

Abstract
Pancreatic ductal adenocarcinoma (PDA) remains a highly lethal disease; new therapeutic modalities are urgently needed. A number of immunotherapies tested in pre-clinical models have shown promise. Early phase clinical trials have demonstrated evidence of immune activation that in some cases correlates with clinical response. Moreover, recent evidence delineates inflammation’s intricate role in PDA, even at its earliest stages. PDA is thus ripe for immunotherapy; however, significant challenges remain before success can be realized. Future studies will need to focus on the discovery of novel PDA antigens, and the identification of the multiple immune suppressive pathways within the PDA tumor microenvironment that inhibit an effective PDA targeted immune response. Technologies are now available to rapidly advance discovery. Rapid translation of new discoveries into scientifically driven clinical trials testing combinations of immune agents will likely continue to shift the procarcinogenic tumor environment towards the most potent anticancer response.
Keywords: pancreatic cancer, immune therapy, cancer vaccine
Introduction
Pancreatic ductal adenocarcinoma (PDA) is the fourth leading cause of cancer related deaths in the United States with an estimated 43,920 new cases and 37,390 deaths in 2012.1 Despite dramatic improvement in mortality following surgical intervention and discovery of new chemotherapy regimens, there has been minimal improvement in disease free and overall survival over the last 30 years. As the field of cancer immunology has grown, a deeper understanding of the immune system’s recognition of tumor cells and their antigens has translated into exciting new treatments for a variety of solid tumors including PDA.
Emerging evidence supports a critical role for the immune system in PDA tumor development, progression and eradication. Clark and colleagues developed a preclinical immune competent mouse model genetically engineered to develop PDA via a point mutation in one Kras allele. This Kras mutation initiates pancreatic intraepithelial neoplasia (PanIN) which eventually progresses to PDA, and recapitulates the stromal and inflammatory cell milieu found in human PDA development.23 Evaluation of these murine PDA and PanINs demonstrated that 50% of the tumor was composed of leukocytes, particularly immunosuppressive cells, including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs), along with a very small percentage of effector T cells. The authors therefore implicated immunosuppressive cells in early PDA tumorigenesis and a possible role in facilitating disease progression.3
Other preclinical studies have recently demonstrated chronic inflammation’s role in PDA carcino genesis. For example, the expression of specific cancer test genes observed in human chronic pancreatitis specimens correlates with expression by their corresponding human PDA tumors.4 As another example, Guerra et al found that Kras mutated mice required a second signal in the form of chronic pancreatitis in order to develop PDA, suggesting that PDA stems from a combination of genetic and nongenetic (e.g., chronic pancreatitis) events.5,6 In addition, infiltrating neutrophils have been shown to contribute to PDA tumorigenesis and immune system evasion in PDA preclinical models by activating angiogenesis in the earliest stages of PDA.7-9 Furthermore, Esposito and colleagues showed that mononuclear inflammatory cells of the innate immune response are recruited to PDAs and may influence the metastatic capacity of the cancer cells.10 Finally, cyclooxygnenase-2 (COX-2) expression is elevated in human PDA and PanIN lesions and increases with increasing PanIN severity when compared to normal pancreas and chronic pancreatitis.11,12
Although PDA was originally thought to be poorly immunogenic, recent data have challenged this presumption. First, a high incidence of tumor specific T lymphocytes is seen in PDA patient bone marrows.13 Second, Fukunga and colleagues analyzed 80 surgically resected PDA tumors looking specifically for CD4+ T cells, CD8+T cells, and dendritic cells within the tumor. They reported that higher levels of CD4+ and CD8+ tumor infiltrating lymphocytes (TILs) in PDAs were associated with longer overall survival after surgical resection. The presence of both CD4+ and CD8+ TILs was an independent favorable prognostic factor in a multivariate analysis.14 However, still not known is whether these T cells are PDA specific.
These data, together with results from early immunotherapy clinical trials (discussed below) support the hypothesis that PDA is able to elicit an anti-tumor immune response. However, this is the exception rather than the rule because there also exists a variety of mechanisms that naturally suppress this response. Importantly, many of these mechanisms likely also support tumor growth and progression. The question is no longer whether or not immunotherapy will improve PDA outcomes, but how do we improve the immune system’s anti-PDA immune response and limit the tumor’s ability to evade immune system detection? This review will discuss our current understanding of tumor antigens, vaccines that are capable of delivering these antigens to induce a PDA specific immune response, and the mechanisms within the tumor microenvironment that inhibit effective immunity. In addition, pre-clinical and clinical studies testing combinatorial immunotherapy approaches that bypass mechanisms of immune tolerance to activate the most potent antitumor immune response will be summarized.
Tumor Antigens and Immune Tolerance
Cancer cells are derived from their normal counterparts after undergoing genetic alterations resulting in their malignant phenotype. These genetic alterations lead to the expression of tumor-associated antigens (TAA) which are misfolded, altered proteins expressed by cancer cells. TAAs are presented by major histocompatibility complex (MHC) molecules (human leukocyte antigen (HLA) in humans) to effector T cells. These T cells contain unique T cell receptors which recognize specific antigen epitopes bound to MHC molecules. This interaction is known as “signal 1”. Signal 1 alone is insufficient for appropriate T cell activation and results in T cell anergy or apoptosis, allowing the tumor to evade immune system detection. The concomitant binding of co-stimulatory molecules such as B7-1 and CD28, on antigen presenting cell (APC) and T cell surfaces respectively, results in “signal 2”, a critical component in T cell activation (Fig 1A).
Figure 1.
