

Abstract
Nitric oxide (NO) plays an important role in acute ischemic preconditioning (IPC). In addition to activating soluble guanylyl cyclase (sGC)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) signaling pathways, NO-mediated protein S-nitros(yl)ation (SNO) has been recently shown to play an essential role in cardioprotection against ischemia–reperfusion (I/R) injury. In our previous studies, we have shown that IPC-induced cardioprotection could be blocked by treatment with either N-nitro-L-arginine methyl ester (L-NAME, a constitutive NO synthase inhibitor) or ascorbate (a reducing agent to decompose SNO). To clarify NO-mediated sGC/cGMP/PKG-dependent or -independent (i.e., SNO) signaling involved in IPC-induced cardioprotection, mouse hearts were Langendorff-perfused in the dark to prevent SNO decomposition by light exposure. Treatment with 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, a highly selective inhibitor of sGC) or KT5823 (a potent and selective inhibitor of PKG) did not abolish IPC-induced acute protection, suggesting that the sGC/cGMP/ PKG signaling pathway does not play an important role in NO-mediated cardioprotective signaling during acute IPC. In addition, treatment with ODQ in IPC hearts provided an additional protective effect on functional recovery, in parallel with a higher SNO level in these ODQ+IPC hearts. In conclusion, these results suggest that the protective effect of NO is not related primarily to activation of the sGC/ cGMP/PKG signaling pathway, but rather through SNO signaling in IPC-induced acute cardioprotection.
Keywords: Nitric oxide, Protein S-nitros(yl)ation, sGC/cGMP/PKG, Ischemic preconditioning
Introduction
Ischemic preconditioning (IPC) is a cellular adaptive phenomenon whereby brief episodes of myocardial ischemia and reperfusion render the heart resistant to subsequent prolonged ischemic injury [1]. Nitric oxide (NO) is an important signaling molecule that mediates IPC-induced acute cardioprotection [2,3]. Early studies have suggested that NO mediates acute IPC-induced protection through the classical soluble guanylyl cyclase (sGC)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) signaling pathways [4–7]. Lochner et al. found that 1H-[1,2,4]oxadiazolo[4,3–a]quinoxalin-1-one (ODQ), an inhibitor of sGC, blocked the IPC-mediated reduction in postischemic contractile dysfunction in a Langendorff-perfused rat heart ischemia–reperfusion (I/R) model [6]. However, in the absence of IPC, ODQ also caused enhanced postischemic contractile dysfunction following I/R, making it difficult to draw conclusions from these studies. Furthermore, the magnitude of the cGMP increase during preconditioning did not correlate with protection [6]. Using a Langendorff-perfused rabbit heart model, Qin et al. reported that IPC was not blocked by ODQ; however, IPC was also unaffected by an NO synthase (NOS) inhibitor, N-nitro-L-arginine methyl ester (L-NAME) [7]. Another in vivo study in enflurane-anesthetized swine hearts reported that IPC-mediated protection was also not affected by a NOS inhibitor [8]. Recently Sips et al. examined acute IPC in hearts from mice lacking the α1 isoform of sGC, the primary isoform expressed in heart. Loss of the α1 isoform of sGC did not block the infarct reduction afforded by IPC. However, they showed that ODQ addition blocked IPC protection in both wild-type and α1 isoform sGC null mice, suggesting that another isoform of sGC might be important for IPC [9]. In addition, sGC/cGMP/PKG has also been evaluated in delayed IPC and postconditioning. Kodani et al. showed that ODQ added on Day 1 did not block the protective effect of delayed preconditioning on Day 2 [10]. Cohen et al. showed that L-NAME but not ODQ blocked protection with postconditioning [11]. However, a recent study by Inserte et al. suggested that cGMP/PKG might contribute to postconditioning protection in part by delaying normalization of pH during reperfusion [12].
Taken together, it is unclear whether there is a requirement for NO-dependent sGC/cGMP/PKG signaling in acute IPC. Most recent studies have shown that IPC-induced cardioprotection is blocked by NOS inhibitors [6,13–16], and that mouse hearts lacking constitutive NOS are not protected by IPC [17,18], suggesting a role of NOS/NO signaling in acute IPC. In addition to activating sGC/cGMP/PKG signaling pathways, it has been shown that NO can directly modify protein sulfhydryl residues through protein S-nitros(yl)ation (SNO), which has emerged as an important posttranslational protein modification in cardiovascular signaling [19,20] and cardioprotection [21–23]. Our recent studies have demonstrated that protein SNO is important for cardioprotection against I/R [22,24]. SNO not only leads to changes in protein structure and function [16,25] but also protects those thiol(s) from further irreversible oxidative modification on reperfusion [21,26].
To further clarify the role of NO-mediated signaling pathways (i.e., protein S-nitros(yl)ation vs sGC/cGMP/PKG) during acute IPC, ODQ and KT5823 (an inhibitor of PKG) were tested in Langendorff-perfused mouse hearts. Interestingly, neither of these inhibitors blocked the protection provided by IPC. In addition, treatment with ODQ enhanced protection with IPC. Furthermore, IPC significantly increased protein SNO, while ODQ treatment prior to and during IPC caused a further increase of SNO. Therefore, NO-mediated protein S-nitros(yl)ation, rather than activation of sGC/cGMP/PKG signaling, appears to play a key role in IPC-induced acute cardioprotection.
Materials and methods
Animals
Male C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME) and were between 12 and 16 weeks old at the time of experimentation. All animals were treated in accordance with National Institutes of Health guidelines and the “Guiding Principles for Research Involving Animals and Human Beings.” This study was reviewed and approved by the Institutional Animal Care and Use Committee of the National Heart Lung and Blood Institute.
