

Summary
Background
Duchenne muscular dystrophy (DMD) is a genetic neuromuscular disorder that affects skeletal muscles and cardiac muscle tissue. In some cases, myocardial injury secondary to hypoxia can lead to dilative cardiomyopathy (DCM). A genetic defect in the dystrophin gene may increase the susceptibility of myocardium to hypoxia. Available data suggest that this may be caused by impaired secretion of NO, which is bound with secretion of VEGF-A.
Material/Methods
Male mice C57BI/10ScSn mdx (animal model of DMD) and healthy mice C57BI/10ScSn were exposed to hypobaric hypoxia in low-pressure chambers. Their hearts were harvested immediately after and 1, 3, 7, and 21 days after exposure to hypoxia. Normobaric mice were used as controls. The expression of VEGF-A in myocardium and cardiac vessel walls was evaluated using immunohistochemistry, Western blotting, and in situ hybridization.
Results
VEGF-A expression in myocardium and vessel walls of healthy mice peaked 24 hours after exposure to hypoxia. The expression of VEGF-A in vessel walls was similar in dystrophic and healthy mice; however, VEGF-A expression in the myocardium of dystrophic mice was impaired, peaking around day 7. In the heart, the total level of VEGF depends on VEGF expression in myocardium, not in vessel endothelium, and our research demonstrates that the expression of VEGF is dystrophin-dependent.
Conclusions
Disordered secretion of VEGF-A in hypoxic myocardium caused the total level of this factor to be impaired in the heart. This factor, which in normal situations protect against hypoxia, promotes the gradual progression of cardiomyopathy.
Keywords: dilative cardiomyopathy, VEGF, hypoxic heart, Duchenne muscular dystrophy
Background
Duchenne muscular dystrophy (DMD) is a genetic neuromuscular disorder. DMD is an inherited X-linked disorder with an incidence of 1 in 3,500 male births, and is due to the absence of dystrophin, a large protein linking the intracellular cytoskeleton to the extracellular matrix [1]. DMD is typically diagnosed in boys 3 to 7 years of age [2,3]. It follows a predictable clinical course marked by progressive skeletal muscle weakness, myocyte hypertrophy, atrophy, and fibrosis. The disease also affects the cardiac muscle. By 13 years of age, 25% of patients demonstrate symptoms of systolic heart failure [2,3]. In general, the involvement of the heart leads to dilative cardiomyopathy (DCM) in 90% of patients. Death usually occurs in the second or third decade of life and is due to respiratory or circulatory failure [3,4]. Over the last 20 years, respiratory care for these patients has improved because of the development of supportive equipment and techniques.
Consequently, DCM is increasing as the major cause of death in people with DMD, so that currently 10–40% of DMD patients (20–30% on average) die due to DCM [1,5]. The classical animal model for human DMD, the mdx mouse, is marked by a mutation genetically similar to the human deletion in the Xp21.1 locus [6,7]. Although the underlying gene defect is the same in humans and the mdx mouse, the clinical picture is quite different. In the mdx mouse, skeletal muscle undergoes an early acute phase of degeneration at about 3–4 weeks of age, followed by a successful regeneration phase. The mdx mouse also develops cardiomyopathy with decreased cardiac function and cardiac fibrosis [8].
The defect in the gene encoding dystrophin – one of the cytoskeletal proteins – may influence the susceptibility of the cardiac tissue to hypoxia [5,6]. Myocardial injury secondary to hypoxia has been thoroughly investigated and described in the setting of myocardial infarction, and reflects local acute ischemic hypoxia and can lead to DCM under certain circumstances.
Hypoxia is an important stimulus for collateral vessel formation in the myocardium [6,9]. Collateral blood vessels supplement normal coronary blood flow and coronary blood flow compromised by coronary artery disease, thereby protecting the myocardium from ischemia. Collateral vessel formation is the result of angiogenesis, which depends upon the appropriate expression of growth factors [10–12]. Hypoxia is the main stimulating factor for the expression of vascular endothelial growth factor (VEGF) [13–16]. VEGF-A, also known as vascular permeability factor, is a potent angiogenic factor and endothelial cell-specific mitogen that is regulated by hypoxia in vitro and in vivo[17,18]. VEGF plays a role in initiating all forms of angiogenesis. It also enhances embryonic and postnatal angiogenesis [12, 19, 20] and modulates vascular remodeling, tubulogenesis, and arteriogenesis [21]. Available data suggest a relationship between impaired VEGF expression, a disturbance in angiogenesis, and the progression of DCM [22–24]. The present study investigated alterations of the secretion of VEGF-A in myocardium and heart vessels between healthy mice and dystrophic mice (C57BL/10ScSn mdx mice), which are the animal model of DMD.