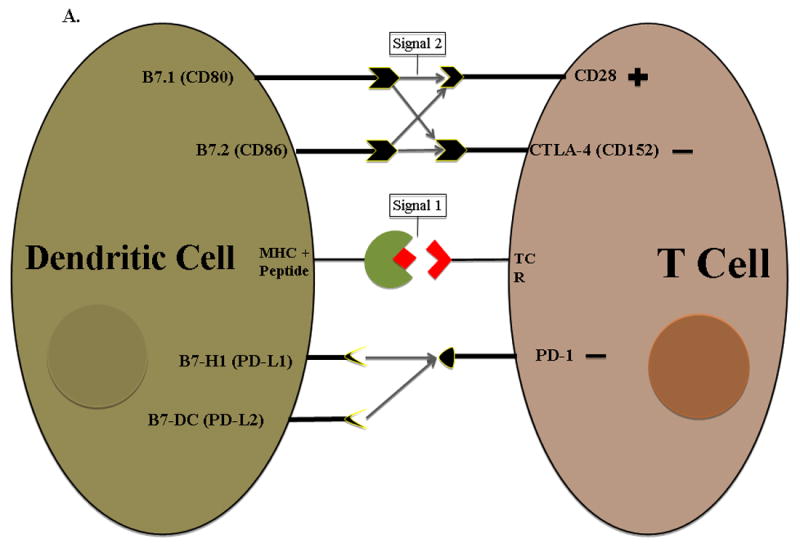
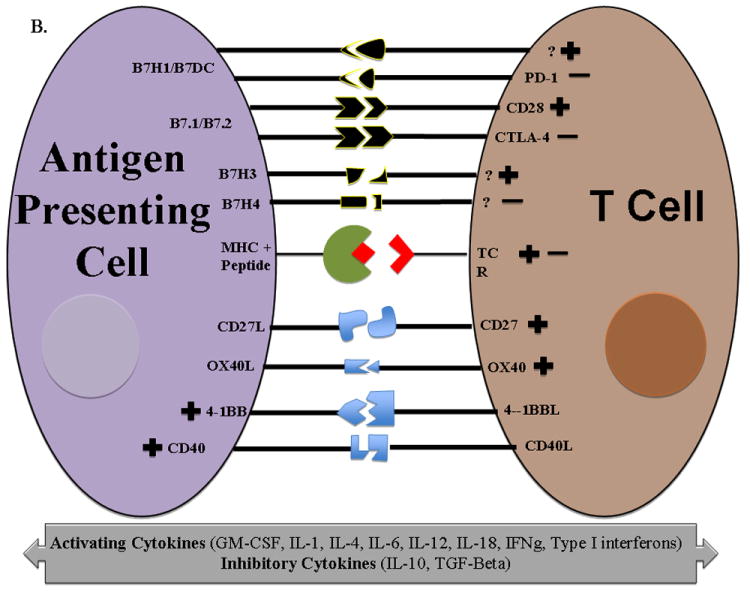
A. TAAs are expressed as small peptide fragments on HLA molecules and presented to T cells for recognition and activation and is referred to as T cell signal 1. However, tumor cells cannot effectively activate T cells alone because they do not express the co-stimulatory molecules such as B7.1 and B7.2 which provide signal 2. Vaccines are designed to deliver TAAs to antigen presenting cells which naturally upregulate both signal 1 and signal 2. Dendritic cells are the most efficient antigen presenting cells due to their ability to readily process tumor antigen and present them on both MHC I and II surface molecules which then bind to T cell receptors on both CD4+ and CD8+ T cells. DCs also upregulate B7-1/B7-2 expression (signal 2) for efficient T cell activation. Members of immune regulatory cell surface receptors on T cells such as CTLA-4 turn off activated T cells by competing with activating receptors (signal 2) for the same ligand, allowing the tumor to evade immune recognition. Cytokines produced by T cells mediate both activating and inhibitory immune responses. B. Examples of different family members of both co-stimulatory and inhibitory signals on antigen presenting cells that interact with T cells. TCR = T cell receptor MHC = Major histocompatibility complex
Most tumor cells lack the necessary surface molecules, i.e. B7-1 (CD80) and B7-2 (CD86), to complete signal 2.15 Professional APCs, for example dendritic cells (DCs), are more efficient T cell activators. Intracellular processing mechanisms unique to DCs enable them to efficiently process tumor antigens and present them on both MHC I and MHC II surface molecules resulting in tumor specific CD8+ and CD4+ T cell activation, respectively. Mature DCs constitutively express co-stimulatory molecules in close proximity to MHC complexes.16 Examples of members of the currently known co-stimulatory families are shown in figure 1B.
Immunotherapy of cancer aims to take advantage of the immune system’s natural ability to recognize and react against new antigens – in the case of cancer, TAAs. However, tumor cells utilize various mechanisms to evade immune system detection. Inflammatory signals, i.e. interferon gamma (IFN-γ), transforming growth factor β (TGF-β), and interleukin 10 (IL-10), play critical roles in both tumor eradication and development (Fig 1A).17 Cell surface molecules such as cytotoxic T lymphocyte antigen-4 (CTLA-4), PD-1, PD-L1 (B7-H1) and PD-L2 (B7-DC) are examples of immune checkpoint signaling pathways which downregulate T cell activation.18 Other families of checkpoint inhibitors are shown in figure 1B.