Compounds
ODQ (Sigma, O3636, St. Louis, MO) is a selective, irreversible, heme-site inhibitor of sGC, and the binding of ODQ is competitive with NO [27]. The inhibitory effect is time dependent with virtually complete inhibition at 10 min. Previous Langendorff-perfused heart studies used ODQ at doses in the range of 1–10 μM [9,11,28,29]. In this study, 10 μM ODQ was used to perfuse hearts, which is based on its inhibitory effect on SNAP (100 μM)-induced increase in coronary flow rate and cGMP production in the myocardium (please see Supplemental Data). KT5823 (Sigma, K1388) is a potent and selective inhibitor of PKG (IC50 ≅ 0.25 μM for PKG). We used 1 μM KT5823, a concentration that has been used previously in Langendorff-perfused rat hearts to inhibit PKG [29].
Langendorff heart perfusion and IPC-ischemia/reperfusion (I/R) protocol
Male C57BL/6J mice were anesthetized with pentobarbital and anticoagulated with heparin. Hearts were excised quickly and placed in ice-cold Krebs-Henseleit buffer (in mM: 120 NaCl, 11 D-glucose, 25 NaHCO3, 1.75 CaCl2, 4.7 KCl, 1.2 MgSO4, and 1.2 KH2PO4). The aorta was cannulated and the heart was perfused in retrograde fashion with Krebs-Henseleit buffer (oxygenated with 95% O2/5% CO2 and maintained at pH 7.4) at a constant pressure of 100 cm of water at 37 °C in the dark to prevent light-induced SNO decomposition. After equilibrium perfusion or IPC (4 cycles of 5 min of ischemia and 5 min of reperfusion), mouse hearts were subjected to 20 min of no-flow ischemia followed by 90 min of reperfusion. Drug administration is illustrated in the IPC-I/R protocol as shown in Fig. 1.
Fig. 1.
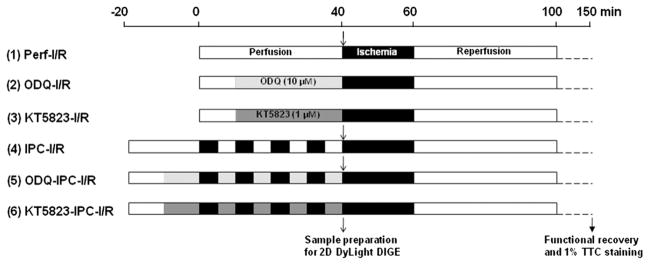
IPC-I/R protocol. Mouse hearts were Langendorff-perfused with Krebs-Henseleit buffer (oxygenated with 95% O2/5% CO2 and maintained at pH 7.4) at a constant pressure of 100 cm of water at 37 °C in the dark. After equilibrium perfusion or IPC (4 cycles of 5 min of ischemia and 5 min of reperfusion), mouse hearts were subjected to 20 min of no-flow ischemia followed by 90 min of reperfusion. Drug administration (10 μM ODQ or 1 μM KT5823) is illustrated for each IPC-I/R protocol.
Hemodynamic and infarct size measurements
To monitor left ventricular developed pressure (LVDP), a latex balloon connected to a pressure transducer was inserted into the left ventricle of Langendorff-perfused hearts. LVDP was recorded and digitized using a PowerLab system (ADInstruments, Colorado Springs, CO). We used the rate pressure product (RPP=LVDP × heart rate) as a measure of function. The postischemic functional recovery was expressed as percentage of the preischemic RPP during the equilibrium period. The time to onset of ischemic contracture was determined. For measurement of myocardial infarct size, hearts were perfused with 1% (w/v) of 2,3,5-triphe-nyltetrazolium chloride (TTC) after 90 min of reperfusion and incubated in TTC at 37 °C for 15 min, followed by fixation in 10% (w/v) formaldehyde. Infarct size was expressed as the percentage of total cross-sectional area of the ventricles.
Total heart homogenate preparation
To prevent SNO decomposition, sample preparations were also carried out in the dark. Total heart homogenate was obtained by grinding snap-frozen mouse heart into powder in liquid nitrogen followed by homogenization with a tight-fitting glass Dounce homogenizer on ice in 1.5 ml homogenate buffer containing (in mM): 300 sucrose, 250 Hepes-NaOH (pH 7.8), 1 EDTA, 0.1 neocuproine (a copper chelating agent). An EDTA-free protease inhibitor tablet (Roche Diagnostics Corporation, Indianapolis, IN) was introduced just before use. Total heart homogenates were saved as aliquots, snap-frozen in liquid nitrogen, and stored at −80 °C.
