

Abstract
The principles of natural protein engineering are obscured by overlapping functions and complexity accumulated through natural selection and evolution. Completely artificial proteins offer a clean slate on which to define and test these protein engineering principles, while recreating and extending natural functions. We introduce this method here with the first design of an oxygen transport protein, akin to human neuroglobin. Beginning with a simple and unnatural helix-forming sequence with just three different amino acids, we assemble a four helix bundle, position histidines to bis-his ligate hemes, and exploit helical rotation and glutamate burial on heme binding to introduce distal histidine strain and facilitate O2 binding. For stable oxygen binding without heme oxidation, water is excluded by simple packing of the protein interior and loops that reduce helical-interface mobility. O2 affinities and exchange timescales match natural globins with distal histidines with the remarkable exception that O2 binds tighter than CO.
It has long been recognized that natural selection and evolution build complexity into natural proteins and biological systems 1-3. This complexity frustrates biochemists seeking to understand structure and function 4 and presents an extraordinary challenge to protein engineers who aim to reproduce or create new functions in proteins. So far, this complexity has severely constrained the ability of protein engineers to approach the efficiency of natural protein catalysts 5-10. However common it may be in Nature, we maintain that complexity is not an essential feature of protein as a material. Neither is it an essential feature of catalysis, as shown by synthetic chemical systems 11. By understanding the origins of complexity and making purposeful efforts to separate multiple utilities and minimize complexity during the design and testing of artificial proteins that are completely independent of natural selection, we show how to progressively build in sophisticated biochemical features that reproduce and exceed natural protein function.
As Jencks made clear 12, protein function requires more than a static structure. In natural proteins, the motion that is part of engineering of protein function is often specific, which can make re-engineering motion for new functions prohibitively difficult. In contrast, artificial proteins offer a full palette of motions, which engineering can edit to facilitate those that are productive and remove those that are unproductive.
The engineering-based design of functional synthetic proteins progresses through 4 stages (Figure 1): 1) assemble a simple, robust generic protein framework, such as a helical bundle, of appropriate size to sustain eventual cofactor binding and catalytic function; 2) insert cofactor binding amino acids, keeping the number of amino acid changes few to control complexity; 3) adjust the sequence for improved structural resolution; 4) iteratively test, redesign and add engineering elements to refine function. This method of designing proteins follows that long used by artists and architects who develop maquettes, simple models that are progressively altered to test and determine the ultimate characteristics of their constructions.
Figure 1.
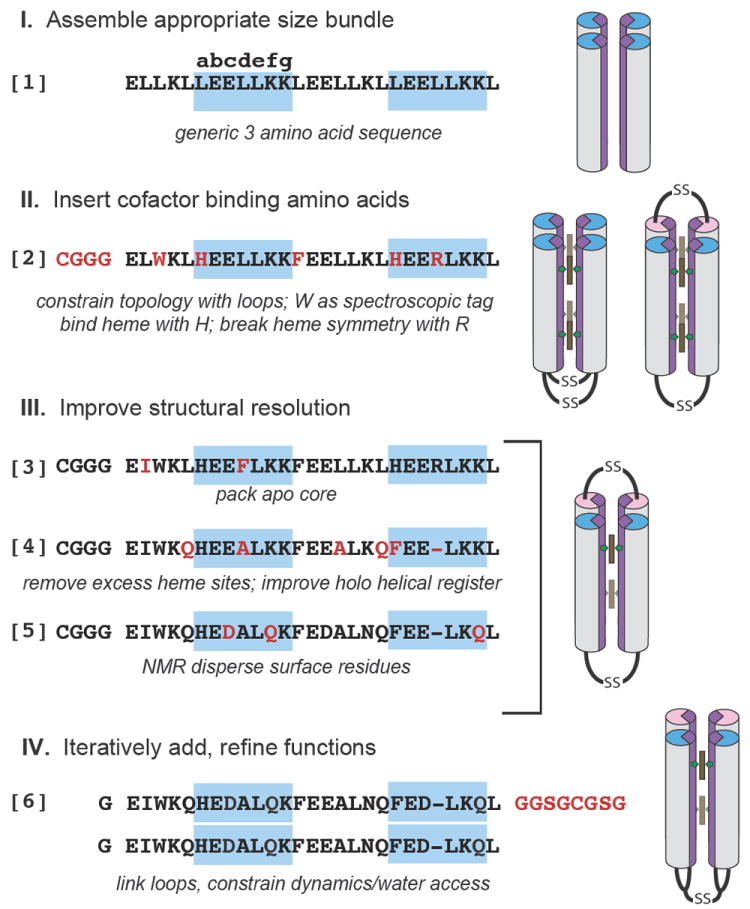
The design of an artificial oxygen transport protein (6) begins with an extremely simple three amino acid near heptad repeat sequence (1) and progresses through the design stages via a series of amino acid changes (red) and trial intermediates (2-5) that are tested to reveal functional properties and clarify the roles of individual amino acids. In 2-5, helical sequences shown are linked by cysteine disulfide loops and self assemble in 4 helix bundles, while in 6 a longer loop (red) unites two identical sequences, with the loops themselves now disulfide linked, as shown at right.
