

An organophosphorus hydrolase from P. pseudoalcaligenes named OPHC2 has been crystallized. Combined with biochemical characterization, it is expected that the structure of this protein will provide insight into the catalytic mechanism of organophosphorus hydrolysis and will highlight the role of key residues involved in substrate specificity.
Keywords: organophosphorus, quorum sensing, phosphotriesterases, lactonases, nerve agents, pesticides, quorum quenching, Pseudomonas pseudoalcaligenes
Abstract
Enzymes that are capable of degrading neurotoxic organophosphorus compounds are of increasing interest because of the lack of efficient and clean methods for their removal. Recently, a novel organophosphorus hydrolase belonging to the metallo-β-lactamase superfamily was identified and isolated from the mesophilic bacterium Pseudomonas pseudoalcaligenes. This enzyme, named OPHC2, is endowed with significant thermal and pH stability, making it an appealing candidate for engineering studies to develop an efficient organophosphorus biodecontaminant. Combined with biochemical studies, structural information will help decipher the catalytic mechanism of organophosphorus hydrolysis by OPHC2 and identify the residues involved in its substrate specificity. Here, the expression, purification, crystallization and X-ray data collection at 2.1 Å resolution of OPHC2 are presented.
1. Introduction
Organophosphorus compounds (OPs) are well known toxic molecules that irreversibly inhibit acetylcholinesterase, a key enzyme in the nerve message transmission system (Singh, 2009 ▶). These compounds have been widely used as agricultural insecticides (Raushel, 2002 ▶) and the most toxic compounds have also been developed as chemical warfare agents (such as tabun, sarin, soman or VX; Gupta, 2009 ▶). Current methods for removing them are slow, expensive and engender ecological concerns (LeJeune et al., 1998 ▶). Novel methods of remediation, such as enzyme-mediated decontamination, are therefore under intensive research (Bigley & Raushel, 2012 ▶; Goldsmith et al., 2012 ▶).
The intensive use of OPs as pesticides starting in the 1950s has resulted in the rapid emergence of microorganisms that are able to degrade OPs and can probably utilize them as carbon and phosphorus sources (Pakala et al., 2007 ▶). Several organophosphorus hydrolases have been identified belonging to different protein families: the prolidases (Cheng et al., 1999 ▶), the paraoxonases (PONs; Ben-David et al., 2012 ▶), the phosphotriesterases (PTEs) and phosphotriesterase-like lactonases (PLLs) from the amidohydrolase superfamily and the organophosphorus hydrolases from the metallo-β-lactamase superfamily (Elias & Tawfik, 2012 ▶). The PTEs isolated from Brevundimonas diminuta (Omburo et al., 1992 ▶) and Agrobacterium radiobacter (Jackson et al., 2006 ▶) are the best characterized organophosphorus hydrolases so far and exhibit near-diffusion-limit rates against the insecticide paraoxon as a substrate (Omburo et al., 1992 ▶). PTEs are believed to have emerged from native lactonases with promiscuous phosphotriesterase activity such as the PLLs (Afriat-Jurnou et al., 2012 ▶; Elias et al., 2008 ▶; Hiblot et al., 2012a ▶).
A novel organophosphorus hydrolase named OPHC2 (GenBank ID AJ605330) has been isolated from Pseudomonas pseudoalcaligenes (Chu et al., 2006 ▶). This enzyme is the second characterized representative of a recently identified organophosphorus hydrolase clade. OPHC2 shares 57.9% sequence identity with methyl-parathion hydrolase (MPH), a protein isolated from Pseudomonas sp. WBC3 (GenBank ID AY251554), a soil bacterium living in organophosphorus-contaminated soil in China (Dong et al., 2005 ▶). The structure of MPH has previously been solved (Dong et al., 2005 ▶) and revealed a metallo-β-lactamase fold containing a bimetallic catalytic site and a bridging water molecule. This feature, also found in PLLs and PTEs, might suggest a similar catalytic mechanism in which the bridging catalytic water molecule is activated by the bimetallic active site and serves as the nucleophile that attacks the phosphorus centre of the bound substrate (Dong et al., 2005 ▶).