Tumor cells, stromal cells, and immune cells within the tumor microenvironment all produce factors that suppress TAA specific T cell responses. TGF-β and the TGF-β receptor interaction results in nuclear translocation of the SMAD4/DPC4 complex.19 The SMAD/DPC4 complex has been shown to suppress tumor growth and is down regulated in over 50% of advanced PDAs.20 TGF-β directly inhibits T cell proliferation and suppresses CD8+ T cell activity via its interactions with SMAD4/DPC4 intracellular protein complex.19 Moreover, TGF-β upregulates FoxP3 expression, a marker for CD4+ CD25+ Tregs which play an integral part in the tumor’s ability to evade the immune system.19
IL-10, produced by Tregs and other immune cells, promotes immune tolerance by directly inhibiting APCs and Th1 cell differentiation and proliferation.21 Analysis of human pancreatic cancer cells after surgical resection demonstrated tumor derived TGF-β and IL-10 acting in conjunction to promote a Th2 cell response and inhibit Th1 cell differentiation and proliferation.22 Th2 predominant immune responses are associated with poor protection against malignant tumors and reduced survival in pancreatic cancer patients when compared with a Th1 response.23,24 This is most likely due to DCs’ inability to activate CTLs in the presence of IL-10 and IL-10’s ability to act directly on tumor cells and downregulate HLA class I expression.21
CTLA-4 is expressed on the activated T cell surface and competes with CD28 to bind B7-1. This interaction turns off the activated T cells by inhibiting B7.1’s intracellular signaling cascade. The CTLA-4 B7.1 interaction limits the immune systems activation response to both foreign and self-antigens. CTLA-4 knockout mice display severe autoimmune disease and have shortened lifespans.18 Although these immunologic checkpoints are important in maintaining self-tolerance, tumor cells utilize them to turn off the anti-tumor immune response.
B7-H1 (PD-L1), and B7-DC (PD-L2) are cell surface ligands expressed by many tumor cells, DCs, activated T cells, B cells and macrophages. They bind to their shared receptor, PD-1, on activated CD4+ and CD8+ T cells. The PD-L1 or PD-L2 and PD-1 interaction downregulates activated CD4+ and CD8+ T cells resulting in cell cycle arrest.25 This tumor specific immunologic checkpoint is yet another mechanism used by tumor cells for immune evasion.
The tumor microenvironment utilizes various other mechanisms and factors such as vascular endothelial growth factor (VEGF), interleukin (IL-6), and cyclooxygenase 2 (COX-2) to impede DC differentiation and maturation.16 Moreover, DC activating cytokines such as granulocyte macrophage colony stimulating factor (GM-CSF) and interleukin 4 (IL-4) are decreased in the tumor microenvironment.16 Stromal cells also secrete cytokines, chemokines, and other tumor promoting factors that inhibit effective tumor killing.17 Thus, the combination of stromal cells, DCs, endothelial cells, macrophages and tumor cells within the tumor microenvironment secrete a combination of cytokines and cellular signals that ultimately determine whether a pro-tumor or anti-tumor immune response is triggered. As discussed below, immunotherapies aim to tip the balance in favor of an antitumor immune response.
Tumor Associated Antigen Identification and Antigen Presentation
The identification of PDA TAAs remains critical to the development of more effective vaccine therapy and the assessment of tumor specific T cell responses. For many years, PDA vaccines targeted a few known proteins that were either the products of oncogenes (mutated Kras) or differentially expressed glycoproteins such as MUC1 and CEA.26 The recent developments in genetics and proteomics have accelerated progress in PDA antigen identification (Fig 2). Differential gene expression analyses enable identification of highly expressed genes in PDA cells when compared with non-neoplastic cells. As an example, using serial analysis of gene expression, Argani and colleagues identified mesothelin as being overexpressed in the majority (close to 100%) of PDAs.27 A follow up studied utilized immunized lymphocytes from PDA patients treated with a granulocyte-macrophage colony stimulating factor (GM-CSF) secreting vaccine (GVAX) to screen mesothelin peptides and demonstrated an increase in post vaccine T cells specific for mesothelin in patients who demonstrated prolonged disease free survival. 28Mesothelin encompasses many of the principles necessary for an optimal tumor antigen in that it has a very low expression profile in normal cells, is highly expressed in PDA cells, and is possibly involved in cancer progression and metastasis.17 Anti-mesothelin antibodies have been used in early phase clinical trials in PDA, mesothelioma and ovarian cancer patients with limited clinical efficacy.29 However, a live attenuated Listeria monocytogenes (LM) recombinant bacterial vaccine expressing mesothelin was administered to metastatic PDA patients with over 37% of patients living longer than 15 months.30 A phase II randomized controlled multicenter trial is currently evaluating the combination of LM expressing mesothelin as a boost following priming with pancreatic GVAX in adults with metastatic PDA (discussed below).
Figure 2. Methods of Tumor Antigen Identification.
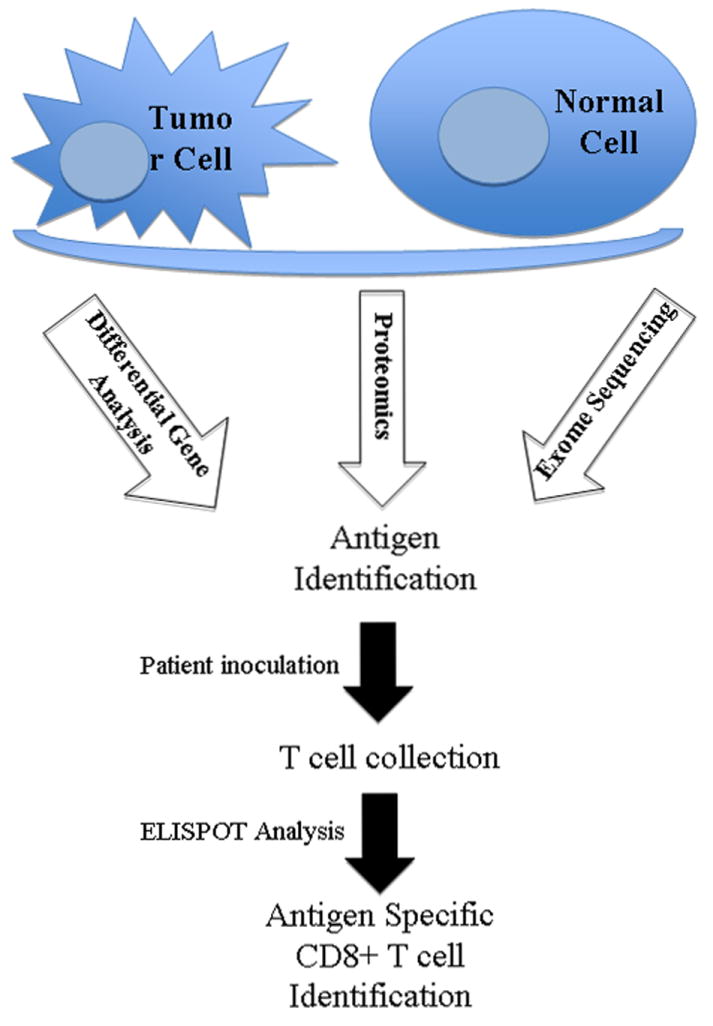
Differential gene analysis, exome sequencing and proteomics are three different methods of identifying tumor TAAs in PDA. Immunoassays conducted on T cells from vaccinated PDA patients determine the presence of antigen specific cytotoxic CD8+ T cells. These antigens can then be used to develop more potent antigen specific immunotherapies.