Identification of SNO proteins by 2D DyLight DIGE
A modified biotin switch method [30] using DyLight-maleimide sulfhydryl-reactive fluors (Pierce Biotechnology, Rockford, IL) was applied to identify SNO proteins [16,25,31]. After the DyLight switch and 2D electrophoresis, each gel was scanned at the unique excitation/emission wavelengths of each DyLight fluor using a Typhoon 9400 imager (GE Healthcare Life Sciences, Piscataway, NJ) at a resolution of 100 μm. Images from each gel were aligned with two internal anchor spots and analyzed using Progenesis Discovery software (Nonlinear Dynamics, Newcastle upon Tyne, UK). The Ettan Spot Handling Workstation (GE Healthcare Life Sciences) performed automated extraction of the selected protein spots followed by in-gel trypsin digestion. We initially picked the Dylight fluorescent spots. The gel was then poststained with SYPRO Ruby (Sigma) and the protein spots that corresponded to the fluorescent DyLight pattern were picked [25]. After sample extraction from the spot handling workstation, each sample was manually desalted using Millipore C18 Ziptips following the manufacturer’s recommendation. For the DyLight fluorescence spot samples, Liquid chromatography–tandem mass spectrometry was performed using an Eksigent nanoLC-Ultra 1D plus system (Dublin, CA) coupled to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using CID fragmentation. The raw file generated from the LTQ Orbitrap Velos was analyzed using Proteome Discoverer v1.3 software (Thermo Fisher Scientific, LLC) using our six-processor Mascot cluster at NIH (v.2.3) search engine. The following search criteria were set: database, Swiss-Prot (Swiss Institute of Bioinformatics); taxonomy, Mus musculus (mouse); enzyme, trypsin; miscleavages, 2; variable modifications, oxidation (M), N-ethylmaleimide (C), deamidation (NQ); MS peptide tolerance was set to 20 ppm and MS/MS tolerance was set as 0.8 Da. The peptide confidence false discovery rate was set to 1%. For those Ruby-stained spots, protein identification was carried out using a MALDI-TOF/TOF instrument (4700 Proteomics Analyzer, Applied Biosystems) in positive-ion mode with a reflector. The peak-list generating software used was GPS Explorer software, set to default parameters (version 3.0, Applied Biosystems). The Mascot (v.2.3) search engine was used for the following search criteria: database, Swiss-Prot (Swiss Institute of Bioinformatics); taxonomy, Mus musculus (mouse); enzyme, trypsin; miscleavages, 2; variable modifications, oxidation (M), N-ethylmaleimide (C), deamidation (NQ); MS peptide tolerance was set to 25 ppm and MS/MS tolerance was set as 0.8 Da. The positive identifications criteria were two or more unique peptides, MS/MS C.I. of 95% or higher, correct molecular mass, and identification from all three 2D gels. Proteins identified in the DyLight spots were included if they were found in 2 out of 3 gels and were at the correct molecular weight. All the proteins that were found in the DyLight fluorescence spots were also validated in the Ruby-stained spot analysis.
Data analysis
Results are expressed as mean±SE. Statistical significance was determined by one-way ANOVA followed by a post hoc Student-Newman-Keuls test.
Results
Inhibition of the sGC/cGMP/PKG pathway does not block IPC-induced cardioprotection
Nadtochiy et al. showed that postischemic cardiac functional recovery in Langendorff-perfused rat hearts was significantly enhanced in the dark, a condition which preserves SNO [32]. Because light exposure causes decomposition of SNO, in this study and our previous studies [16,25,31], hearts were perfused in the dark.
Langendorff-perfused mouse hearts were treated with 10 μM ODQ or 1 μM KT5823 10 min prior to and during 4 cycles of IPC (Fig. 1). As shown in the Supplemental Data, 10 μM ODQ was sufficient to block sGC activity in perfused mouse hearts. Perfusion of hearts with ODQ or KT5823 alone in non-IPC hearts did not change cardiac hemodynamics (Table 1). In addition, in the absence of IPC, the drug treatment did not lead to changes in postischemic functional recovery (Table 1, Fig. 2A) or infarct size (Fig. 2B). IPC resulted in a significant reduction in postischemic contractile dysfunction and a decrease in myocardial infarction. However, treatment with ODQ or KT5823 prior to and during IPC did not block IPC-induced protection (Fig. 2).
Table 1.
Hemodynamic parameters.
| Heart samples | (n) | BW (g) | Predrug equilibration
|
Postdrug prior to IPC
|
Time to ischemic contracture (min)onset | End of reperfusion
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FR (ml/min) | HR (bpm) | LVDP (cm H2O) | +dp/dt | −dp/dt | FR (ml/min) | HR (bpm) | LVDP (cm H2O) | +dp/dt | −dp/dt | FR (ml/min) | HR (bpm) | LVDP (cm H2O) | +dp/dt | −dp/dt | |||||
| (cm H2O/ms) | (cm H2O/ms) | (cm H2O/ms) | |||||||||||||||||
| Perf-I/R | 10 | 24.7±0.3 | 2.3±0.2 | 351±16 | 127±9 | 6.3±0.4 | −4.8±0.2 | 8.6±0.2 | 2.0±0.2 | 340±16 | 54±9 | 4.1±0.2 | −3.6±0.2 | ||||||
| IPC-I/R | 9 | 24.9±0.4 | 2.2±0.3 | 357±14 | 124±9 | 6.4±0.4 | −4.6±0.3 | 5.8±0.2** | 1.9±0.2 | 348±16 | 79±9* | 4.8±0.2* | −4.1±0.3* | ||||||
| ODQ-I/R | 5 | 25.1±0.4 | 2.2±0.3 | 362±13 | 131±9 | 6.2±0.5 | −4.7±0.4 | 2.2±0.4 | 352±15 | 126±9 | 6.1±0.4 | −4.5±0.4 | 8.3±0.5 | 1.8±0.3 | 351±15 | 62±8 | 3.9±0.3 | −3.4±0.3 | |
| ODQ-IPC-I/R | 8 | 24.6±0.4 | 2.1±0.2 | 352±12 | 130±8 | 6.3±0.4 | −4.5±0.3 | 2.0±0.2 | 341±19 | 127±8 | 6.1±0.4 | −4.3±0.3 | 3.7±0.1**,# | 1.7±0.2 | 341±16 | 111±12**,# | 5.2±0.3* | −4.5±0.2* | |
| KT5823-I/R | 5 | 24.1±0.5 | 2.1±0.2 | 349±17 | 121±10 | 6.0±0.6 | −4.4±0.5 | 2.0±0.3 | 347±14 | 120±9 | 5.9±0.5 | −4.3±0.4 | 9.0±0.3 | 1.7±0.1 | 339±17 | 49±8 | 4.3±0.2 | −3.9±0.4 | |
| KT5823-IPC-I/R | 6 | 25.6±0.3 | 2.0±0.2 | 342±15 | 126±10 | 6.1±0.5 | −4.3±0.4 | 2.0±0.1 | 340±17 | 121±9 | 6.0±0.5 | −4.2±0.4 | 5.9±0.1** | 1.7±0.2 | 333±18 | 81±7* | 4.9±0.3* | −4.2±0.2* | |
Values are mean±SE; (n), number of hearts; BW, body weight; FR, flow rate; HR, heart rate (beats per min, bpm); LVDP, left ventricular developed pressure; ±dp/dt, rates of pressure rise and fall, respectively.