Material and Methods
Animals and the experimental model
All mice were handled according to the guidelines of the Institutional Animal Care and Use Committee. Consent No. 128/99 was granted by the Bioethics Committee at the CM UMK. C57Bl/10ScSn and C57BL/10ScSn mdx male mice, weighing 20–30 grams, were purchased from Jackson Laboratory (Bar Harbor, MA). All mice were housed in an individually vented cage system with a 12-hour light/dark cycle and received standard mouse chow and water ad libitum. The temperature was maintained at approximately 20°C. Adult mice C57BI/10ScSn (homozygotes +/+ in the locus encoding dystrophin, referred to herein as “normal”) and adult mice C57BI/10ScSn mdx (homozygotes −/− in the locus encoding dystrophin, referred to herein as “dystrophic” or “mdx”) were used in the experiment. The animals came from the laboratory of the Military Institute of Aviation Medicine.
The animals were divided into experimental and control groups. Each group contained of 25 representatives of 1 strain. Initially, each group was placed in a low-pressure chamber for 1 h. The change from normal air pressure to low pressure corresponded to a rise from the altitude of 113 meters above sea level (P=1000 hPa, pO2=210 hPa) to 7000 m above sea level (P=410.25 hPa; pO2= 86.15 hPa). The target pressure was achieved within 10 minutes, and the initial pressure was restored within the same timeframe.
In a previous study [25] we showed that 7000 meters corresponds to the sublethal hypobaric hypoxia conditions, and that the dystrophin-deficient mdx mice were more susceptible to acute hypobaric hypoxia than were normal mice of the same strain.
Hearts were harvested from the control group (normobaric mice) and mice exposed to low pressure directly after and 1, 3, 7, and 21 days following the hypobaric exposure and fixed in a 4% solution of paraformaldehyde (PFA) in 0.01 M phosphate buffer (PBS, pH=7.4). The heart specimens were dipped in paraffin and cut into 6 μm thick sections along the plane perpendicular to the long axis of the heart (transverse sections through the ventricles). Some of the material was frozen in liquid nitrogen and stored at −70°C for later analysis. Specimens were subjected to Western blot analysis, in situ hybridization, and immunohistochemical analysis.
Western blot analysis
VEGF-A protein expression was determined using 100 mg of homogenized cardiac tissue suspended in 1 ml of mM Tris-HCl buffer (pH 7.4) and centrifuged at 10 000 rpm for 20 min. In order to select proper parameters for electrophoresis, the quantity of protein in the supernatant was assessed by the biuret reaction (according to Mejbaum-Katzenellenbogen) [26]. Electrophoresis was carried out on a 12% polyacrylamide gel, chosen according to the molecular weight of VEGF-A (46 kDa). Coomassie BR dye was used for protein detection. The proteins were transferred (semidry transfer) to a nitrocellulose membrane (0.45 mm pore; PROTRAN, Schleicher & Schnell BioScience) using the MINI Semi-Dry-Blot device (ROTH) at 1–2 mA/cm over 1–2 h and at room temperature. Immunodetection of VEGF-A was performed using the ABC kit (Santa Cruz Biotechnology). The membranes were blocked in 0.05%Tween 20 for 1 h, then incubated with primary anti-VEGF-A (Santa Cruz Biotechnology) at 3 μl/ml in 0.05% Tween 20 in a humid chamber with orbital shaking for 1.5 h, followed by incubation with the secondary biotinylated antibody under similar conditions for 1 h. Proteins were visualized using the horseradish peroxidase-diaminobenzidine system (DAB Substrate Kit for Peroxidase; SK-4100, Vector Laboratories).