OPHC2 has been shown to exhibit methyl-parathion hydrolysing activity (Chu et al., 2006 ▶). While OPHC2 originates from a mesophilic bacterium (P. pseudoalcaligenes), its temperature optimum for catalysis is 338 K. Possible explanations for this feature have been hypothesized, such as a putative disulfide bridge and a higher number of surface salt bridges compared with MPH (Chu et al., 2010 ▶). Given its organophosphorus hydrolase activity, combined with thermal and pH stability (Chu et al., 2006 ▶), OPHC2 represents an interesting target for attempts to develop an efficient OP biodecontaminant. The structure of OPHC2 will thus help to decipher the structural determinants that account for its thermal stability. Moreover, the comparative analysis of MPH and OPHC2 structures and their careful biochemical characterization will lead to the identification of key residues involved in substrate binding and enzymatic specificity. This information will serve to engineer and improve OPHC2 catalytic efficiency against organophosphorus compounds. In this report, we describe the expression, purification, crystallization and X-ray data collection of OPHC2.
2. Cloning, expression and purification of OPHC2
The full gene encoding for OPHC2 with its N-terminus periplasmic signal peptide (UniProt ID Q5W503) was optimized for Escherichia coli expression and synthesized by GeneArt (Life Technologies, France). The gene was subsequently cloned into a custom version of pET22-b(+) (Novagen) containing an N-terminal streptavidin peptide (for affinity chromatography purification) and a tobacco etch virus protease (TEV) cleavage site (for removal of the tag; Gotthard et al., 2011 ▶) using NdeI and XhoI as cloning sites to avoid adding the plasmid’s pelB leader sequence. Recombinant OPHC2 protein was overexpressed using a protocol similar to that used for the PLLs SsoPox and SisLac (Hiblot et al., 2012a ▶,b ▶). Briefly, recombinant OPHC2 protein was overproduced in E. coli BL21(DE3)-pGro7/GroEL (TaKaRa). Protein expression was performed in 2 l ZYP medium (100 µg ml−1 ampicillin, 34 µg ml−1 chloramphenicol) inoculated with a 50 ml overnight preculture. The culture was grown at 310 K until it reached an OD600nm of 1.0. The induction of the protein was conducted by consumption of the lactose in ZYP medium, a temperature transition to 298 K during 20 h and the addition of 0.2 mM CoCl2. Cells were harvested by centrifugation (4500g, 277 K, 15 min). Pellets were resuspended in lysis buffer (50 mM HEPES pH 8, 150 mM NaCl, 0.2 mM CoCl2, 0.25 mg ml−1 lysozyme, 10 µg ml−1 DNAse, 0.1 mM PMSF) and stored at 193 K for 2 h. Suspended frozen cells were thawed at 310 K for 15 min and disrupted by three steps of 30 s of sonication (QSonica sonicator Q700; amplitude at 50). Cell debris was removed by centrifugation (12 500g, 277 K, 30 min).
Attempts to purify the protein using streptavidin affinity chromatography failed, indicating that the natural signal peptide of OPHC2 was recognized, processed and the protein subsequently exported to the periplasm by the E. coli cell machinery, which also removes the affinity tag. Consequently, another purification strategy was used: the thermal stability of OPHC2 (Chu et al., 2006 ▶) was exploited for purification in combination with differential ammonium sulfate precipitation. After 30 min incubation at 342 K, host proteins that precipitated were removed by a centrifugation step (12 000g, 277 K, 30 min). Remaining contaminant proteins were precipitated by ammonium sulfate (2 h incubation at 277 K with 291 g l−1 ammonium sulfate) and discarded after a centrifugation step (12 000g, 277 K, 30 min). OPHC2 was then concentrated by 36 h incubation with a final ammonium sulfate concentration of 476 g l−1, followed by centrifugation (15 min, 277 K, 12 000g) and resuspension in activity buffer (50 mM HEPES pH 8, 150 mM NaCl, 0.2 mM CoCl2). The remaining ammonium sulfate was removed by overnight dialysis against the activity buffer. The protein was concentrated using a centrifugation device (Amicon Ultra MWCO 10 kDa; Millipore, Ireland) prior to a size-exclusion chromatography step (Superdex 75 16/60, GE Healthcare). Although the affinity tag was probably removed by the E. coli cell machinery while processing the signal peptide, the fractions containing the protein were pooled and submitted to a tag-removal step in order to minimize the heterogeneity of the sample. We used TEV protease [1:13(w:w) ratio for 6 h at 303 K in activity buffer] to remove the tag (van den Berg et al., 2006 ▶). Precipitated TEV protease was removed by centrifugation (12 000g, 277 K, 10 min). The OPHC2 crystal structure did not show any tag or signal peptide. The sample was concentrated and subsequently reloaded onto a size-exclusion chromatography column in activity buffer (Superdex 75 16/60, GE Healthcare). Fractions containing pure protein were pooled and concentrated prior to crystallization trials using a centrifugation device (Amicon Ultra MWCO 10 kDa; Millipore, Ireland). The yield of production was about 8 mg per litre of culture. The purity of the protein was checked with Coomassie-stained 15% SDS–PAGE (Fig. 1 ▶).