In recent years, proteomic based approaches such as serological analysis of recombinant cDNA expression libraries, serological proteome analysis, and protein microarray technology have all been used as an alternative approach to identify tumor antigens. Annexin A2 was recently identified as a PDA antigen, utilizing human sera to screen the proteins expressed by the PDA GVAX which served as the proteome. Subsequently, pre- and post treatment sera were analyzed from 60 patients treated with GVAX and demonstrated a post-treatment induction of annexin A2 antibodies in the majority of patients with prolonged disease free survival.31 This suggested that antibody responses might be inhibiting annexin A2 mediated tumor progression. Pre clinical studies demonstrated that annexin A2 plays a critical role in pancreatic cancer invasion and metastasis, making it unlikely that annexin A2 expression will be lost during cancer progression.31 Moreover, annexin A2 directed therapy prevented tumor metastases in two mouse models of PDA, suggesting a critical role for annexin A2 in PDA metastatic activity. Anti-annexin antibodies and vaccines are currently in development for future clinical testing.32
Exome sequencing is a type of high-throughput dideoxy sequencing analysis that specifically focuses on the coding portion of the genome and is capable of identifying most types of coding genetic alterations in human cancer.33 Jones and colleagues analyzed 24 pancreatic cancers and found that they contain an average of 63 genetic alterations.34 These alterations identified a core set of 12 cellular signaling pathways including Kras signaling, TGF-β signaling and hedgehog signaling pathways, which were modified in 67 to 100% of the tumors.34 More and more PDA tumors have been sequenced using exome sequencing. This type of high output and efficient gene analysis provides the opportunity to move from generic vaccines to patient specific vaccines. It is now possible to quickly sequence individual patient tumors and identify patient specific mutations that may serve as unique vaccine targets. These new mutation derived TAAs are likely to be critical to that particular tumor’s growth and differentiation, and also a relatively new change in the tumor microenvironment thereby decreasing the likelihood of immune tolerance to these unique antigens. One study has already shown that specific mutation identification using exome sequencing can lead to the discovery of novel and unique epitopes with higher affinity to HLA complexes thereby enhancing antigen presentation.35 Thus, future pancreatic cancer vaccine therapy should feasibly be tailored to the individual patient based on their own tumor characteristics.
Vaccine Therapy
The most potent vaccine therapy potentiates antigen presentation by DCs activating pathogen specific effector and memory T cells. Cancer vaccines were first approved for hepatoma and cervical cancer prevention. More recently, the first vaccine (Provenge, Sipuleucel-T) was approved for the treatment of prostate cancer.36
Antigen Specific Vaccines
Antigen specific vaccines target known tumor antigens thereby eliciting a tumor specific immune response. Peptide or protein vaccines use identified immunodominant tumor epitopes to stimulate T cell anti-tumor responses. In PDA, the natural starting point for these vaccines became tumor markers such as carcinoembryonic antigen (CEA) and MUC-1, as well as mutated proteins that play a prominent role in PDA such as Ras and telomerase.37 The advantages of peptide vaccines include ease of manufacturing and administration but they are also poorly immunogenic and less able to create immunological memory.38 The first clinically tested PDA vaccine was a mutant Ras peptide vaccine designed to stimulate an anti mutant Ras response. Of the 5 PDA patients in this trial, two lived over 8 months and immunologic analysis demonstrated the presence of tumor and Ras mutation specific CD4+ and CD8+T cells in both of these patients.39,40 However, follow up studies failed to show clinically significant immunity to peptide delivered Kras.
Evidence suggested that the treatment failure of these early vaccines was due to the immune system’s inability to respond appropriately to these antigens.41 This led to the discovery that local cytokines were crucial in eliciting a proper immune response and that GM-CSF was found to be the most effective at stimulating the activation of both CD4+ and CD8+ T cells in murine tumor models.41,42 Subsequently, peptide vaccination was administered in combination with GM-CSF, eliciting more effective dendritic cell activation and antigen presentation. A second mutated ras peptide vaccine clinical trial was conducted where 48 histologically confirmed PDA patients received mutant ras peptide vaccine, this time in combination with intradermal GM-CSF. Over 50% of patients demonstrated a tumor specific immune response associated with their Ras mutant vaccine. Moreover, these patients demonstrated a statistically significant improved median overall survival versus their non-responding counterparts (148 days vs. 61 days, p=0.0002) (Table 1).43
Table 1.