P<0.05,
P<0.01, vs Perf-I/R;
P<0.05 vs IPC-I/R.
Fig. 2.

ODQ and KT5823 treatment did not block IPC-induced cardioprotection. Inset (n), number of animals in each group. (A) Postischemic left ventricular RPP functional recovery. (B) Infarct size, measured at the end of reperfusion by 1% TTC staining. ** P<0.01 vs Perf-I/R; ## P<0.01 and # P<0.05 vs IPC-I/R by one-way ANOVA post hoc Student-Newman-Keuls test.
ODQ treatment enhances IPC-mediated protection
In contrast with KT5823 treatment, which had no effect on IPC, ODQ treatment prior to and during IPC led to a significant increase in postischemic cardiac functional recovery compared to IPC alone (Fig. 2A). Infarct size was also smaller with ODQ+IPC (8.7±2.1%) versus IPC alone (11.3±1.0%, Fig. 2B). KT5823 inhibits PKG but does not affect upstream NO/sGC/cGMP signaling. In contrast, ODQ competitively inhibits NO binding to the heme site of sGC, and therefore could increase the bioavailability of NO to enhance formation of SNO, which might lead to an additional SNO-mediated improvement in postischemic functional recovery.
Similar to previous findings [33,34], IPC reduced the time to onset ischemic contracture (5.8±0.2 min vs 8.6±0.2 min in Perf-I/R as shown in Table 1). ODQ treatment alone did not change the time to onset ischemic contracture, i.e., 8.3±0.5 min. However, ODQ treatment in IPC hearts further reduced the time to onset ischemic contracture (3.7±0.1 min).
ODQ treatment during IPC increases protein S-nitros(yl)ation
To test whether ODQ-treated IPC hearts might show increased levels of SNO over that observed with IPC alone, total heart homogenates were subjected to 2D DyLight DIGE proteomic analysis for SNO detection in IPC±ODQ and perfusion control hearts. A representative 2D DyLight DIGE gel is shown in Fig. 3A. SNO proteins in perfusion control hearts were labeled by DyLight 549 (yellow), IPC hearts were labeled with DyLight 488 (green), and ODQ-treated IPC hearts were labeled with DyLight 649 (red). As shown in Fig. 3A and Table 2, IPC significantly increased SNO levels compared to perfusion control (overlay image of DyLight 488/549 in Fig. 3A). ODQ+IPC hearts also had significantly higher SNO levels compared to perfusion control (overlay image of DyLight 649/549 in Fig. 3A). Compared to IPC hearts, ODQ+IPC hearts had a larger increase of SNO in selected proteins such as mitochondrial complex I-75 KDa, electron transfer flavoprotein dehydrogenase, electron transfer flavoproteins α/β, and creatine kinase S/M type (Fig. 3B, Table 2). These data are consistent with the hypothesis that increased bioavailability of NO leads to higher levels of SNO in ODQ+IPC hearts compared to IPC hearts and that this increased SNO might result in additional improvement of postischemic cardiac functional recovery.
Fig. 3.

Differences in protein S-nitros(yl)ation in total heart homogenate isolated from perfusion control and IPC hearts with and without ODQ treatment. (A) Top panels: a representative 2D DyLight DIGE gel was scanned at each of the distinct wavelengths of the DyLight fluors, showing a pattern of protein SNO for that particular treatment group. Bottom left panel: overlaid images of DyLight 488 (IPC, green) vs 549 (Perfusion, yellow). Bottom right panel: overlaid images of DyLight 649 (ODQ+IPC, red) vs 549 (Perfusion, yellow). (B) The overlaid images of DyLight 649 (ODQ+IPC, red) vs 488 (IPC, green) showed that there was still an increase of S-nitros(yl)ation for some proteins in ODQ+IPC vs IPC hearts. The protein spots were picked for MS/MS analysis, and are listed in Table 2.
Table 2.
SNO levels of identified proteins in the 2D DyLight DIGE studies.
