod. 69, 612–616 10.1095/biolreprod.103.017012 (doi:10.1095/biolreprod.103.017012) [DOI] [PubMed] [Google Scholar]
- 87.Eddy EM. 1975. Germ plasm and the differentiation of the germ cell line. Int. Rev. Cytol. 43, 229–280 10.1016/S0074-7696(08)60070-4 (doi:10.1016/S0074-7696(08)60070-4) [DOI] [PubMed] [Google Scholar]
- 88.Chuma S, Hosokawa M, Tanaka T, Nakatsuji N. 2009. Ultrastructural characterization of spermatogenesis and its evolutionary conservation in the germline: germinal granules in mammals. Mol. Cell Endocrinol. 306, 17–23 10.1016/j.mce.2008.11.009 (doi:10.1016/j.mce.2008.11.009) [DOI] [PubMed] [Google Scholar]
- 89.Kotaja N, Bhattacharyya SN, Jaskiewicz L, Kimmins S, Parvinen M, Filipowicz W, Sassone-Corsi P. 2006. The chromatoid body of male germ cells: similarity with processing bodies and presence of Dicer and microRNA pathway components. Proc. Natl Acad. Sci. USA 103, 2647–2652 10.1073/pnas.0509333103 (doi:10.1073/pnas.0509333103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Meikar O, Da Ros M, Liljenback H, Toppari J, Kotaja N. 2010. Accumulation of piRNAs in the chromatoid bodies purified by a novel isolation protocol. Exp. Cell Res. 316, 1567–1575 10.1016/j.yexcr.2010.02.023 (doi:10.1016/j.yexcr.2010.02.023) [DOI] [PubMed] [Google Scholar]
- 91.Watanabe T, et al. 2011. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 332, 848–852 10.1126/science.1203919 (doi:10.1126/science.1203919) [DOI] [PMC free article] [PubMed] [Google Scholar]