In situ hybridization
Sections were attached to glass slides, deparaffinization, and rehydrated. Specimens were blocked using 1% solution of H2O2 and proteinase K (200 μg/ml) was added for cell permeabilization. In order to deactivate any remaining RNase and proteinase K, the sections were fixed a second time in 0.4% PFA at 4°C for 20 min. Prehybridization was carried out in the humid chamber at 45°C for 60 min. The samples were hybridized with the VEGF-A mRNA probe (5’ biotin-TGG GTG CAG CCT GGG ACC ACT TGG CAT GGT GGA GGT A-biotin-3’; DNA-Gdansk, Poland) overnight under similar conditions. Parallel negative control reactions were performed without probe. After hybridization, the slides were washed 2×5 min in 4× SSC/30% formamide, 2×5 min in 2×SCC/30% formamide, 1×15 min in 0.1% TRITON X-100 in TBS with albumin.
The ABC Staining System Kit (Santa Cruz Biotechnology, sc-2017) was used to detect the hybrids. Then, the preparations were dehydrated using a set of alcohol solutions and xylene, and mounted in Mountex medium.
Immunohistochemistry
Heart tissue slices on glass slides were deparaffinization and hydrated. Endogenous peroxidase was blocked by incubating samples in 0.1% H2O2 for 30 min, and then incubated with monoclonal mouse anti-VEGF antibody (Santa Cruz Biochemistry, sc-7269) at 3 μl/ml in 1.5% normal goat serum/PBS at room temperature for 30 min. Primary antibody was detected using the ABC Staining System Kit (Santa Cruz Biochemistry, sc-2017). Subsequently, the sections were incubated with biotinylated secondary antibody for 30 min and visualized using the DAB-horseradish peroxidase system.
Nuclei were stained with Gill’s hematoxylin No. 2 (Sigma), and then sections were dehydrated using a standard set of alcohol solutions and mounted in Mountex medium. Adjacent sections used as negative controls were processed in the same manner except that primary antibodies were omitted.
Visual analysis
The slides were evaluated by optical microscopy using a Nikon Eclypse 80i equipped with a digital camera DS-5Mc (5 megapixel) and software for digital analysis (NIS-Elements, Nikon; Japan). Mean optical density was determined under constant light conditions according to a gray scale ranging from 0 to 255, where 0 = black and 255 = white. Mean brightness/gray value is the statistical mean of brightness values of pixels. NIS-Elements uses brightness/gray calibration curve evaluation of this parameter, and shows brightness/density ranges that correspond to the range of gray values recalculated via brightness/density calibration curves.
Five samples from each group were chosen for analysis, and 6 preparations from each mouse were examined. Five fields of myocardium measuring 70×50 μm and not containing blood vessels were evaluated in each preparation (Figure 1). The result recorded for each mouse was the mean of the values obtained for each sample. Endothelial specimens on similar field sizes that contained cardiac vessels only were evaluated, and the mean value of all results was calculated for every group of 5 mice.
Figure 1.
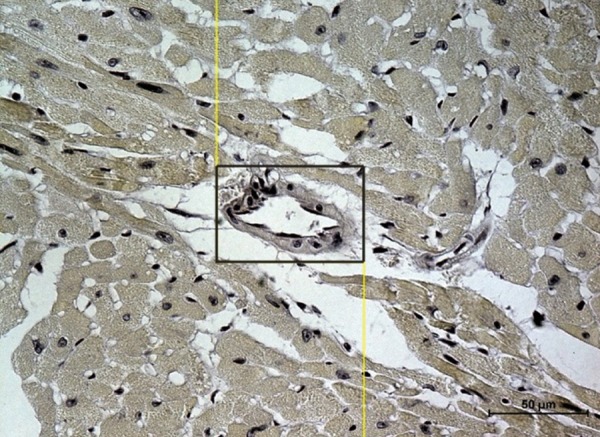
Heart’s specimen from a control mouse. The measuring frame of 70×50 μm is marked. In the black frame – vessel. Scale bar = 50 μm.
For the control group, the mean optical density was defined as 100% of the change in optical density saturation for preparations containing both myocardium and vessels (both arterioles/venules and capillaries), without distinguishing between these 2 compartments. This allowed us to calculate the mean value for the whole organ (Figure 1). The results obtained in the experimental groups are expressed as percentages of the results for the control group.
Statistical analysis
All data are presented as the mean ± standard deviation (SD). Statistical analysis was performed using STATISTICA 7.1. The values obtained from in situ hybridization analysis were analyzed using the unpaired Student’s T test. The values obtained from Western analysis of VEGF were analyzed using the Wilcoxon signed rank test. Differences were considered statistically significant at p<0.05.