Pancreatic cancer vaccine trials
| Study | Patient Population/Stage of Disease | Vaccine | Immunologic Analysis | Median Survival | Reference | |
|---|---|---|---|---|---|---|
| Peptide Vaccines | Gjertsen, 1995 | 5 patients with histologically confirmed PDA | Mutated KRAS Peptide | 2/5 with vaccine-induced ras specific CD4+ and CD8+ T cells | 2 immune responders showed longer survival | [39] |
| Gjertsen, 2001 | 48 patients, 10 surgically resected PDA & 38 with advanced PDA | Mutated KRAS Peptide with GM-CSF | Mutant KRAS specific response in 58% of patients | 148 days in responders versus 61 days in non- responders | [43] | |
| Bernhardt, 2006 | 48 patients with unresectable PDA | Telomerase Peptide (GV1001) with GM-CSF | Immune response measured as DTH and in vitro T cell proliferation in 24 of 38 patients (75% of intermediate dose group) | 8.6 months in intermediate dose group | [46] | |
| Gilliam, 2012 | 154 patients with advanced PDA, unwilling or unable to take chemotherapy | Gastrin peptide vaccine (G17DT) versus placebo | Anti-G17DT response in 78% of patients in G17DT arm | 151 days (G17DT) vs. 82 days (placebo), P= 0.03 | [44] | |
| Recombinant Vaccines | Kaufman, 2007 | 10 patients with advanced stage PDA | TRICOM, MUC-1 & CEA in poxvirus with GM-CSF | Antigen specific T cell responses in 5 of 8 evaluated patients | 15.1 months in responders vs. 3.9 months in non-responders (p=0.002), 6.3 months overall | [49] |
| Le, 2012 | 28 patients with mesothelioma, lung, pancreatic, or ovarian cancer liver metastases | Live attenuated Listeria vaccine (ANZ-100) (n=9) vs. Live attenuated mesothelin expressing Listeria vaccine (CRS-207) (n=17) | LLO and Mesothelin specific T cell responses seen in CRS-207 arm | 37% of patients in CRS-207 arm alive after 15 months | [30] | |
| Dendritic Cell Vaccine | Lepisto, 2008 | 12 patients with resected pancreatic and biliary cancers | MUC-1 pulsed autologous DC vaccine | Not reported | 26 months | [56] |
| Whole Cell Vaccines | Jaffee, 2001 | 14 patients with resected PDA | GM-CSF secreting whole cell vaccine in combination with standard chemoradiation | 3 of 14 patients developed DTH to autologous tumor cells | NR (3 of 14 patients with prolonged disease free survival) | [61] |
| Laheru, 2008 | 50 patients with advanced PDA, having failed at least 2 chemotherapy regimens | Arm A: GM-CSF whole cell vaccine alone (n=20) vs. Arm B: GM-CSF whole cell vaccine & Cy (n=30) | Enhanced mesothelin specific T cell responses associated in Arm B | Arm A: 2.3 months Arm B: 4.3 months | [63] | |
| Hardacre, 2010 | 62 patients with resected PDA | HyperAcute-Pancreas Vaccine plus standard gemcitabine/5-FU based chemotherapy | NR | NR | [66] | |
| Lutz, 2011 | 60 patients with resected PDA | GM-CSF secreting whole cell vaccine in combination with standard chemoradiation | Induction of CD8+ mesothelin specific T cells correlating with overall survival | 24.8 months | [62] | |
| Combinatorial Immunotherapy | Le, 2012 | 30 patients with locally advanced, treatment refractory or metastatic PDA | Ipilimumab (IPI) alone vs. IPI plus GM-CSF whole cell vaccine | IRAEs (p=0.037), mesothelin T cell responses (p=0.012), and enhancement of the T cell repertoire (p=0.026) associated with OS in both arms | IPI alone: 3.3 months IPI plus GM-CSF vaccine: 5.5 months | [71] |
PDA pancreatic ductal adenocarcinoma; vs. versus; DTH delayed type hypersensitivity responses; GM-CSF granulocyte macrophage colony stimulating factor; DC dendritic cell; LAK lymphokine activated killer; Cy cyclophosphamide; LLO listeriolysin o; OS overall survival; IRAE immune related adverse effects
Another early phase clinical trial used a gastrin based vaccine in 154 patients with advanced stage PDA. This multi-institution double blinded placebo controlled trial demonstrated a nearly two fold increase in median overall survival in the treatment versus the placebo group (151 days versus 82 days, respectively, p = 0.03).44 Anti-gastrin immune responses were noted in 73.8% of the patients and correlated with longer overall median survival versus non responders and placebo (176 vs. 63 vs. 83 days respectively, p = 0.003).44
Telomerase, which is reactivated in over 85% of PDA cells, has also been used to develop PDA peptide vaccines. Phase I and II clinical trials involving patients with locally advanced and metastatic PDA also demonstrated prolonged survival in immune responders versus non responders in early phase clinical trials.45,46 These studies were encouraging, demonstrating evidence of immune induction that was associated with survival benefits. However, the very modest survival benefits were likely due to the suboptimal ability of peptide vaccines to adequately activate DCs and induce the most potent immune response.46
Recombinant vaccines contain bacterial and viral antigen carriers thereby increasing DC activation and improving antigen presentation. They stimulate the innate immune response while efficiently recruiting and activating DCs. The most common carriers include Bacille Calmette-Guerin (BCG), Listeria monocytogenes (LM), Salmonella and Poxviruses. Clinical trials using CEA and MUC1 recombinant vaccines have shown little efficacy in PDA patients. However, the failure of this approach was likely due to the lack of immunogenicity of the TAAs that were targeted and to the advanced stage of the PDA patients in the study.47-50 LM based vaccines have been widely studied, given LM’s intracellular lifecycle and subsequent ability to target MHC class I and II pathways.51 Additionally, LM is simple to grow, treat, and easily engineered to express antigens making it an ideal vector for tumor immunotherapy.51
Genetically engineered LM vaccines carrying tumor antigens have been studied in murine breast and colon cancer models. LM vaccination provided tumor regression and overall survival improvement versus placebo.52,53 In a pre-clinical metastatic colon cancer model, an attenuated LM strain expressing the immunodominant murine colon cancer antigen AH1 was shown to increase overall survival. LM was noted to preferentially infect the liver and promote a peritumoral proinflammatory environment leading to the influx of tumor specific T cells and IFN-γ production.54 These findings sparked the first human studies using antigen expressing LM based vaccines.