Figure 1.
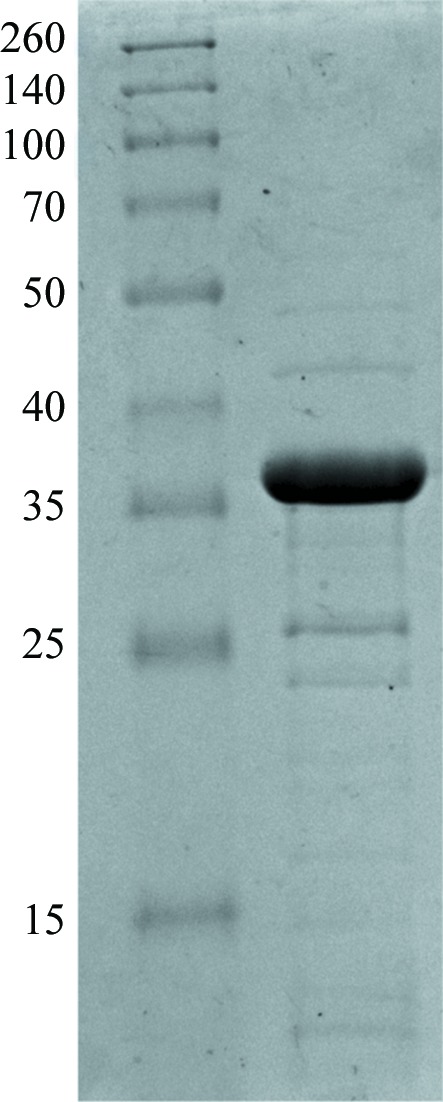
15% SDS–PAGE of OPHC2 protein stained with Coomassie Blue. Left lane, molecular-weight markers (Thermo Scientific Spectra Multicolor broad range protein ladder; labelled in kDa). Right lane, 11 µg OPHC2 protein.
3. Kinetic experiment
Methyl-paraoxon (∊405 nm = 17 000 M −1 cm−1) hydrolysis by OPHC2 (2 µl at 725 µg ml−1 in a 200 µl reaction volume) was performed in triplicates, at 298 K, in activity buffer, recorded with a microplate reader (Synergy HT, BioTek, USA) and monitored by the Gen5.1 software in a 6.2 mm path-length cell in 96-well plates, as described previously (Hiblot et al., 2012a ▶,b ▶). The specific activity of OPHC2 was evaluated using Excel software (Microsoft, USA). The measured specific activity against methyl-paraoxon as substrate (1 mM) is 0.632 ± 0.088 µmol mg−1 min−1. This value is slightly lower than the previously published specific activity against another substrate, methyl-parathion (1.982 µmol min−1 mg−1 at 310 K against 1.9 mM substrate; Chu et al., 2006 ▶). This difference could be explained by the difference in the nature of the substrates, their different concentrations and the temperature of the assays, but also, as proposed previously (Ng et al., 2011 ▶), by the different nature of the protein-expression system: E. coli in our case and Pichia pastoris in the previous work (Chu et al., 2006 ▶).
4. Protein crystallization
OPHC2 was concentrated to 16.4 mg ml−1 for crystallization trials. Crystallization assays were performed using a sitting-drop vapour-diffusion method setup in a 96-well plate and the commercial screen conditions Wizard I and II (Emerald BioSystems). The plates were incubated at 293 K and monitored using a Rock Imager and Rock Maker system (Formulatrix Inc., USA). Reproducible crystals appeared after 3 months at 293 K in a condition consisting of 10% PEG 8000, 100 mM Tris buffer pH 7, 200 mM MgCl2. Crystals grew in drops containing 2:1 and 1:1 protein:precipitant ratios (respective volumes of 200 nl:100 nl and 100 nl:100 nl; 200 µl reservoir volume; Fig. 2 ▶).
Figure 2.

A typical crystal of OPHC2 (average dimensions of 170 × 80 × 40 µm).
5. Data collection
A cryoprotectant solution consisting of the crystallization solution supplemented with 20%(v/v) glycerol was added to the crystal-containing drops (1 µl cryoprotectant was added to the 200 or 300 nl drops). The crystals were then transferred in a drop (1 µl) containing the cryoprotectant solution for 1 min and flash-cooled in liquid nitrogen. X-ray diffraction intensities were collected on the PROXIMA-1 beamline at SOLEIL (Gif-Sur-Yvette, France) using a wavelength of 0.980 Å and a PILATUS 6M detector with 0.15 s exposures. Diffraction data were collected from 1200 images using the fine-slicing method; individual frames consisted of 0.15° steps over a range of 180° (Fig. 3 ▶).