Results
There were significant differences in cardiac VEGF expression in response to hypoxia between mdx and normal mice (Figure 2). During the first days following hypoxia, expression of VEGF in the hearts of the control group did not differ from that observed in the hearts of the normal and mdx mice (102±3% vs. 99±4%, respectively) (p>0.05) (Figure 2C). Immediately after exposure to hypoxia, VEGF expression was 98±5% and 96±6% in normal and mdx mice, respectively (p>0.05). One day later, the changes in VEGF concentrations were similar in normal (168±8%) and mdx mice (172±9%) (p>0.05). Statistically significant differences became apparent on the third day following hypoxia (p<0.05). The decrease in VEGF expression was markedly greater among the mdx mice (113±7%) than the normal mice (133±5%) (p<0.05). This trend reversed on day 7 – VEGF expression in normal mice continued to decrease (107±5%), whereas it increased in mdx mice, reaching 147±6% (p<0.05). The differences between the groups were statistically significant. After day 21, VEGF expression began to decline in both groups, reaching its initial concentration in normal mice (104±6%) and falling below the initial concentration in mdx mice (70±4%) (p<0.05).
Figure 2.

(A) Western analysis of VEGF expression in the heart following hypoxia normal mice. Control group (1); immediately after (2); 1 day after (3); 3 days after (4); 7 days after (5), and 21 days after (6) hypoxia. (B) Western analysis of VEGF expression in the heart following hypobaric hypoxia in dystrophic mice. Control group (1); immediately after (2); 1 day after (3); 3 days after (4); 7 days after (5), and 21 days after (6) hypoxia. (C) Quantitative analysis of western blot signals in normal and mdx mice. Note the difference in the timing of maximum VEGF expression between normal and mdx mice. The control group signal is set to 100%. * Statistically significant compared to healthy mice. In each group was 10 mice.
Effects on VEGF mRNA expression are depicted in Figure 3A for the cardiac vessel endothelium cells and in Figure 3B for the myocardium. In the case of cardiac endothelium cells, there was no difference in VEGF-A mRNA expression between the control groups for normal and mdx mice (101±8% vs. 99±7%, respectively) (p>0.05) or between the normal and dystrophic mice immediately following exposure to hypoxia (103±9% vs. 98±8%, respectively) (p>0.05), on day 1 (228±13% vs. 233±14%) (p>0.05), and on day 3 (144±10% vs. 133±8%) (p>0.05). However, on day 1 there was a marked increase in VEGF-A mRNA expression in normal and mdx mice exposed to hypoxia as compared to normobaric control mice. By day 7, VEGF-A mRNA expression levels had returned to their initial levels (106±10% vs. 93±13%, respectively) (p>0.05).
Figure 3.

In situ hybridization analysis of VEGF mRNA expression in cardiac vessel endothelial cells (A) and in cardiac myocytes (B) from normal (C57BI/10ScSn) and mdx (C57BI/10ScSn) mice (values are optical densities expressed as percentage of the signal for control mice). N – 5 samples from each group, 5 fields of myocardium in 6 preparations from each mouse were examined together 150 analysis. * Statistically significant compared to normal – healthy mice.
The difference in VEGF-A mRNA expression in myocardium between normal and dystrophic animals was significant (p>0.05) (Figure 3B). After day 1, normal mice expressed relatively more VEGF mRNA than did mdx mice (234±15 vs. 123±9%, respectively) (p<0.05). On day 3, VEGF-A mRNA expression in normal mice was similar to that in mdx mice (132±9% vs. 122±8%) (p>0.05). By day 7, VEGF-A mRNA expression in normal mice decreased to the initial level (83±6%), whereas in mdx mice, the level of mRNA remained stable and twice as high as that in the control group (119%±8%) (p<0.05). Twenty-one days following hypoxia, the expression of VEGF-A mRNA was similar in both groups and similar to the initial levels (57±5% vs. 43±7%) (p>0.05). The expression of VEGF-A mRNA in control myocardium was lower than that in endothelial cells (60% vs. 100%, respectively).