Patients diagnosed with hepatic metastases from mesothelioma, ovarian cancer, non-small cell lung cancer, and PDA were administered a live attenuated Listeria strain expressing mesothelin, a cell surface molecule overexpressed by these tumors. The vaccine was well tolerated with minimal adverse effects.30 Thirty-seven percent of patients survived over 15 months. Half of these patients were those with metastatic PDA and immunologic analysis revealed listeriolysin O and mesothelin specific T cell responses.30
Viral vectors have also been modified to express TAAs such as prostate specific antigen (PSA), and human papillomavirus (HPV) antigens. A poxvirus PDA vaccine expressing both MUC-1 and CEA combined with GM-CSF called PANVAC-V has also been developed. A phase I trial involving patients with advanced treatment refractory PDA reported a median overall survival of 6.3 months.49 Immunologic analysis revealed a MUC-1 or CEA specific T cell response in 62.5% of patients and was associated with a significant survival advantage over non-responders (15.1 months versus 3.9 months, respectively, p = 0.002).49 A phase III randomized controlled clinical trial of 255 metastatic pancreatic cancer patients was conducted where PANVAC-V was compared with standard gemcitabine therapy, and regrettably the vaccine failed to demonstrate an improvement in overall survival.50
As discussed above, DCs play a critical role in creating an efficient anti-tumor immune response. DC vaccines attempt to exploit these characteristics by isolating DCs and loading them with tumor associated antigens or tumor mRNA while in culture and subsequently reinfusing them in patients.55 DC vaccine therapy has been mostly studied in prostate cancer, breast cancer, melanoma and renal cell cancer where it has been generally well tolerated but with underwhelming clinical efficacy.17 Twelve patients with surgically resected pancreatic and biliary cancer patients were treated with MUC-1 loaded DC vaccine in an early phase clinical trial. Thirty-three percent of these patients were alive four years after treatment although immunological analysis did not demonstrate elevated anti-MUC1 antibodies.56 In a separate clinical trial, DC vaccine therapy combined with standard gemcitabine in patients with inoperable PDA refractory to standard treatment failed to demonstrate a statistically significant survival advantage.57 The reason for this failure could be due to the lack of immunogenicity of the TAA that was targeted, to the combination with gemcitabine which can induce lymphopenia and dampen the immune system’s ability to respond, or to the advanced stage of the patients treated.
Unfortunately we are often unable to ascertain which specific tumor antigen will lead to the most robust immune response in each particular patient. The ideal antigen will not be susceptible to pre-existing tolerance, will be critical to the tumor’s malignant phenotype and will be expressed solely by the tumor.17 Given the tumor’s adaptive ability and ever-changing genome, it is unlikely that a single antigen will fulfill all of these criteria.
Whole Cell Vaccines
Whole cell vaccines avoid the need for antigen specification since they include irradiated whole tumor cells. These tumor cells are transduced with GM-CSF genes and more efficiently prime the immune system by enhancing antigen presentation by DCs. Multiple tumor antigens can then be processed from the irradiated tumor cells in the presence of these activated DCs.
The first clinical application of GM-CSF transduced whole cell vaccines was attempted in late stage renal cell carcinoma patients. The vaccine was well tolerated at all doses with minimal clinical side effects. Delayed type hypersensitivity reactions were noted in patients receiving the GM-CSF autologous vaccine and correlated with their clinical response.58
Autologous whole cell vaccines have subsequently been tested in melanoma, prostate and non-small cell lung cancer. Unfortunately, the processing and development of autologous vaccines is not feasible for all cancers, including PDA. An alternative approach is the use of allogeneic whole cell tumor vaccines. The potential drawback when compared with autologous vaccines is that patients are no longer being vaccinated against their own specific tumor antigens. Evidence suggests, however, that antigen presentation by DCs is host derived and therefore the vaccine cell’s sole role is to present antigens.41 Additionally, of the tumor antigens we have identified in melanoma, over 50% are shared amongst all tumor cell lines making allogeneic vaccination a reasonable approach.41
Allogeneic GM-CSF vaccine therapy, called GVAX, has been tested in a variety of early phase clinical trials. For the most part, it has been well tolerated with minimal adverse effects.59,60 The first phase I study of pancreatic GVAX was performed on 14 patients with stage 2 or 3, surgically resected PDA. The vaccine was administered in three escalating doses, the first one being 8 weeks after surgical resection. Patients then received standard adjuvant chemoradiation for 6 months followed by 3 additional monthly vaccinations. Post vaccination delayed type hypersensitivity responses to autologous tumor cells were noted in 3 of the 14 patients, all three of which are alive more than 12 years after their diagnosis.61
Mesothelin has been considered as a PDA tumor antigen since it is upregulated in most pancreatic tumor cells. Immunologic analysis of the phase I pancreatic GVAX trial demonstrated induction of CD8+ T cells to multiple HLA-A2, A3, and A24-restricted mesothelin epitopes in the three patients experiencing DTH responses.28 None of the tumor cells used in GVAX were shown to express HLA-A2, A3, or A24 thus providing the first clear cut evidence that tumor specific CD8+ T cells can be generated by an immunotherapy approach designed to recruit APCs to the vaccination site.28
A phase II GVAX study was subsequently conducted in 60 patients with surgically resected PDA. Again, patients received their first vaccine 8 weeks after surgical resection followed by standard chemoradiation. If patients were disease free at that point, they then received three additional monthly vaccines. In contrast to the phase I study, patients in this phase II trial received an additional and final fifth vaccine 6 months after their fourth vaccine. The median disease free survival and overall survival was17.3 months (95% CI, 14.6–22.8) and 24.8 months (95% CI, 21.2–31.6) respectively.62 The vaccine was well tolerated with minimal adverse effects. Moreover, immunologic analysis revealed mesothelin specific CD8+ T cells that correlated with prolonged disease free survival.62