Assemble scaffold and cofactors
Figure 1 describes our starting maquette comprising polar glutamate (E) and lysine (K), and non-polar leucine (L). These have high α-helical forming propensities 13 which, when arranged in a near repeating heptad sequence LEELLKK LEELLKL, spontaneously assemble into a water-soluble four-α-helical bundle with E and K exposed and L buried in a molten globular interior 14. To make a bundle of a length typical of natural proteins, we use almost four heptad near repeats (1 in Figure 1). Such a bundle is free to associate with helices parallel or anti-parallel. Adding an N-terminal CGGG sequence for disulfide-mediated dimerization restricts helical topologies to syn or anti. Completing this stage, we replaced an internal leucine at e-position 7 of each helix with tryptophan to facilitate optical detection of the protein.
In the second stage of design, we found that replacing leucines at internal a-positions 10 and 24 with histidine was sufficient to anchor up to 4 hemes in the bundle15. We also replaced a-position leucine 17 between the hemes with phenylalanine, a common amino acid near hemes in natural proteins. To allow us to discriminate between the heme binding sites, we replaced another interior leucine with arginine (2 in Figure 1). No effort was made to design core cavities that accommodate heme; reliance was put on interior histidine-iron ligation to position the heme in a malleable hydrophobic interior.
Structural resolution is helpful in keeping progressive design on track. Singular structures in these bundles can be engineered without computation; introduction of ®-branched aliphatic or aromatic residues along with a polar bond across helices provided by the histidines (3 in Figure 1) confers tertiary structure to the four-α-helix interior of the apo-protein as seen by NMR and X-ray crystallography 16,17.
While various functions can be added to this basic heme binding protein, for oxygen transport we elected to simplify the design by lowering the heme capacity from 4 to 2 through replacing histidine at position 24 with phenylalanine. Inspection of model apo-structures showed that a substantial rotation of >50° around the helical axes was required on heme binding to accommodate the histidine rotamers typical of natural bis-histidine heme-binding proteins (supplementary figure S1) 18. This rotation exports hydrophobic interior residues into, and imports polar residues from, the aqueous phase. Modelling identified four amino acids for substitution to interfacially compatible alanines or glutamines 19 and one for deletion to more favourably realign the binary pattern following heme addition yielding 4 (Figure 1). However, three inwardly rotating b-position glutamates at 11, 18 and 25 were deliberately left in place 18 to apply strain to weaken one of the two histidine-heme iron ligation interactions as occurs in neuroglobin. This creates an entatic state 20. FTIR shows that these strain-inducing glutamates have pK values elevated by >2 units (supplementary figure S2) and, as in natural heme proteins, are strongly electrostatically coupled to heme oxidation-reduction, changing the pK by more than 3 units 21. NMR showed that addition of two hemes B transformed unstructured apo-4 into a well-defined tertiary structure 22(supplementary figure S3). It appears that the histidine-iron polar bond can also provide a nucleus for interior packing that promotes a singular structure around different porphyrin cofactors 22. To ease NMR structure determination and assignment of 90% of the peptide backbone (supplementary figure S4), external residues were also diversified at this time, yielding 5 (Figure 1).
Exclude water for oxy-ferrous stability
None of the molecules discussed so far, indeed no artificial heme proteins so far examined, 23-25 bind O2 stably at room temperature. This is not because the oxy-ferrous state cannot be formed. 2, 3, 4 and 5 all include glutamate-based helical strain, which weakens the histidine ligation and allows competition from other ligands. Each form an oxy-ferrous state at near 100% yield at -15C by first exposing the heme proteins to carbon monoxide and then illuminating at low temperature to initiate ligand exchange for O2, as Chance did with cytochrome oxidase 26. The likely reason for room temperature instability is the access of water and protons to the heme binding site which facilitates electron transfer from reduced heme to oxygen. This is clear from mutant studies of globins and the relative stability of the oxy-ferrous state in chemical systems in water-free solvents 11,27,28.
To engineer water exclusion we constrained the culpable protein motion. The crystal and NMR structures of 3 reveal that one of the two interhelical interfaces, that which lies between the helices not constrained by heme binding, has an unusually low degree of surface complementarity and a high degree of inter-helix motions. Indeed, sequence 2 can be modified to introduce large scale syn-anti flipping 29. To constrain motion, the loops were reconfigured to link the helices across the most mobile interface. This also allowed the loops to be linked into a monomeric ‘candelabra’ structure, further constraining motion (6 in Figure 1). NMR confirmed the water restricting effects of loop reconfiguration and monomerization. H/D analysis of hydrogen exchange protection factors of 4 and 6 at pH 7 and 25°C (supplementary figure S5) shows complete exchange within 15 minutes for 4 and much slower exchange rates for 6 with several core residue backbone amides, including some close to the heme exchanging on the several hours time scale.
Each maquette 2 - 6 displays ferric and ferrous visible spectra indicative of six-coordinate bis-histidine ligated heme B, characteristic of cytochromes b, deoxy-neuroglobin and cytoglobin, and quite distinct from the five-coordinate myoglobin and hemoglobin. Because the <1 nM KD for binding of the first heme is much tighter than 50 nM KD for the second heme 22, we simplify spectral analysis by binding one heme per bundle (Figure 2A). NMR assignments unambiguously identify the first heme B to bind at H7 positions at the open end of the candelabra structure of 6. Only ferrous heme 6 shows rapid and complete conversion of the ferrous heme into the oxyferrous heme with a halftime of ~50 milliseconds measured by stopped-flow spectroscopy. This oxyferrous spectrum is remarkably similar to native neuroglobin (supplementary figure S6). The oxyferrous state is stable for tens of seconds before single electron transfer from ferrous heme to O2 appears to generate superoxide.