| Spots | Protein name | Accession number | MW (kDa) | Protein pI | SNO (arbitrary ratio of DyLight intensity)
|
||
|---|---|---|---|---|---|---|---|
| IPC vs Perf | ODQ+IPC vs Perf | ODQ+IPC vs IPC | |||||
| 1 | Aconitate hydratase, mitochondrial | Q99KI0 | 85.4 | 8.08 | 2.3±0.3* | 2.9±0.4* | 1.2±0.2 |
| 2 | Trifunctional enzyme subunit α | Q8BMS1 | 82.6 | 9.24 | 2.2±0.3* | 2.4±0.3* | 1.1±0.1 |
| 3 | Mitochondrial complex I-75 KDa | Q91VD9 | 79.7 | 5.51 | – | – | 2.0±0.1* |
| 4 | Electron transfer flavoprotein dehydrogenase | Q921G7 | 68.1 | 7.34 | – | – | 1.9±0.1* |
| 5 | ATP synthase subunit α | Q03265 | 59.7 | 9.22 | 2.5±0.3* | 2.7±0.4* | 1.1±0.1 |
| 6 | Heat shock protein 60 kDa | P63038 | 61.0 | 5.91 | 2.5±0.2* | 2.8±0.2* | 1.2±0.1 |
| 7 | α-Enolase | P17182 | 47.4 | 6.37 | 2.4±0.3* | 2.6±0.3* | 1.1±0.1 |
| 8 | Creatine kinase S-type | Q6P8J7 | 47.8 | 8.64 | 2.5±0.2* | 3.8±0.3* | 1.6±0.1* |
| 9 | Creatine kinase M-type | P07310 | 43.0 | 6.58 | 2.4±0.4* | 3.7±0.1* | 1.6±0.2* |
| 10 | α-Cardiac muscle actin | P68033 | 42.3 | 5.23 | 2.9±0.4* | 3.4±0.5* | 1.2±0.2 |
| 11 | Pyruvate dehydrogenase E1 subunit α | P35486 | 35.8 | 8.25 | 2.9±0.8* | 3.1±0.8* | 1.1±0.1 |
| 12 | Malate dehydrogenase, cytoplasmic | P14152 | 36.5 | 6.16 | 2.2±0.1* | 2.5±0.2* | 1.2±0.1 |
| 13 | Electron transfer flavoprotein α | Q99LC5 | 35.0 | 8.62 | 2.2±0.2* | 2.8±0.3* | 1.3±0.1 |
| 14 | Electron transfer flavoprotein β | Q9DCW4 | 27.8 | 8.24 | 2.1±0.1* | 3.5±0.4* | 1.7±0.1* |
| 15 | Myosin Light chain 1 | P09542 | 22.5 | 5.03 | 2.7±0.3* | 2.9±0.5* | 1.1±0.1 |
| 16 | Myoglobin | P04247 | 17.1 | 7.07 | 2.4±0.1* | 2.7±0.4* | 1.1±0.1 |
Positive identifications from three independent experiments consisted of two peptides or more, a best ion score of 95% or higher, and a correct molecular mass position.
“–“, indicates proteins detected only in IPC or ODQ+IPC hearts but undetectable in perfusion control hearts.
P<0.05, significant differences between two groups (n=3).
Discussion
Recent data suggest that protein S-nitros(yl)ation plays an important role in cardioprotection [16,22,25]. Our previous studies have demonstrated that IPC results in an increase in SNO, and that SNO leads to altered protein activity and protection against further oxidation [22,25,26]. Previous studies reported that IPC was blocked by reducing agents such as N-acetylcysteine [35], N-2-mercaptopropionyl glycine [7,36], and glutathione [15], suggesting that a redox-sensitive mechanism is involved in the protection afforded by IPC. Ascorbate, a reducing agent that specifically decomposes SNO [30], has been found to block IPC in both in vivo [37,38] and in vitro [16] studies. Our recent study in Langendorff-perfused mouse hearts showed that activation of NO signaling is important for IPC-induced cardioprotection, and that treatment with L-NAME (a NOS inhibitor) 10 min prior to and during four cycles of IPC blocked the protection provided by IPC [16]. Taken together these studies suggest that NO-mediated redox-based SNO signaling plays a role in IPC-induced acute cardioprotection.
A key criterion used to classify a response to NO or an NO donor as cGMP-dependent is to test whether the effect is prevented by the selective inhibitor of sGC, ODQ. In this study, neither ODQ nor KT5823 caused any changes in cardiac hemodynamic parameters under basal conditions (Table 1), suggesting that the blockade of sGC/cGMP/PKG has little or no effect on the vascular response (coronary flow rate) or cardiac function in Langendorff-perfused mouse hearts. Treatment of perfused mouse hearts with ODQ or KT5823 before a sustained period of ischemia did not alter postischemic functional recovery (Table 1, Fig. 2A) or infarct size (Fig. 2B) in non-IPC hearts. However, mouse hearts treated with ODQ or KT5823 were still protected by IPC, suggesting that blockade of the sGC/cGMP/PKG pathway does not abolish NO-dependent IPC-induced cardioprotection. To our surprise, instead of blocking IPC, ODQ treatment provided additional improvement in postischemic functional recovery in IPC hearts (Fig. 2A), suggesting that a NO/SNO-mediated effect might also contribute to a better preservation of contractile function after ischemia and reperfusion [39–41].
IPC has been previously shown to shorten the time to ischemic contracture [33,34,42]. It has been proposed that the more rapid time to ischemic contracture observed with IPC is due to earlier inhibition of glycolysis, due either to inhibition or to depletion of glycogen, and the earlier inhibition of glycolysis is consistent with the reduced ischemic acidosis observed with IPC [42]. The reduction in ischemic acidosis has been suggested to contribute to cardioprotection by reducing the rise in Na+ (by reducing Na+–H+ exchanger) and thereby reducing Ca2+ overload during ischemia and early reperfusion via a reduction in Ca2+ entry via Na+–Ca2+ exchanger [43]. The additional reduction in time to onset ischemic contracture observed when ODQ is added with IPC would be consistent with earlier inhibition of glycolysis perhaps secondary to SNO-mediated inhibition of glycolysis.