A second phase II clinical trial looked at the combination of GVAX therapy with immune modulating doses of cyclophosphamide (Cy). It was also the first study where GVAX therapy was used to treat patients with metastatic PDA. Eligible patients were those with stage 4 PDA who had failed gemcitabine therapy. Fifty patients were divided without randomization into two arms: 30 patients received GVAX alone and 20 patients received intravenous cyclophosphamide one day prior to GVAX therapy. Median survival was 2.3 months in the GVAX alone versus 4.7 months in the GVAX plus Cy cohort.63 Although not statistically significant, the presence of higher avidity mesothelin specific T cells showed a strong trend toward prolonged progression free survival.63 The Cy and GVAX combination was well tolerated with minimal adverse effects. The authors concluded that vaccine therapy in combination with immune modulating doses of cyclophosphamide is safe, feasible and acceptable treatment for patients who have progressed on first line chemotherapy regimens.63
Our group is currently conducting a phase II study to assess how long after initial vaccination PDA patients should continue receiving boosting vaccinations. Eligible patients for this study include disease free survivors from the aforementioned phase I and II GVAX trials and vaccine naïve patients who have received four primary monthly vaccinations. The boosting vaccination protocol calls for vaccination every 6 months as long as the patient remains disease free.
Despite the encouraging safety and survival data from these phase I and II trials, tumor tolerance continues to limit vaccine therapy. Animal studies suggest that Cy does not have a durable effect in depleting Tregs but repetitive administration of metronomic Cy may lead to durable Treg inhibition.64 An ongoing randomized controlled clinical trial is now testing GVAX in combination with Cy, including metronomic Cy, in patients with resectable PDA in the neoadjuvant and adjuvant setting. Eligible patients are randomized into one of three arms: GVAX alone, GVAX in combination with Cy given one day prior to each vaccination or GVAX in combination with repetitive administration of metronomic Cy. Primary endpoints include the assessment of the immune cell infiltrates, particularly Tregs, CD4+ and CD8+ effector T cells in the resected tumors following the neoadjuvant vaccination, as well as the assessment of the changes in the number and function of peripheral mesothelin-specific CD8+ T cells. Participants receive their first vaccination two weeks prior to undergoing pancreatic resection (the Whipple procedure) and a second vaccination in 6-10 weeks after surgical resection followed by standard adjuvant chemoradiation. If patients remain disease free 1-2 months after standard adjuvant therapy, they receive the vaccine once every 28 days for a total of six vaccinations.
Administering the vaccine in the neoadjuvant setting for the first time has given insight into the tumor’s ability to evade the immune system and suppress vaccine therapy. Immunohistochemistry staining of the resected tumors has revealed peri and intratumoral lymphoid aggregates for the first time. Moreover, PD-1 and B7-H1 positive immune cells are abundantly present within these aggregates, suggesting that a combination of PD-1 and B7-H1 blockade may further enhance the anti-tumor effect of GVAX therapy (unpublished data).
More recently, HyperAcute vaccines, which are allogeneic whole cell vaccines composed of irradiated cancer cells that have been genetically modified to add α(1,3)-Galactosyl (αGal) residues to their cell-surface have been utilized in early phase clinical trials with promising results. Human cells lack a key enzyme in a common αGal epitope pathway. Chronic stimulus from gut flora leads to high levels of circulating anti-αGal antibodies.65 The complement-mediated destruction of αGal labeled tumor cells results in an enhanced anti-tumor immune response targeting tumor-associated antigens. A HyperAcute-Pancreas vaccine phase II clinical trial of patients with previously resected PDA has been conducted. Hardacre et al reported 90% (18/20) overall survival for patients with approximately 1 year of follow-up.66 Median disease free survival (DFS) had not been reached but the data suggested a pending significant increase in median DFS when compared with historical controls.66 Moreover, they reported no significant grade 3 or 4 toxicities.66 A phase III clinical trial is currently being performed.
Immune modulating agents and combinations under development for PDA
Pancreatic cancer immunotherapy will benefit greatly from PDA specific tumor antigen identification but tumor cells still have the capability of utilizing immune checkpoints to evade detection. CTLA-4 and PD-1 are well characterized as inducing T cell apoptosis and anergy when bound to their ligands. Moreover, PD-1 and B7-H1 are highly expressed in peritumoral lymphoid aggregates seen with GVAX therapy suggesting that this immunologic checkpoint is used to evade anti-tumor immune response. Single agent anti-PD1 and anti-CTLA4 therapy has evoked promising results but is most likely insufficient individual treatment regimens for PDA. Mesothelin specific CD8+ T cell responses are upregulated when GVAX is administered in combination with anti-PD1 antibodies versus GVAX alone in murine models (unpublished data). Moreover, the combination of GVAX therapy and anti-PD1 has demonstrated a survival advantage in pre-clinical metastatic pancreatic cancer models (unpublished data).
Anti-CTLA-4 monoclonal antibody therapy has produced durable tumor regression in murine melanoma models and early phase clinical trials in patients with advanced melanoma.18,67-69 Despite these promising results, anti-CTLA-4 therapy is associated with a nearly 30% incidence of grade III/IV autoimmune toxicity.69 These adverse effects include colitis and encephalitis but also correlated positively with clinical response.69 In an early phase clinical trial of 27 patients with locally advanced or metastatic pancreatic cancer, single agent ipilimumab, a human anti-CTLA-4 monoclonal antibody, did not have any responders but one subject did experience a significantly delayed response.70 As discussed above, this was predicted from preclinical models that support combinations of immune modulators with vaccine as being more effective than single agent immune modulators administered alone.