Figure 2.
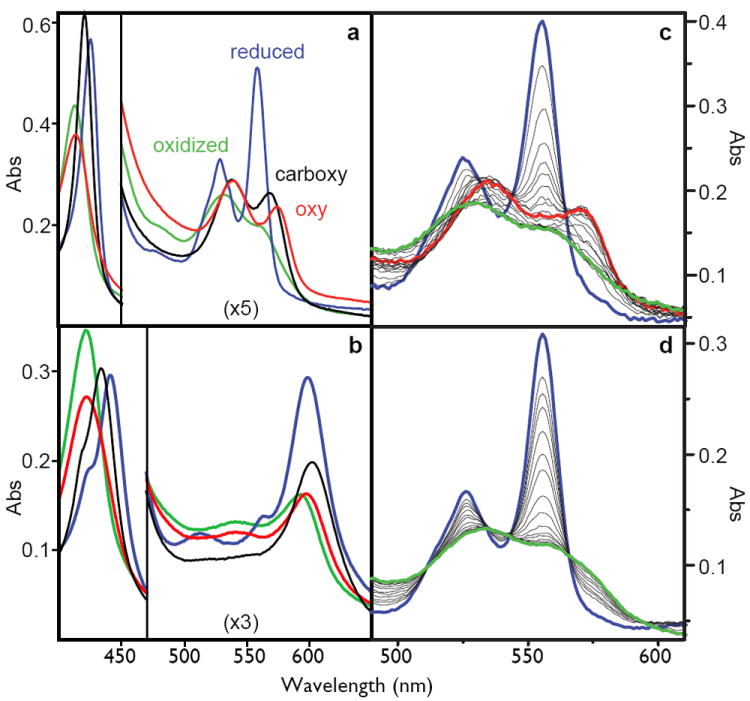
Left: the spectra of the oxidized (green), reduced (blue), carboxy-ferrous (black) or oxy-ferrous (red) artificial oxygen transport protein 6 with either heme B (A) or heme A as the cofactor (B). These spectra are obtained at -15C where these spectra are stable for more than an hour. Right: stopped-flow spectral changes for mixing the reduced heme B proteins with oxygen at 15C. The fully designed oxygen transport protein 6 (C), shows the transformation of the reduced heme (blue) to the oxy-ferrous state (red) which eventually becomes oxidized (green), while the early intermediate 2 (D) proceeds directly and rapidly to the oxidized form.
A similar experiment with CO shows rapid displacement of one histidine to generate an indefinitely stable carbonmonoxy-ferrous heme species in a 400 milliseconds halftime (not shown). After CO photolysis, CO rebinding to 6 is multiphasic (Figure 3, supplementary figure S7) as observed in the hexacoordinate globins; detailed analysis of this data allows the estimation of the histidine on- and off-rates (Table 1). CO rapid mixing shows slower binding than expected from photolysis, indicating the presence of CO relatively accessible and inaccessible conformations, termed “open” and “closed” in analysis of hexacoordinate hemoglobins 30. Saturation of the observed CO binding rate allows estimates of the opening and closing rates (supplementary figure S8). Competition between CO recombination and oxidation by added ferricyanide also allows an estimate of the rate of thermal CO dissociation. Table 1 presents the kinetic constants (also shown in Figure 3A) and equilibrium binding constants that have been determined for 6 and contrasts them with several natural oxygen-binding proteins. While the oxygen off-rate is similar to human neuroglobin 31, the on-rate is almost 100 times slower and resembles Ascaris hemoglobin. These artificial proteins are robust and adaptable enough to redesign to test hypotheses that a large hydrophobic pocket, as in neuroglobin 32, can speed O2 binding, while proximal strain, as in Ascaris hemoglobin 33, slows O2 binding.
Figure 3.
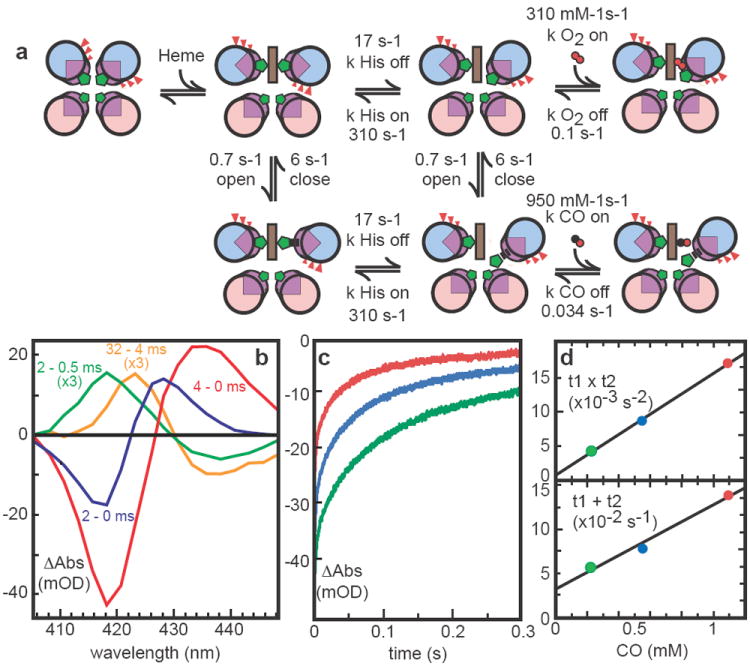
Modeling kinetics of heme ligand binding and release. A) Heme binding rotates helices but incurs strain by burying glutamates (red). Like some natural hemoglobins, at least two conformations are present; closed form cannot bind CO. His release with helical rotation precedes gaseous ligand binding. CO photolysis difference spectra (B) of 6 heme B show a rapid microsecond relaxation (yellow) followed by slower His and CO binding (green) and even slower displacement of His by CO (blue). Subsequent biphasic recombination kinetics (characteristic times t1 and t2) at 418 nm (C) as a function of CO concentration (D) determine His on/off rates.