There were also differences in the expression of VEGF-A protein expression between the normal and mdx mice. In the case of endothelium, there were no difference in VEGF-A concentrations in normal and mdx mice (Figure 4A). In the case of myocardial cells, the initial control levels (99±7% vs. 92±6%, respectively) and those measured immediately following hypoxia (104±7% vs. 96±4%, respectively) did not differ between normal and mdx mice (p>0.05) (Figure 4B). Differences in VEGF-A protein concentration became apparent 1 day after hypoxia in the normal group. In these animals, there was an increase in optical density up to 168% (±9%) of the control level (Figure 5A). In dystrophic mdx mice, the protein concentration remained constant at 95±7% (p<0.05) (Figure 5B). After 3 days, the concentration of VEGF-A in normal mice fell to 120% (±7%), but remained elevated relative to the initial level. In the mdx mice, the concentration of VEGF remained stable at 86±8% (p<0.05). After 7 days, the VEGF-A concentration in normal mice returned to the initial value (99±4%) (Figure 6A), whereas in the mdx mice, the level of VEGF-A protein began to increase, reaching 135±9% (p<0.05) (Figure 6B). After 21 days, there was a clear difference between the 2 groups in terms of VEGF concentration. In control mice, the VEGF concentration remained at its initial level (103±7%) but had decreased in mdx mice (76±8%) (p<0.05).
Figure 4.

Immunohistochemical analysis of VEGF expression in cardiac vessel endothelial cells (A) and in cardiac myocytes (B) from normal (C57BI/10ScSn) and mdx (C57BI/10ScSn) mice (values are optical densities expressed as percentage of the signal for control mice).). N – 5 samples from each group, 5 fields of myocardium in 6 preparations from each mouse were examined together 150 analysis. * Statistically significant compared to normal – healthy mice.
Figure 5.

(A) Heart’s specimen from normal mice obtained 1 day after exposure to hypoxia. Increased expression of VEGF in both myocardium and vessel walls. (B) Heart’s specimen from mdx mice obtained 1 day after exposure to hypoxia. Low expression of VEGF in myocardium and an enhanced signal in vessel walls. Scale bar =100 μm.
Figure 6.

(A) Heart’s specimen from normal mice obtained 7 day after exposure to hypoxia. The expression of VEGF returns to initial levels. (B) Heart’s specimen from mdx mice obtained 7 day after exposure to hypoxia. The expression of VEGF in myocardium remains elevated but returns to normal in vessel walls. Scale bar =100 μm.
Discussion
Hypoxia is the primary cause of ischemic cardiomyopathy, DCM [22,27,28]. During ischemia, certain factors, such as FGF-2 [29] or VEGF and NO, are released and cause the collateral vessels to open [9,30]. In dystrophic mdx mice, the low concentration of NO leads to ischemic cardiomyopathy [28,31,32]. In normal muscular tissue, hypoxia induces an increase in VEGF expression through increased transcription of VEGF mRNA and improved protein stability [10]. Some reports have shown that dystrophinopathy is caused by lesions in the myocardium (tissue) but not in endothelial cells, in vessels [31]. Ischemia is the main factor responsible for the tissue damage. The increased expression of VEGF enhances both the quantity and density of vessels, and not only in heart – similar effects are observed in brain [33,34]. The VEGF expression disorders in dystrophic mdx mice described in this paper, with the initial fall in the third day and a rise in the seventh day, are no exception. In their experiment with prolonged hypoxia of the brain, Kuo et al [33] described similar changes in VEGF expression. After a fall on day 4, VEGF expression increased on day 7 [34].