Figure 3.

A diffraction pattern from a crystal of OPHC2. The edge of the diffraction frame is at 1.63 Å resolution.
6. Results and conclusions
X-ray diffraction data were integrated, scaled and merged using the XDS program (Kabsch, 1993 ▶; Table 1 ▶). The OPHC2 crystals belonged to the monoclinic space group C2, with unit-cell parameters a = 109.9, b = 63.8, c = 221.3 Å, β = 101.8°. With a molecular weight of 35 kDa for OPHC2, the calculated Matthews coefficient suggests between four and five monomers per asymmetric unit (2.71 and 2.17 Å3 Da−1, corresponding to 54.7 and 43.38% solvent content, respectively). Initial molecular replacement (MR) using a monomer of MPH as a model (PDB entry 1p9e; Y. Dong, L. Sun, M. Bartlam, Z. Rao & X. Zhang, unpublished work) was performed using Phaser (McCoy et al., 2007 ▶). Only two molecules could initially be placed in the asymmetric unit (R free = 50.9%), while the crystal packing was clearly incomplete and residual density corresponding to other monomers could be observed. Attempts to place additional monomers using the initial solution as a fixed input failed. The initial solution was then submitted to ARP/wARP (Morris et al., 2003 ▶) for automated model construction. After 50 cycles of ARP/wARP, peptide fragments belonging to two new monomers were built and the R free factor decreased to 39.1%, yielding electron-density maps that were sufficiently informative to evaluate the model. We identified two fragments of the protein (fragment 1, amino acids 26–166; fragment 2, amino acids 214–295) that were well defined in the initially placed monomers. However, the residues between these parts were missing from the electron-density maps, and the conformation of the equivalent residues in the starting model (PDB entry 1p9e) was incompatible with the observed crystal packing. We thus performed new MR searches by using fragments 1 and 2 as models. A total of four fragments 1 and three fragments 2 were placed using Phaser (R free = 35.4%), revealing a total of four monomers in the asymmetric unit. The quality improvement of the maps allowed extension of the fragments using Coot (Emsley & Cowtan, 2004 ▶). The incomplete monomer revealed by the positioning of a fragment 1 was reconstructed by superposition of a complete monomer. Another round of MR was performed using fragments 1 and 2 as models with the previous solution as a fixed input. This allowed us to identify a fifth monomer (R free = 31.7%) that forms a dimer with a symmetry-related monomer through a twofold crystallographic axis. After manual building and reconstruction of the five monomers, the current R free factor is 30.21%. The asymmetric unit contains two homodimers of OPHC2 and one monomer. Notably, despite the sequence identity (57%) between MPH and OPHC2, the molecular replacement was not straightforward. Indeed, a significant fragment from OPHC2 (167–213) differs from the MPH model and is poorly defined in the OPHC2 structure. The fact that the conformation of the corresponding fragment in MPH is incompatible with the observed packing of the OPHC2 crystal explains why the MR searches with the complete MPH model failed. This structural difference between the enzymes may denote a functional difference and different substrate specificity; some residues belonging to this fragment are second-shell active-site residues in MPH (e.g. Phe178, Trp179, Asp190, Asp193, Phe196 and Phe197). Complete biochemical and kinetic characterization of OPHC2, as well as the construction of the structure, refinement, interpretation of the structure and the identification of the metal cations using anomalous scattering, are in progress.
Table 1. Data-collection statistics.
Values in parentheses are for the last bin.
| Beamline | PROXIMA-1 |
| Wavelength (Å) | 0.980 |
| Detector | PILATUS 6M |
| Oscillation (°) | 0.15 |
| No. of frames | 1200 |
| Resolution (Å) | 2.1 (2.2–2.1) |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 109.9, b = 63.8, c = 221.3, β = 101.8 |
| No. of observed reflections | 252270 (24317) |
| No. of unique reflections | 82530 (9469) |
| Completeness (%) | 93.7 (82.6) |
| R meas † (%) | 6.5 (50.1) |
| 〈I/σ(I)〉 | 13.67 (3.07) |
| Multiplicity | 3.06 (2.57) |
| Mosaicity (°) | 0.508 |
R
meas = 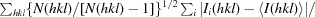
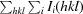 .
.