Table 1.
Heme iron ligand on and off rates and equilibrium constants in natural and artificial proteins
| Heme protein | Ligation | kHis on (s-1) | kHis off (s-1) | kCO on (uM-1 s-1) | kCO off (s-1) | KdCO (nM) | kO2 on (uM-1 s-1) | kO2 off (s-1) | KdO2 (nM) | KdO2/ KdCO |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 (heme B) | bis-his | 310 | 17 | 0.034 | 0.1 | |||||
| Single His (Apparent)* | 0.95 (0.006) |
36 (6000) |
0.31 (0.017) |
32 (600) |
(0.1) | |||||
| Neuroglobin31 | bis-his | 2000 | 4.5 | 65 (0.15) |
0.014 | 0.21 (93) |
250 (0.56) |
0.8 | 3.2 (1400) |
(15) |
| Myoglobin 47 | his-aquo (distal his) | n/a | n/a | 0.5 | 0.019 | 37 | 14 | 12 | 860 | 23 |
| Human hemoglobin 48 | his-aquo (distal his) | n/a | n/a | 2.2 | 0.009 | 4 | 19 | 15 | 770 | 190 |
| Ascaris hemoglobin 49 | his-aquo (distal tyr) | n/a | n/a | 0.2 | 0.018 | 90 | 1.5 | 0.004 | 3 | 0.03 |
| Microperoxidase50 | his-aquo | n/a | n/a | 20 | 0.01 | 0.5 | ||||
| Combinatorial bundles44 | mixed | ? | ? | (3-11) (kapp) | 0.03-0.11 | (6-25) |
Apparent binding rates (in parentheses) are slowed by bis-histidine ligation. Corresponding rates not in parentheses are estimated binding rates without histidine impedance facilitating comparison with proteins with different fractions of bis-histidine ligation.
Test engineering elements
To test if the helical rotation model of histidine strain was indeed operating to promote CO and O2 binding, we changed the b-position glutamates of 6 to alanines. As anticipated, CO binding slows by more than an order of magnitude (supplementary figure S9). Moreover, O2 fails to form detectable oxyferrous heme. Instead, the heme undergoes oxidation within the mixing time, plausibly via an outer-sphere electron transfer. Thus, removal of the modelled entatic state 20 by conversion of interfacial glutamates to alanines disables the histidine-O2 or CO exchange-gated rotational mechanism.
In exploring the effectiveness of loops to control mobility of free helices and so exclude water from the interior, we found that the oxy-ferrous heme is not stabilized by looping together helices already linked by heme (5) but is stabilized by looping together helices not linked by heme (6). Disulfide linkage of the loops in 6 proves unessential, since eliminating the disulfide through Cys to Ser substitution maintains oxy-ferrous heme stability.
Our development of helical strain and motion constraint to promote O2 binding to ferrous heme is not constrained to one site, but extends along the 4-helix bundle to hemes at other positions. A di-heme variant of 6 with histidine at positions 7 and the complementary position 42, or a single-heme variant with histidine only at positions 42 each display oxy-ferrous heme properties similar to that observed in 6 (supplementary figure S10).
Heme and substrate specificity
The interior of 6 adopts a unique structure not only around heme B but also around other porphyrins including heme A (supplementary figures S11, S12). Heme A has markedly different peripheral substitutions (Figure 2B) that result in different redox and spectral properties. The Em8.0 of bound heme A is -100 mV (vs NHE, data not shown), about 200 mV more positive than heme B and hence less favoured to reduce O2 to O2-.. Accordingly, the oxy-ferrous heme A (spectrally analogous to the cytochrome oxidase Compound A described by Chance 26) persists longer, about half a minute, before electron transfer.
In all natural hemoglobins with distal histidines, either preferentially bound to the heme iron, as in neuroglobin, or displaced from the iron, as in myoglobin or human hemoglobin, CO is a poison that binds more tightly than O2. Our work shows this is not an essential property. The binding of CO to 6 is weaker than to natural globins (Table 1). This results in a net 10-fold discrimination favouring O2 binding over CO binding, the largest observed for any distal histidine heme-protein complex, and comparable to distal tyrosine sites with extreme O2 affinity. It seems likely that the strained, distal histidine in 6 remains available to stabilize bound oxygen, as is seen in hemoglobins, myoglobins and horseradish peroxidase 34.