Since VEGF expression disorders occur only in mdx mice and are not seen in normal mice, they do not have to be related to prolonged ischemia. In our experiment, VEGF expression disorders in normal and dystrophic mdx mice were limited to inappropriate expression and transcription of VEGF in myocardial cells. It follows that expression of endothelial VEGF would have an impact on total VEGF expression in the heart only on day 1 after hypoxia (Figures 2, 3A, 4A); after that, the level of VEGF expression in ischemic (hypoxic) cardiac tissue is the consequence of changes in the myocardium. In fact, on the third day, the expression of VEGF protein and VEGF mRNA are decreased (Figures 3B, 4B), leading to dissimilarities in VEGF expression between normal and dystrophic hearts (Figure 2). Differences are more profound on the following days. In the heart, total VEGF expression is higher on the seventh day (Figure 1). Increased expression of VEGF mRNA (Figure 4) and protein (Figure 4B) in the myocardium influence this situation. These increases could be due to increased VEGF mRNA stability, as well as increased transcription. The situation changes between days 1 and 7, when the relative expression of VEGF mRNA is higher. By day 21, mRNA expression is reduced. During the same period, VEGF protein expression levels vary as well. However, the pattern of changes in VEGF protein expression does not mirror that of VEGF mRNA expression. Up to the 3rd day after hypoxia, the protein expression level remains unchanged relative to the control group. By day 7, protein expression increases remarkably but falls again by day 21. In the heart, the total level of VEGF depends on VEGF expression in myocardium, not in vessel endothelium, and our research demonstrates that the expression of VEGF is dystrophin-dependent. The relationships between vascularization, other growth factors such as bFGF [35], PDGF [36], and dystrophin disorders have been documented. The reason for the discrepancies between the levels of VEGF protein and mRNA expression in myocardium seems to be associated with decreased stimulation of VEGF mRNA expression in myocytes. In myocardium, higher levels of VEGF expression are not associated with increased protein stability, as has been reported [10]. Instead, the secretion of VEGF from dystrophic myocardium is compromised and VEGF is retained in cells [29,30]. Even if the expression of VEGF protein is increased only marginally, a negative feedback mechanism blocks secretion of VEGF from the cell, resulting in an elevated level of VEGF within the cell. Thus, the dramatic reduction in VEGF mRNA transcription on day 21 that we observed in our experiments was the result of the cells receiving incorrect (inflated) information about of the level of available VEGF.
Our results suggest that high-altitude hypoxia causes abnormalities in cardiac muscle functioning in dystrophic mdx mice. The mutation in the dystrophin gene results in aberrant VEGF secretion, which ultimately leads to ischemia. This aberrant reaction is present only in the myocardium, not in the endothelium. The disorders localized to non-vascular cardiac tissues related to dystrophic cardiomyopathy have been also described by other authors [31].
Myocardium is the main reservoir of VEGF for the heart [15]. Impaired myocardial expression of VEGF causes a significant loss of VEGF to the whole organ. These disturbances are not only quantitative – the timing and speed of expression are affected as well. Therefore, in such a setting, the heart is not properly protected against hypoxia, which could promote the gradual progression of cardiomyopathy, eventually manifesting as heart failure [23,37–39].
Conclusions
Our study investigated whether increased cardiac muscle susceptibility to ischemia was related to the efficacy of hypoxia-induced VEGF expression. We have provided new support for the role of dystrophin in angiogenesis through its interactions with other growth factors. These data may aid in the development of new therapeutic options for dystrophin-related disorders [8,37].
Acknowledgements
D. Nowak and H. Kozlowska contributed equally to this study. The authors thank Mrs. Alina Kiestrzyn-Wojcik, Mrs. Mariola Rzeszowska, and Mr. Mariusz Szczepanski for their technical support. There are no relationships with industry.
Abbreviations
- bFGF
basic fibroblast growth factor
- DCM
dilative cardiomyopathy