Acknowledgments
This research was supported by a grant to EC from Délégation Générale pour l’Armement (REI #2009 34 0045). GG and JH are PhD students granted by Délégation Générale pour l’Armement. DG is a PhD student granted by AP-HM (Marseille, France). We thank the AFMB-CNRS UMR 6098 for full access to the expression and crystallization platform. We are grateful to Agnes Toth-Petroczy for critical reading of the manuscript.
References
- Afriat-Jurnou, L., Jackson, C. J. & Tawfik, D. S. (2012). Biochemistry, 51, 6047–6055. [DOI] [PubMed]
- Ben-David, M., Elias, M., Filippi, J. J., Duñach, E., Silman, I., Sussman, J. L. & Tawfik, D. S. (2012). J. Mol. Biol. 418, 181–196. [DOI] [PubMed]
- Berg, S. van den, Löfdahl, P. A., Härd, T. & Berglund, H. (2006). J. Biotechnol. 121, 291–298. [DOI] [PubMed]
- Bigley, A. N. & Raushel, F. M. (2012). Biochim. Biophys. Acta, doi: 10.1016/j.bbapap.2012.04.004. [DOI] [PMC free article] [PubMed]
- Cheng, T.-C., DeFrank, J. J. & Rastogi, V. K. (1999). Chem. Biol. Interact. 119–120, 455–462. [DOI] [PubMed]
- Chu, X.-Y., Tian, J., Wu, N.-F. & Fan, Y.-L. (2010). Appl. Microbiol. Biotechnol. 88, 125–131. [DOI] [PubMed]
- Chu, X., Wu, N., Deng, M., Tian, J., Yao, B. & Fan, Y. (2006). Protein Expr. Purif. 49, 9–14. [DOI] [PubMed]
- Dong, Y.-J., Bartlam, M., Sun, L., Zhou, Y.-F., Zhang, Z.-P., Zhang, C.-G., Rao, Z. & Zhang, X.-E. (2005). J. Mol. Biol. 353, 655–663. [DOI] [PubMed]
- Elias, M., Dupuy, J., Merone, L., Mandrich, L., Porzio, E., Moniot, S., Rochu, D., Lecomte, C., Rossi, M., Masson, P., Manco, G. & Chabriere, E. (2008). J. Mol. Biol. 379, 1017–1028. [DOI] [PubMed]
- Elias, M. & Tawfik, D. S. (2012). J. Biol. Chem. 287, 11–20. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Goldsmith, M., Ashani, Y., Simo, Y., Ben-David, M., Leader, H., Silman, I., Sussman, J. L. & Tawfik, D. S. (2012). Chem. Biol. 19, 456–466. [DOI] [PubMed]
- Gotthard, G., Hiblot, J., Elias, M. & Chabrière, E. (2011). Acta Cryst. F67, 354–357. [DOI] [PMC free article] [PubMed]
- Gupta, R. C. (2009). Handbook of Toxicology of Chemical Warfare Agents London: Academic Press.
- Hiblot, J., Gotthard, G., Chabriere, E. & Elias, M. (2012a). PLoS One, 7, e47028. [DOI] [PMC free article] [PubMed]
- Hiblot, J., Gotthard, G., Chabriere, E. & Elias, M. (2012b). Sci. Rep. 2, 779. [DOI] [PMC free article] [PubMed]
- Jackson, C. J., Carr, P. D., Kim, H.-K., Liu, J.-W., Herrald, P., Mitić, N., Schenk, G., Smith, C. A. & Ollis, D. L. (2006). Biochem. J. 397, 501–508. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst. 26, 795–800.
- LeJeune, K. E., Wild, J. R. & Russell, A. J. (1998). Nature (London), 395, 27–28. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Morris, R. J., Perrakis, A. & Lamzin, V. S. (2003). Methods Enzymol. 374, 229–244. [DOI] [PubMed]
- Ng, F. S. W., Wright, D. M. & Seah, S. Y. K. (2011). Appl. Environ. Microbiol. 77, 1181–1186. [DOI] [PMC free article] [PubMed]
- Omburo, G. A., Kuo, J. M., Mullins, L. S. & Raushel, F. M. (1992). J. Biol. Chem. 267, 13278–13283. [PubMed]
- Pakala, S. B., Gorla, P., Pinjari, A. B., Krovidi, R. K., Baru, R., Yanamandra, M., Merrick, M. & Siddavattam, D. (2007). Appl. Microbiol. Biotechnol. 73, 1452–1462. [DOI] [PubMed]
- Raushel, F. M. (2002). Curr. Opin. Microbiol. 5, 288–295. [DOI] [PubMed]
- Singh, B. K. (2009). Nature Rev. Microbiol. 7, 156–164. [DOI] [PubMed]