Protein engineering: control complexity and utility
Management of complexity and multiple-utility is critical to engineering protein design. Natural proteins accumulate complexity in a way analogous to that described in genetic systems as “Muller’s ratchet” 2: a change is made, typically for minor or even no selective advantage, which becomes essential as new changes come to depend on this old change. The resulting interdependency between amino acids is the source of protein complexity. Not only does one amino acid commonly affect the function of nearby or allosterically remote amino acids, any amino acid typically supports multiple functions or utilities, in a way analogous to the organism scale description of Darwin 1,35,36.
Amino acid interdependency and multiple-utility of amino acids presents a serious challenge to re-engineering a natural protein for a new purpose. When more than a few amino acids are ‘rationally’ introduced to natural proteins by site directed mutagenesis, protein stability and activity are often deeply compromised 37. Limited adaptability of natural proteins to accommodate new catalytic site functionalities has been the experience to date with a variety of powerful approaches that includes abzymes 38, computational design combined with directed evolution6 and recombination through domain swapping 39.
Maquette design that begins with simple protein sequences never exposed to natural selection, and tests the effects of amino acid changes through the progressive stages of design, does not eliminate complexity or multiple utility, but it allows them to be more clearly seen and moderated. This approach offers reconciliation of each amino acid with specific roles in structure and function, the choice to eliminate redundancies, and the freedom to organize assembly and mechanistic elements while incorporating the next stage of development. As a result, it takes relatively few amino acid changes to incorporate the function of oxygen transport into a generic three amino acid bundle. That such a modest redesign succeeds supports the view that many protein-engineering elements do not require atomistic precision and that exacting mimics of natural protein sites is neither necessary nor good engineering. Indeed, despite the widespread fragility of enzymes to multiple active site mutations, it appears that relatively simple and unsophisticated engineering principles underlie natural protein functions such as electron transfer 40 and even catalysis 41,42.
One of these key engineering principles is the creation and control of protein motion. The long range helical motion and strain that facilitates ligand exchange for oxygen transport, while still permitting relatively secure heme binding via bis-his ligation, would not have been possible merely by targeting the static structure of the final oxyferrous state in a natural heme protein. Any static structure-focused design 6,7,38 fails to address molecular choreography often critical for function. On the other hand, liberal motion is equally unworkable. Our balance of allowing inter-helical protein motion but constraining it through bonded and loop contacts allows small ligands to penetrate and bind while excluding the water that so profoundly affects active site properties. In different maquette variants, limiting water access slowed the rate for heme-oxygen electron transfer by 4 orders of magnitude, suggesting the raising of a catalytic barrier by >5 kcal.
By keeping the maquette design simple and complexity low, we maintain the capacity to intentionally insert more complex functional elements such as allostery and cooperativity. The helical rotation built into this system introduces a long-range negative cooperativity in the two heme binding sites. Versions of 6 that modify the histidines at one or the other binding site clearly show that on their own both heme binding sites are high affinity, while together, rotation on binding the first heme weakens the second. Such long-range helical coupling is a prospective means to build a maquette with positive cooperativity between the heme sites for oxygen binding, recreating the vital oxygen transport regulation for which hemoglobin is so well known.
The creation of the first completely artificial oxygen transport protein allows us to reconsider the design of natural oxygen transport globins. Despite the common view that natural globins are full of exquisitely refined functional properties reflected in the globin fold 43, it is clear that transport function is readily achieved without such a fold. Indeed, the results presented here reinforce the developing view that the physical chemistry of heme in an oxygen transport protein can accommodate a wide range of histidine, oxygen and carbon monoxide ligand exchange rates including, as we have seen here, the preference for oxygen over carbon monoxide binding. The discrimination favouring O2 over CO binding comes about without added engineering. Taking into account the impedance from the 95% bis-histidine ligation in 6 expected from the His on and off rate ratio, binding of CO and O2 ligands are both relatively slow compared to most natural globins, but the thermal release rates are towards the fast and slow ends of the natural range, respectively (Table 1) 44. Traditionally, slow O2 off rates are explained in terms of nearby polar residues providing a hydrogen bond to the O2; simple modeling of 6 shows the distal histidine could play this role here. Yet no special design was required beyond giving this histidine sufficient mobility via the helical strain arising from glutamate burial. The ease with which globin-like properties can be reproduced in a completely unrelated and simply engineered maquette suggests that the relatively complex globin fold is for the most part unremarkable, and may be common in nature not because of a uniquely capable design for oxygen binding, but simply because it is good enough.
Additional Methods
Typical CO photolysis conditions are 10 μM synthetic protein in 20 mM borate, pH 9.0, 100 mM KCl and CO/Ar gas mixtures ranging from 4% to 100% CO gas saturation with a slight excess of sodium dithionite monitored in the UV to assure the heme is reduced.
Table 1 reports the direct, apparent CO and O2 binding rates as well as estimates of the faster rates when not bis-histidine ligated. Apparent CO binding rates from photolysis are ~ 20 times faster than that reported by stopped flow, suggesting that shortly after photolysis, bis-histidine ferrous 6 may be in a relatively unrelaxed “open” state, prone to histidine dissociation and CO rebinding, compared to the dark, equilibrium resting “closed” state 30.
Stable spectra of the oxy-ferrous form are also obtained by direct bubbling of ferrous heme protein with oxygen at -15C without using CO as an intermediate. Competition between CO and O2 in a range of mixtures of the two gases reaches equilibrium over 30 minutes after initial photolysis at -15 C and indicates that O2 binds with 10 times greater affinity than CO (e.g. a 91:9 ratio of CO:O2 gives 50:50 mix of bound CO and O2).