A role for sGC and PKG is commonly included in the signaling pathway leading to activation of the mitochondrial KATP channel [44], mitochondrial permeability transition pore [44,45], and cardioprotection [4,46–48]. A role for sGC in cardioprotection has been demonstrated with bradykinin-mediated protection, as the addition of ODQ blocked bradykinin-induced protection [29]. Interestingly, in the same paper it was reported that ODQ did not block the protection induced by ouabain, suggesting that not all cardioprotective pathways require sGC. Qin et al. showed that protection provided by the NO donor SNAP was blocked by the addition of ODQ, again suggesting an important role for sGC [7]. ODQ has also been shown to block the IPC-mediated protection [6,9]. However, in the study by Lochner et al., in the absence of IPC, ODQ alone also cause enhanced postischemic contractile dysfunction [6]. Sips et al. reported that IPC was not blocked with loss of the α1 isoform of sGC, the primary isoform expressed in heart. However, they showed that ODQ addition blocked IPC-induced protection in both wild-type and α1 isoform sGC null mice, suggesting that another isoform of sGC might be important for IPC [9]. The protection provided by postconditioning has also been shown to be blocked by ODQ [12,49,50], but interestingly a recent paper by Cohen et al. showed that protection provided by the addition of SNAP at reperfusion was not blocked by ODQ [11]. Although sGC has been shown in many models to be required for protection, not all protection requires sGC. For example, ouabain-induced protection was not blocked by ODQ [29]. Sips et al. also showed that a different isoform of sGC is important for protection [9]. A role for a cGMP-independent NO-induced cardioprotection against I/R injury has also been demonstrated in studies using isolated cardiomyocytes [51,52]. Therefore, the variable results could be due to the differences in animal species, IPC protocols, and whether or not ODQ would elicit some vascular response such as a decrease in coronary flow rate. In this study, ODQ treatment did not change flow rate (Table 1), suggesting little or no vascular response induced by ODQ.
Because ODQ competitively inhibits NO binding to the heme site of sGC, ODQ treatment could increase the bioavailability of NO during IPC, resulting in higher levels of SNO. Using 2D DyLight DIGE proteomics, we found an increase in SNO for several proteins in ODQ+IPC hearts compared to IPC hearts (Fig. 3, Table 2). It is important to emphasize that our perfused heart studies were performed in the dark to prevent the decomposition of SNO during IPC-I/R. Interestingly, it has been shown that sGC can undergo S-nitros(yl)ation in vascular cells, and S-nitros(yl)ation of sGC results in decreased responsiveness to NO [53]. The SNO proteins identified in this study are mostly mitochondrial (Table 2), and because of dynamic range issues, most of the proteomic methods are biased toward detection of high-abundance proteins [22]. Since IPC has been shown to increase protein S-nitros(yl)ation [16,25,26], it will be interesting to know whether IPC could possibly lead to S-nitros(yl)ation of sGC, thus shifting downstream NO signaling pathways toward SNO signaling.
In summary, treatment with ODQ or KT5823 to inhibit sGC/ cGMP/PKG signaling did not block the protection provided by IPC. In addition, IPC hearts treated with ODQ enhanced protection concomitant with a higher SNO level. In conclusion, these results suggest, at least in some models of cardioprotection, that NO-mediated cardioprotection is regulated by protein S-nitros(yl)ation rather than through activation of the sGC/cGMP/PKG signaling.
Supplementary Material
Acknowledgments
This work was supported by the NIH Intramural Program (J.S., A.A., and E.M.), NIH Grants 1F32HL096142 (M.K.) and 5R01HL039752 (C.S.), and the China Scholarship Council No. 2011659007 (G.T.).
Abbreviations
- cGMP
cyclic guanosine monophosphate
- 2D DIGE
two-dimensional difference gel electrophoresis
- IPC
ischemic preconditioning
- I/R
ischemia/reperfusion
- NO
nitric oxide
- PKG
protein kinase G
- sGC
soluble guanylyl cyclase
- SNO
S-nitros(yl)ation
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2012.09.005.
Footnotes
The authors declare that they have no conflict of interests.
References
- 1.Murry C, Jennings R, Reimer K. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 2.Cohen MV, Yang X-M, Downey JM. Nitric oxide is a preconditioning mimetic and cardioprotectant and is the basis of many available infarct-sparing strategies. Cardiovasc Res. 2006;70:231–239. doi: 10.1016/j.cardiores.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 3.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Costa ADT, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, Critz SD. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res. 2005;97:329–336. doi: 10.1161/01.RES.0000178451.08719.5b. [DOI] [PubMed] [Google Scholar]
- 5.Cuong DV, Kim N, Youm JB, Joo H, Warda M, Lee J-W, Park WS, Kim T, Kang S, Kim H, Han J. Nitric oxide-cGMP-protein kinase G signaling pathway induces anoxic preconditioning through activation of ATP-sensitive K+ channels in rat hearts. Am J Physiol. 2006;290:H1808–H1817. doi: 10.1152/ajpheart.00772.2005. [DOI] [PubMed] [Google Scholar]