- DMD
Duchenne muscular dystrophy
- MCP-1
monocyte chemotactic protein-1
- mdxmouse
dystrophic mouse
- m-RNA
messenger RNA
- NGF
nerve growth factor
- NO
Nitric Oxide
- PBS
0.01 M phosphate buffer
- PDGF
platelet-derived growth factor
- PFA
paraformaldehyde
- VEGF
vascular endothelial growth factor
- VEGF-A
vascular endothelial growth factor A
Footnotes
Source of support: This work was financed by the scientific grants the BW 29/00 and 17/03 (CM UMK)
References
- 1.Emery AE. Population frequencies of inherited neuromuscular diseases-a world survey. Neuromuscul Disord. 1991;1(1):19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 2.Baxter P. Treatment of the heart in Duchenne muscular dystrophy. Dev Med Child Neurol. 2006;48(3):163. doi: 10.1017/S0012162206000351. [DOI] [PubMed] [Google Scholar]
- 3.de Kermadec JM, Becane HM, Chenard A, et al. Prevalence of left ventricular systolic dysfunction in Duchenne muscular dystrophy: an echocardiographic study. Am Heart J. 1994;127(3):618–23. doi: 10.1016/0002-8703(94)90672-6. [DOI] [PubMed] [Google Scholar]
- 4.Eagle M, Baudouin SV, Chandler C, et al. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12(10):926–29. doi: 10.1016/s0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- 5.Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003;99(1):1–19. doi: 10.1159/000068446. [DOI] [PubMed] [Google Scholar]
- 6.Danialou G, Comtois AS, Dudley R, et al. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001;15(9):1655–57. doi: 10.1096/fj.01-0030fje. [DOI] [PubMed] [Google Scholar]
- 7.Bulfield G, Siller WG, Wight PA, Moore KJ. X-chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–92. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinlan JG, Hahn HS, Wong BL, et al. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14(8–9):491–96. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Schaper W. Collateral circulation: past and present. Basic Res Cardiol. 2009;104(1):5–21. doi: 10.1007/s00395-008-0760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy AP, Levy NS, Goldberg MA. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem. 1996;271(5):2746–53. doi: 10.1074/jbc.271.5.2746. [DOI] [PubMed] [Google Scholar]
- 11.Hoeben A, Landuyt B, Highley MS, et al. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56(4):549–80. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 12.Tallquist MD, Soriano P, Klinghoffer RA. Growth factor signaling pathways in vascular development. Oncogene. 1999;18(55):7917–32. doi: 10.1038/sj.onc.1203216. [DOI] [PubMed] [Google Scholar]
- 13.Poltorak Z, Cohen T, Neufeld G. The VEGF splice variants: properties, receptors, and usage for the treatment of ischemic diseases. Herz. 2000;25(2):126–29. doi: 10.1007/pl00001950. [DOI] [PubMed] [Google Scholar]
- 14.Carmeliet P. Basic Concepts of (Myocardial) Angiogenesis: Role of Vascular Endothelial Growth Factor and Angiopoietin. Curr Interv Cardiol Rep. 1999;1(4):322–35. [PubMed] [Google Scholar]
- 15.Giordano FJ, Gerber HP, Wiliams SP, et al. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA. 2001;98(10):5780–85. doi: 10.1073/pnas.091415198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frelin C, Ladoux A, D’angelo G. Vascular endothelial growth factors and angiogenesis. Ann Endocrinol (Paris) 2000;61(1):70–74. [PubMed] [Google Scholar]
- 17.Leung DW, Cachianes G, Kuang WJ, et al. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246(4935):1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 18.Sharma HS, Wünsch M, Brand T, et al. Molecular biology of the coronary vascular and myocardial responses to ischemia. J Cardiovasc Pharmacol. 1992;20(Suppl 1):S23–31. [PubMed] [Google Scholar]
- 19.Hashimoto E, Ogita T, Nakaoka T, et al. Rapid induction of vascular endothelial growth factor expression by transient ischemia in rat heart. Am J Physiol. 1994;267(5 Pt 2):H1948–54. doi: 10.1152/ajpheart.1994.267.5.H1948. [DOI] [PubMed] [Google Scholar]
- 20.Banai S, Shweiki D, Pinson A, et al. Upregulation of vascular endothelial growth factor expression induced by myocardial ischaemia: implications for coronary angiogenesis. Cardiovasc Res. 1994;28(8):1176–79. doi: 10.1093/cvr/28.8.1176. [DOI] [PubMed] [Google Scholar]
- 21.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–95. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 22.Tham E, Wang J, Piehl F, Weber G. Upregulation of VEGF-A without angiogenesis in a mouse model of dilated cardiomyopathy caused by mitochondrial dysfunction. J Histochem Cytochem. 2002;50(7):935–44. doi: 10.1177/002215540205000707. [DOI] [PubMed] [Google Scholar]