Materials
Peptide synthesis reagents were purchased from Perseptive Biosystems (Foster City, CA). Hemin was from Fluka (Buchs, Switzerland). The chloride salt of Fe3+-protoporphyrin III was synthesized by literature methods (51). Deuterium oxide, 15N-ammonium chloride, 13C-glucose and (trimethylsilyl)-propionate were from Cambridge Isotope Laboratories (Andover, MA). All other solvents and reagents were either from Fisher Scientific (Springfield, NJ) or Sigma (St. Louis, MO).
Peptide synthesis and purification
Solid phase syntheses of 1-5 were performed on a continuous flow PerSeptive Biosystems (Framingham, MA) as described previously. Crude peptides were purified to homogeneity by reversed phase C18 HPLC using aqueous-acetonitrile gradients containing 0.1% (vol/vol) trifluoroacetic acid. Peptide homogeneity and composition were assayed by analytical HPLC and laser desorption mass spectrometry.
The gene encoding 6 was designed using the program Amplify (52), the gene constructed using assembly PCR (53) and ligated into the overexpression vector pET 32d(+) (Novagen, Inc.) modified to replace the enterokinase cleavage site with a TEV-protease cleavage site, yielding the plasmid pHP7-TEV. The plasmid was transformed into DH5α cells and sequenced in the region of interest to verify the presence of the intact gene and the plasmid was transformed into BL21(DE3) cells. Mutations to 6 were created using the quick change protocol (Stratagene, Inc.)
For unlabelled expression, these were grown in TPP medium (54) at 37°C to an OD600 of 1.0 and induced with IPTG at a final concentration of 1 mM for 2 hours before harvesting. For 15N-labeled expression, cells were grown at 37°C in minimal media containing 1 g/L 15N ammonium chloride to an OD600 of ~1.0, induced with 1 mM IPTG, and shaken at 37°C for an additional 3 hours. For 13C,15N-double labeling, cells were grown in minimal media containing 1% w/v 13C glucose, 1 g/L 15N ammonium chloride and 8 mL/L 13C,15N Bioexpress medium (Cambridge Isotope Labs). Cells were grown at 37°C to an OD600 of ~1.0, induced with 1 mM IPTG, and shaken at 37°C for an additional 2 hours.
Cells were collected by centrifugation, broken open using a French press, and purified on a Ni-nitrilotriacetic acid column (Qiagen, Inc.) according to manufacturers instructions. The fusion protein was dialyzed into 50 mM Tris-HCl, 1 mM DTT, pH 8.0 and then cleaved overnight with His6-tagged TEV protease (Invitrogen, Inc.). The reaction mixture was filtered through Ni-nitrilotriacetic acid resin, and the purified 6 concentrated by lyophilization. Purified peptides were dissolved in 20 mM K2HPO4, 100 mM KCl pH 8.0 and cysteine residues were air oxidized to the symmetric disulfides overnight. Disulfide formation was followed by analytical C18 HPLC.
FTIR
6 was washed by dilution with 100 mM potassium phosphate and 100 mM KCl at pH 6.0 or 8.0 and concentrated to ~1 mM with Vivascience centrifugal concentrators. 100 μM benzyl viologen was added as a redox mediator and the sample was placed in a microelectrochemical chamber above an ATR-FTIR microprism (3 bounce silicon; 3mm diameter; SensIR). The chamber had a 5 mm diameter glassy carbon working electrode within 1 mm of the prism surface and the sample solution was connected to a counter electrode and an Ag/AgCl reference electrode via an ion-permeable frit. Potential was switched cyclically between 0 mV and −450 mV (versus SHE). Before each potential change, a reference scan was recorded. The potential was then switched and a sample spectrum was recorded after 10 minutes to allow for redox equilibration. Data at pH 6.0 averages 50 reductive minus 50 oxidative cycles of 500 interferograms each at 4 cm-1 resolution; data at pH 8 averages 40 reduced minus oxidized cycles; the double difference spectrum pH 6 minus 0.58 pH 8 reflected appropriate protein concentration dependent weighting at the two pH values.
Supplementary Material
Acknowledgments
We thank A. J. Wand for assistance in NMR measurements, P. R. Rich for FTIR measurements, M. S. Hargrove for neuroglobin reference spectra and helpful discussions and D. Hilvert for valuable suggestions. This work was supported by grants from DOE, NIH and NSF.
Footnotes
Author contributions
R.L.K. and J.L.R.A. both designed proteins and performed the bulk of the measurements; K.S.R. made initial spectroscopic observations and L.A.S. contributed to spectroscopic measurements; C.C.M. designed and performed spectroscopic measurements and analysis; paper preparation was largely conducted by C.C.M. and P.L.D.