- 6.Lochner A, Marais E, Du Toit E, Moolman J. Nitric oxide triggers classic ischemic preconditioning. Ann N Y Acad Sci. 2002;962:402–414. doi: 10.1111/j.1749-6632.2002.tb04084.x. [DOI] [PubMed] [Google Scholar]
- 7.Qin Q, Yang X-M, Cui L, Critz SD, Cohen MV, Browner NC, Lincoln TM, Downey JM. Exogenous NO triggers preconditioning via a cGMP- and mitoKATP-dependent mechanism. Am J Physiol. 2004;287:H712–H718. doi: 10.1152/ajpheart.00954.2003. [DOI] [PubMed] [Google Scholar]
- 8.Post H, Schulz R, Behrends M, Gres P, Umschlag C, Heusch G. No involvement of endogenous nitric oxide in classical ischemic preconditioning in swine. J Mol Cell Cardiol. 2000;32:725–733. doi: 10.1006/jmcc.2000.1117. [DOI] [PubMed] [Google Scholar]
- 9.Sips P, Brouckaert P, Ichinose F. The alpha1 isoform of soluble guanylate cyclase regulates cardiac contractility but is not required for ischemic preconditioning. Basic Res Cardiol. 2011;106:635–643. doi: 10.1007/s00395-011-0167-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kodani E, Xuan Y-T, Takano H, Shinmura K, Tang X-L, Bolli R. Role of cyclic guanosine monophosphate in late preconditioning in conscious rabbits. Circulation. 2002;105:3046–3052. doi: 10.1161/01.cir.0000019408.67709.b5. [DOI] [PubMed] [Google Scholar]
- 11.Cohen MV, Yang X-M, Liu Y, Solenkova NV, Downey JM Cardioprotective PKG. independent NO signaling at reperfusion. Am J Physiol. 2010;299:H2028–H2036. doi: 10.1152/ajpheart.00527.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inserte J, Barba I, Poncelas-Nozal M, Hernando V, Agullo L, Ruiz-Meana M, Garcia-Dorado D. cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion. J Mol Cell Cardiol. 2011;50:903–909. doi: 10.1016/j.yjmcc.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Marina Prendes M, Gonzalez M, Savino E, Varela A. Role of endogenous nitric oxide in classic preconditioning in rat hearts. Regul Pept. 2007;139:141–145. doi: 10.1016/j.regpep.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 14.Andelová E, Pancza BM, Styk D, Ravingerová JT. The role of NO in ischemia/reperfusion injury in isolated rat heart. Gen Physiol Biophys. 2005;24:411–426. [PubMed] [Google Scholar]
- 15.Novalija E, Hogg N, Kevin LG, Camara AKS, Stowe DF. Ischemic preconditioning: triggering role of nitric oxide-derived oxidants in isolated hearts. J Cardiovasc Pharmacol. 2003;42:593–600. doi: 10.1097/00005344-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Sun J, Kohr MJ, Nguyen T, Aponte AM, Connelly PS, Esfahani SG, Gucek M, Daniels MP, Steenbergen C, Murphy E. Disruption of caveolae blocks ischemic preconditioning-mediated S-nitrosylation of mitochondrial proteins. Antioxid Redox Signal. 2012;16:45–56. doi: 10.1089/ars.2010.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Talukder MAH, Yang F, Shimokawa H, Zweier JL. eNOS is required for acute in vivo ischemic preconditioning of the heart: effects of ischemic duration and sex. Am J Physiol. 2010;299:H437–H445. doi: 10.1152/ajpheart.00384.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X-M, Zhang G-X, Yu Y-Q, Kimura S, Nishiyama A, Matsuyoshi H, Shimizu J, Takaki M. The opposite roles of nNOS in cardiac ischemia-reperfusion-induced injury and in ischemia preconditioning-induced cardioprotection in mice. J Physiol Sci. 2009;59:253–262. doi: 10.1007/s12576-009-0030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulman IH, Hare JM. Regulation of cardiovascular cellular processes by S-nitrosylation. Biochim Biophys Acta. 2012;1820:752–762. doi: 10.1016/j.bbagen.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lima B, Forrester MT, Hess DT, Stamler JS. S-Nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun J, Steenbergen C, Murphy E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid Redox Signal. 2006;8:1693–1705. doi: 10.1089/ars.2006.8.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun J. Protein S-nitrosylation: a role of nitric oxide signaling in cardiac ischemic preconditioning. Acta Physiol Sin. 2007;59:544–552. [PubMed] [Google Scholar]
- 24.Murphy E, Kohr M, Sun J, Nguyen T, Steenbergen C. S-Nitrosylation: a radical way to protect the heart. J Mol Cell Cardiol. 2012;52:568–577. doi: 10.1016/j.yjmcc.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun J, Morgan M, Shen R-F, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 26.Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- 28.Oldenburg O, Qin Q, Krieg T, Yang X-M, Philipp S, Critz SD, Cohen MV, Downey JM. Bradykinin induces mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP channel opening and leads to cardioprotection. Am J Physiol. 2004;286:H468–H476. doi: 10.1152/ajpheart.00360.2003. [DOI] [PubMed] [Google Scholar]
- 29.Pasdois P, Quinlan CL, Rissa A, Tariosse L, Vinassa B, Costa ADT, Pierre SV, Dos Santos P, Garlid KD. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am J Physiol. 2007;292:H1470–H1478. doi: 10.1152/ajpheart.00877.2006. [DOI] [PubMed] [Google Scholar]
- 30.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;86(PL1) doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 31.Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-β activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation. 2009;120:245–254. doi: 10.1161/CIRCULATIONAHA.109.868729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J Mol Cell Cardiol. 2007;42:812–825. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King LM, Opie LH. Does preconditioning act by glycogen depletion in the isolated rat heart? J Mol Cell Cardiol. 1996;28:2305–2321. doi: 10.1006/jmcc.1996.0224. [DOI] [PubMed] [Google Scholar]