- 23.Abraham D, Hofbauer R, Schafer R, et al. Selective downregulation of VEGF-A(165), VEGF-R(1), and decreased capillary density in patients with dilative with not ischemic cardimyopathy. Circ Res. 2000;87(8):644–47. doi: 10.1161/01.res.87.8.644. [DOI] [PubMed] [Google Scholar]
- 24.Mosqueira M, Willmann G, Ruohola-Baker H, Khurana TS. Chronic hypoxia impairs muscle function in the Drosophila model of Duchenne’s muscular dystrophy (DMD) PLoS One. 2010;5(10):e13450. doi: 10.1371/journal.pone.0013450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowinski J, Kozlowska H, Rejment E. Sensitivity to acute altitude hypoxia and mutation in the dystrophin-coding gene – experimental study on mdx mice. Pol Przegl Med Lotn. 1997;3:207–14. [in Polish] [Google Scholar]
- 26.Mejbaum-Katzenellenbogen W, Mochnacka I. Kolorymetryczne oznaczanie białek. Metoda biuretowa. Kurs Praktyczny z Biochemii. PWN Warszawa. 1966:167–68. [in Polish] [Google Scholar]
- 27.Harre JM. The Dilative, Restrictive, and Infiltrative Cardiomiopathies. In: Libby P, Bonow RO, Mann DL, Zipes DP, editors. Braunwald’s Heart Disease A Textbook of Cardiovascular Medicine. 8th Editions. Saunders Elsevier; Philadelphia: 2008. pp. 1739–60. [Google Scholar]
- 28.Zheng QS, Guo WG, Lu ZF, et al. Dystrophin: from non-ischemic cardiomyopathy to ischemic cardiomyopathy. Med Hypotheses. 2008;71(3):434–38. doi: 10.1016/j.mehy.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Schaper W. Collateral vessel growth in the human heart. Role of fibroblast growth factor-2. Circulation. 1996;94(4):600–1. doi: 10.1161/01.cir.94.4.600. [DOI] [PubMed] [Google Scholar]
- 30.Hainsey TA, Senapati S, Kuhn DE, Rafael JA. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul Disord. 2003;13(4):294–302. doi: 10.1016/s0960-8966(02)00286-9. [DOI] [PubMed] [Google Scholar]
- 31.Straino S, Germani A, Di Carlo A, et al. Enhanced arteriogenesis and wound repair in dystrophin-deficient mdx mice. Circulation. 2004;110(21):3341–48. doi: 10.1161/01.CIR.0000147776.50787.74. [DOI] [PubMed] [Google Scholar]
- 32.Matsunaga T, Warltier DC, Weihrauch DW, et al. Ischemia-induced coronary collateral growth is dependent on vascular endothelial growth factor and nitric oxide. Circulation. 2000;102(25):3098–103. doi: 10.1161/01.cir.102.25.3098. [DOI] [PubMed] [Google Scholar]
- 33.Kuo NT, Benhayon D, Przybylski RJ, et al. Prolonged hypoxia increases vascular endothelial growth factor mRNA and protein in adult mouse brain. J Appl Physiol. 1999;86(1):260–64. doi: 10.1152/jappl.1999.86.1.260. [DOI] [PubMed] [Google Scholar]
- 34.Nico B, Mangieri D, Crivellato E, et al. HIF activation and VEGF overexpression are coupled with ZO-1 up-phosphorylation in the brain of dystrophic mdx mouse. Brain Pathol. 2007;17(4):399–406. doi: 10.1111/j.1750-3639.2007.00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D’Amore PA, Brown RH, Jr, Ku PT, et al. Elevated basic fibroblast growth factor in the serum of patients with Duchenne muscular dystrophy. Ann Neurol. 1994;35(3):362–65. doi: 10.1002/ana.410350320. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y, Haginoya K, Sun G, et al. Platelet-derived growth factor and its receptors are related to the progression of human muscular dystrophy: an immunohistochemical study. J Pathol. 2003;201(1):149–59. doi: 10.1002/path.1414. [DOI] [PubMed] [Google Scholar]
- 37.Zhou YF, Stabile E, Walker J, et al. Effects of gene delivery on collateral development in chronic hypoperfusion: diverse effects of angiopoietin-1 versus vascular endothelial growth factor. J Am Coll Cardiol. 2004;44(4):897–903. doi: 10.1016/j.jacc.2004.05.046. [DOI] [PubMed] [Google Scholar]
- 38.Bouchentouf M, Benabdallah BF, Bigey P, et al. Vascular endothelial growth factor reduced hypoxia-induced death of human myoblasts and improved their engraftment in mouse muscles. Gene Ther. 2008;15(6):404–14. doi: 10.1038/sj.gt.3303059. [DOI] [PubMed] [Google Scholar]
- 39.Zisa D, Shabbir A, Mastri M, et al. Intramuscular VEGF repairs the failing heart: role of host-derived growth factors and mobilization of progenitor cells. Am J Physiol Regul Integr Comp Physiol. 2009;297(5):r1503–15. doi: 10.1152/ajpregu.00227.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]