References
- 1.Darwin C. Origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. 6. John Murray; London: 1872. [PMC free article] [PubMed] [Google Scholar]
- 2.Muller HJ. The relation of recombination to mutational advance. Mutation Res. 1964;1:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- 3.Csete ME, Doyle JC. Reverse engineering of biological complexity. Science. 2002;295(5560):1664–1669. doi: 10.1126/science.1069981. [DOI] [PubMed] [Google Scholar]
- 4.Kraut DA, Carroll KS, Herschlag D. Challenges in enzyme mechanism and energetics. Annual Review of Biochemistry. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 5.Bolon DN, Mayo SL. Enzyme-like proteins by computational design. P Natl Acad Sci USA. 2001;98(25):14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319(5868):1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008 doi: 10.1038/nature06879. in press. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan J, DeGrado WF. De novo design of catalytic proteins. P Natl Acad Sci USA. 2004;101(32):11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moffet DA, et al. Peroxidase activity in heme proteins derived from a designed combinatorial library. Journal of the American Chemical Society. 2000;122(31):7612–7613. doi: 10.1021/ja0036007. [DOI] [PubMed] [Google Scholar]
- 10.Monien BH, et al. Detection of heme oxygenase activity in a library of four-helix bundle proteins: Towards the de novo synthesis of functional heme proteins. Journal of Molecular Biology. 2007;371(3):739–753. doi: 10.1016/j.jmb.2007.05.047. [DOI] [PubMed] [Google Scholar]
- 11.Collman JP, Boulatov R, Sunderland CJ, Fu L. Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem Rev. 2004;104(2):561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 12.Jencks WP. Binding-Energy, Specificity, and Enzymic Catalysis - Circe Effect. Advances in Enzymology. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 13.Chou PY, Fasman GD. Empirical Predictions of Protein Conformation. Annual Review of Biochemistry. 1978;47:251–276. doi: 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- 14.Regan L, Degrado WF. Characterization of a Helical Protein Designed from 1st Principles. Science. 1988;241(4868):976–978. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 15.Robertson DE, et al. Design and Synthesis of Multi-Heme Proteins. Nature. 1994;368(6470):425–431. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 16.Gibney BR, et al. Iterative protein redesign. Journal of the American Chemical Society. 1999;121(21):4952–4960. [Google Scholar]
- 17.Huang SS, et al. X-ray structure of a maquette scaffold. J Mol Biol. 2003;326:1219–1225. doi: 10.1016/s0022-2836(02)01441-9. [DOI] [PubMed] [Google Scholar]
- 18.Huang SS, et al. The HP-1 maquette: From an apoprotein structure to a structured hemoprotein designed to promote redox-coupled proton exchange. P Natl Acad Sci USA. 2004;101(15):5536–5541. doi: 10.1073/pnas.0306676101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marshall SA, Mayo SL. Achieving stability and conformational specificity in designed proteins via binary patterning. Journal of Molecular Biology. 2001;305(3):619–631. doi: 10.1006/jmbi.2000.4319. [DOI] [PubMed] [Google Scholar]
- 20.Vallee BL, Williams RJP. Metalloenzymes - Entatic Nature of Their Active Sites. P Natl Acad Sci USA. 1968;59(2):498–505. doi: 10.1073/pnas.59.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shifman JM, et al. Functionalized de novo designed proteins: Mechanism of proton coupling to oxidation/reduction in heme protein maquettes. Biochemistry. 1998;37(47):16815–16827. doi: 10.1021/bi9816857. [DOI] [PubMed] [Google Scholar]
- 22.Koder RL, et al. Native-like structure in designed four helix bundles driven by buried polar interactions. Journal of the American Chemical Society. 2006;128(45):14450–14451. doi: 10.1021/ja064883r. [DOI] [PubMed] [Google Scholar]
- 23.Isogai Y, et al. Design and synthesis of a globin fold. Biochemistry. 1999;38(23):7431–7443. doi: 10.1021/bi983006y. [DOI] [PubMed] [Google Scholar]
- 24.Gibney BR, et al. Self-assembly of heme A and heme B in a designed four-helix bundle: Implications for a cytochrome c oxidase maquette. Biochemistry. 2000;39(36):11041–11049. doi: 10.1021/bi000925r. [DOI] [PubMed] [Google Scholar]
- 25.Zhuang JY, et al. Design of a five-coordinate heme protein maquette: A spectroscopic model of deoxymyoglobin. Inorganic Chemistry. 2004;43(26):8218–8220. doi: 10.1021/ic048502r. [DOI] [PubMed] [Google Scholar]
- 26.Chance B, Saronio C, Leigh JS. Functional Intermediates in Reaction of Cytochrome-Oxidase with Oxygen. P Natl Acad Sci USA. 1975;72(4):1635–1640. doi: 10.1073/pnas.72.4.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shikama K. The molecular mechanism of autoxidation for myoglobin and hemoglobin: A venerable puzzle. Chem Rev. 1998;98(4):1357–1373. doi: 10.1021/cr970042e. [DOI] [PubMed] [Google Scholar]
- 28.Wang JH. Hemoglobin Studies .2. A Synthetic Material with Hemoglobin-Like Property. Journal of the American Chemical Society. 1958;80(12):3168–3169. [Google Scholar]