- 34.Kolocassides KG, Galinanes M, Hearse DJ. Preconditioning accelerates contracture and ATP depletion in blood-perfused rat hearts. Am J Physiol. 1995;269:H1415–H1420. doi: 10.1152/ajpheart.1995.269.4.H1415. [DOI] [PubMed] [Google Scholar]
- 35.Chen W, Gabel S, Steenbergen C, Murphy E. A redox-based mechanism for cardioprotection induced by ischemic preconditioning in perfused rat heart. Circ Res. 1995;77:424–429. doi: 10.1161/01.res.77.2.424. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka M, Fujiwara H, Yamasaki K, Sasayama S. Superoxide dismutase and N-2-mercaptopropionyl glycine attenuate infarct size limitation effect of ischaemic preconditioning in the rabbit. Cardiovasc Res. 1994;28:980–986. doi: 10.1093/cvr/28.7.980. [DOI] [PubMed] [Google Scholar]
- 37.Skyschally A, Schulz R, Gres P, Korth H-G, Heusch G. Attenuation of ischemic preconditioning in pigs by scavenging of free oxyradicals with ascorbic acid. Am J Physiol. 2003;284:H698–H703. doi: 10.1152/ajpheart.00693.2002. [DOI] [PubMed] [Google Scholar]
- 38.Tsovolas K, Iliodromitis EK, Andreadou I, Zoga A, Demopoulou M, Iliodromitis KE, Manolaki T, Markantonis SL, Kremastinos DT. Acute administration of vitamin C abrogates protection from ischemic preconditioning in rabbits. Pharmacol Res. 2008;57:283–289. doi: 10.1016/j.phrs.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Paolocci N, Ekelund UEG, Isoda T, Ozaki M, Vandegaer K, Georgakopoulos D, Harrison RW, Kass DA, Hare JM. cGMP-independent inotropic effects of nitric oxide and peroxynitrite donors: potential role for nitrosylation. Am J Physiol. 2000;279:H1982–H1988. doi: 10.1152/ajpheart.2000.279.4.H1982. [DOI] [PubMed] [Google Scholar]
- 40.Heusch G, Post H, Michel MC, Kelm M, Schulz R. Endogenous nitric oxide and myocardial adaptation to ischemia. Circ Res. 2000;87:146–152. doi: 10.1161/01.res.87.2.146. [DOI] [PubMed] [Google Scholar]
- 41.Rassaf T, Poll LW, Brouzos P, Lauer T, Totzeck M, Kleinbongard P, Gharini P, Andersen K, Schulz R, Heusch G, Modder U, Kelm M. Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J. 2006;27:1699–1705. doi: 10.1093/eurheartj/ehl096. [DOI] [PubMed] [Google Scholar]
- 42.Kolocassides KG, Seymour A-ML, Galinanes M, Hearse DJ. Paradoxical effect of ischemic preconditioning on ischemic contracture? NMR studies of energy metabolism and intracellular pH in the rat heart. J Mol Cell Cardiol. 1996;28:1045–1057. doi: 10.1006/jmcc.1996.0097. [DOI] [PubMed] [Google Scholar]
- 43.Steenbergen C, Perlman ME, London RE, Murphy E. Mechanism of preconditioning. Ionic alterations. Circ Res. 1993;72:112–125. doi: 10.1161/01.res.72.1.112. [DOI] [PubMed] [Google Scholar]
- 44.Costa ADT, Garlid KD. Intramitochondrial signaling: interactions among mitoKATP, PKCε, ROS, and MPT. Am J Physiol. 2008;295:H874–H882. doi: 10.1152/ajpheart.01189.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borutaite V, Morkuniene R, Arandarcikaite O, Jekabsone A, Barauskaite J, Brown G. Nitric oxide protects the heart from ischemia-induced apoptosis and mitochondrial damage via protein kinase G mediated blockage of permeability transition and cytochrome c release. J Biomed Sci. 2009;16:70. doi: 10.1186/1423-0127-16-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costa ADT, Pierre SV, Cohen MV, Downey JM, Garlid KD. cGMP signalling in pre- and post-conditioning: the role of mitochondria. Cardiovasc Res. 2008;77:344–352. doi: 10.1093/cvr/cvm050. [DOI] [PubMed] [Google Scholar]
- 47.Burley DS, Ferdinandy P, Baxter GF. Cyclic GMP and protein kinase-G in myocardial ischaemia-reperfusion: opportunities and obstacles for survival signaling. Br J Pharmacol. 2007;152:855–869. doi: 10.1038/sj.bjp.0707409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122:216–238. doi: 10.1016/j.pharmthera.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Penna C, Cappello S, Mancardi D, Raimondo S, Rastaldo R, Gattullo D, Losano G, Pagliaro P. Post-conditioning reduces infarct size in the isolated rat heart: role of coronary flow and pressure and the nitric oxide/cGMP pathway. Basic Res Cardiol. 2006;101:168–179. doi: 10.1007/s00395-005-0543-6. [DOI] [PubMed] [Google Scholar]
- 50.Yang X-M, Philipp S, Downey JM, Cohen MV. Postconditioning’s protection is not dependent on circulating blood factors or cells but involves adenosine receptors and requires PI3-kinase and guanylyl cyclase activation. Basic Res Cardiol. 2005;100:57–63. doi: 10.1007/s00395-004-0498-4. [DOI] [PubMed] [Google Scholar]
- 51.Garreffa AM, Woodman OL, Cao AH, Ritchie RH. Sodium nitroprus-side protects adult rat cardiac myocytes from cellular injury induced by simulated ischemia: role for a non-cGMP-dependent mechanism of nitric oxide protection. J Cardiovasc Pharmacol. 2006;47:1–8. doi: 10.1097/01.fjc.0000189601.12276.8b. [DOI] [PubMed] [Google Scholar]
- 52.Iwase H, Robin E, Guzy RD, Mungai PT, Vanden Hoek TL, Chandel NS, Levraut J, Schumacker PT. Nitric oxide during ischemia attenuates oxidant stress and cell death during ischemia and reperfusion in cardiomyocytes. Free Radic Biol Med. 2007;43:590–599. doi: 10.1016/j.freeradbiomed.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 53.Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc Natl Acad Sci USA. 2007;104:12312–12317. doi: 10.1073/pnas.0703944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.