- 29.Grosset AM, et al. Proof of principle in a de novo designed protein maquette: An allosterically regulated, charge-activated conformational switch in a tetra-alpha-helix bundle. Biochemistry. 2001;40(18):5474–5487. doi: 10.1021/bi002504f. [DOI] [PubMed] [Google Scholar]
- 30.Trent JT, Hvitved AN, Hargrove MS. A model for ligand binding to hexacoordinate hemoglobins. Biochemistry. 2001;40(20):6155–6163. doi: 10.1021/bi0100790. [DOI] [PubMed] [Google Scholar]
- 31.Dewilde S, et al. Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family. Journal of Biological Chemistry. 2001;276(42):38949–38955. doi: 10.1074/jbc.M106438200. [DOI] [PubMed] [Google Scholar]
- 32.Pesce A, et al. Human brain neuroglobin structure reveals a distinct mode of controlling oxygen affinity. Structure. 2003;11(9):1087–1095. doi: 10.1016/s0969-2126(03)00166-7. [DOI] [PubMed] [Google Scholar]
- 33.Peterson ES, et al. A comparison of functional and structural consequences of the tyrosine B10 and glutamine E7 motifs in two invertebrate hemoglobins (Ascaris suum and Lucina pectinata) Biochemistry. 1997;36(42):13110–13121. doi: 10.1021/bi971156n. [DOI] [PubMed] [Google Scholar]
- 34.Borovik AS. Bioinspired hydrogen bond motifs in ligand design: The role of noncovalent interactions in metal ion mediated activation of dioxygen. Accounts of Chemical Research. 2005;38(1):54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 35.Mclendon G. Control of Biological Electron-Transport Via Molecular Recognition and Binding - the Velcro Model. Structure and Bonding. 1991;75:159–174. [Google Scholar]
- 36.Page CC, Moser CC, Dutton PL. Mechanism for electron transfer within and between proteins. Curr Op Chem Biol. 2003;7:1–6. doi: 10.1016/j.cbpa.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Brannigan JA, Wilkinson AJ. Protein engineering 20 years on. Nature Reviews Molecular Cell Biology. 2002;3(12):964–970. doi: 10.1038/nrm975. [DOI] [PubMed] [Google Scholar]
- 38.Hilvert D. Critical analysis of antibody catalysis. Annual Review of Biochemistry. 2000;69:751–793. doi: 10.1146/annurev.biochem.69.1.751. [DOI] [PubMed] [Google Scholar]
- 39.Carbone MN, Arnold FH. Engineering by homologous recombination: exploring sequence and function within a conserved fold. Current Opinion in Structural Biology. 2007;17(4):454–459. doi: 10.1016/j.sbi.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Moser CC, et al. Nature of Biological Electron-Transfer. Nature. 1992;355(6363):796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 41.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301(5637):1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 42.Warshel A. Computer simulations of enzyme catalysis: Methods, progress, and insights. Annual Review of Biophysics and Biomolecular Structure. 2003;32:425–443. doi: 10.1146/annurev.biophys.32.110601.141807. [DOI] [PubMed] [Google Scholar]
- 43.Frauenfelder H, McMahon BH, Fenimore PW. Myoglobin: The hydrogen atom of biology and a paradigm of complexity. P Natl Acad Sci USA. 2003;100(15):8615–8617. doi: 10.1073/pnas.1633688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moffet DA, et al. Carbon Monoxide Binding by de Novo Heme Proteins Derived from Designed Combinatorial Libraries. Journal of the American Chemical Society. 2001;123(10):2109–2115. doi: 10.1021/ja0036007. [DOI] [PubMed] [Google Scholar]
- 45.Hargrove MS. A flash photolysis method to characterize hexacoordinate hemoglobin kinetics. Biophysical Journal. 2000;79(5):2733–2738. doi: 10.1016/S0006-3495(00)76512-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ansari A, et al. The Role of Solvent Viscosity in the Dynamics of Protein Conformational-Changes. Science. 1992;256(5065):1796–1798. doi: 10.1126/science.1615323. [DOI] [PubMed] [Google Scholar]
- 47.Springer BA, Sligar SG, Olson JS, Phillips GN. Mechanisms of Ligand Recognition in Myoglobin. Chem Rev. 1994;94(3):699–714. [Google Scholar]
- 48.Mathews AJ, et al. The Effects of E7 and E11 Mutations on the Kinetics of Ligand-Binding to R-State Human-Hemoglobin. Journal of Biological Chemistry. 1989;264(28):16573–16583. [PubMed] [Google Scholar]
- 49.Goldberg DE. Oxygen-avid hemoglobin of Ascaris. Chem Rev. 1999;99(12):3371–3378. doi: 10.1021/cr970152l. [DOI] [PubMed] [Google Scholar]
- 50.Sharma VS, Schmidt MR, Ranney HM. Dissociation of CO from Carboxyhemoglobin. Journal of Biological Chemistry. 1976;251(14):4267–4272. [PubMed] [Google Scholar]
- 51.Smith KM, Parish DW, Inouye WS. Methyl deuteration reactions in vinylporphyrins: protoporphyrins IX, III, and XIII. J Org Chem. 1986;51:666–671. [Google Scholar]
- 52.Engels WR. Contributing Software to the Internet - the Amplify Program. Trends Biochem Sci. 1993;18:448–450. doi: 10.1016/0968-0004(93)90148-g. [DOI] [PubMed] [Google Scholar]
- 53.Stemmer WPC, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-Step Assembly of a Gene and Entire Plasmid from Large Numbers of Oligodeoxyribonucleotides. Gene. 1995;164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 54.Moore JT, Uppal A, Maley F, Maley GF. Overcoming inclusion body formation in a high-level expression system. Protein Expression And Purification. 1993;4:160–163. doi: 10.1006/prep.1993.1022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.