The combination of vaccine therapy and immune checkpoint blockade is currently being tested in early phase clinical trials. Le and colleagues conducted a phase Ib trial where 30 subjects with previously treated, locally advanced or metastatic PDA were randomized to one of two arms: ipilimumab (IPI) alone or IPI in combination with GVAX. Median overall survival was 3.3 months in IPI alone versus 5.5 months in IPI plus GVAX (p=0.12). Twelve month overall survival was 7% versus 27% in the IPI group versus IPI plus GVAX group respectively.71 Immune related adverse effect profiles were similar and were associated with increased overall survival in both arms.71 Although underpowered, this study suggests a possible role for combinatorial immunotherapy in advanced PDA.
The anti-tumor immune response is determined within the tumor microenvironment. It is here where the complex interactions between tumor cells and the adjacent immune cell network dictate tumor tolerance or eradication. Multiple reports have documented the presence of immunosuppressive cells at the early points of tumor development including Tregs, MDSCs and TAMs.72 This has lead to extensive efforts to modify the tumor microenvironment in a manner that limits its immunosuppressive capabilities.
Beatty and colleagues used an agonist CD40 antibody in both a pre-clinical model to demonstrate that CD40-activated macrophages rapidly infiltrate tumors, become tumoricidal, and facilitate the depletion of tumor stroma in a pancreatic cancer murine model.73 They also tested the agonist CD40 antibody in combination with gemcitabine in patients who were chemotherapy naïve and surgically unresectable. Of the 21 patients who received the antibody, 4 had a partial response, 4 had progressive disease and 11 had stable disease.73 Two patients were excluded from the analysis due to a cerebrovascular accident and inability to receive post therapy imaging.73 The median progression free survival was 5.6 months and median overall survival was 7.4 months.73 Compared to historical controls receiving standard gemcitabine therapy, anti-CD40 therapy revealed promising clinical efficacy in this early phase clinical trial.
B7-H1, the ligand for the immune T cell expressed immune checkpoint PD-1, is expressed on DCs and its expression is upregulated by certain tumor cells.25 PD-1 is expressed by tumor associated T cells, including Tregs. The PD-1/ B7-H1 interaction decreases IL-12 production by DCs and the anti-tumor immune response is limited.25 Monoclonal antibodies directed at B7-H1 and PD-1 in murine models have led to increased cytotoxic T cell mediated tumor specific immunity and are currently being investigated as single agent therapies in early phase clinical trials.74 Its adverse effect profile is considerably lower than that seen with ipilimumab. Anti-PD-L1 therapy in patients with treatment refractory non-small cell lung cancer, melanoma and renal cell cancer was observed to have an objective response rate of 6-17%.75 Anti-PD1 therapy was noted to have an objective response rate of 20-25% in a similar patient population.76 Both therapies were associated with grade III/IV autoimmune toxicity in 9-14% of patients. 75,76
In addition to expressing PD-1, Tregs have a variety of mechanisms which limit the anti tumor immune response.77 Tumors have been shown to induce rapid expansion of Tregs in both human and murine tumor models, leading to delayed rejection of immunogenic tumors.78-80 Although they are generally present in small numbers, Treg presence is elevated in the tumor microenvironment. They inhibit CD4+ and CD8+ T cells through direct cell to cell contact and by secreting immune mediators such as IL-10 and TGF-β.17
Hiraoka and colleagues analyzed 198 PDA tumor specimens along with 84 associated PanIN lesions, comparing tumor T cell populations with non-neoplastic pancreatic tissue. They reported significantly higher Treg populations in PDA tumors when compared with non-neoplastic inflammation (p< 0.0001).81 Moreover, a higher Treg prevalence within the PDA tumor specimens was associated with a worse prognosis independent of other survival factors (p<0.0001).81 Anti-Treg therapy with monoclonal antibodies targeting Treg molecules such as glucocorticoid-induced tumor necrosis factor and LAG-3 are being studied in animal models.82 Immune modulatory doses of cyclophosphamide suppress Treg populations and augment anti-tumor immune response in PDA patients when used in combination with vaccine therapy.83
Chemotherapy and Immunotherapy
Recent reports have demonstrated chemotherapy’s ability to augment the anti-tumor immune response. For example, doxorubicin induced tumor cell apoptosis increases antigen presentation when compared with mitomycin.84 Additionally, docetaxel and paclitaxel have been shown to improve GM-CSF vaccine anti-tumor immunity through its ability to increase DC presence and activation.85,86 Moreover, Plate and colleagues demonstrated that PDA patients undergoing gemcitabine therapy had decreased memory T cells and increased de novo T cell activation.87
Studying the relationship between immunotherapy and chemotherapy demonstrates that the timing and dose of chemotherapy can result in a synergistic combination. Metronomic Cy is immune modulating as opposed to high dose Cy which is lympho depleting and immunosuppressive.88 Cy increases antitumor immunity by decreasing the number and function of Tregs.89,90 Moreover, metronomic Cy contributes to the transformation of CD4+ T cells into Th1 and Th17 lymphocytes thereby increasing effector T cell population.91 The dose and timing of Cy in relation to vaccine therapy for optimal immunomodulatory effects remains under investigation. Emens and colleagues reported a phase I clinical trial looking at dose ranges of an allogeneic Her-2 positive GM-CSF secreting tumor vaccine in combination with Cy and doxorubicin in metastatic breast cancer patients. They concluded that the immunomodulatory activity of low dose Cy has a narrow therapeutic window, not exceeding 200 mg/m2.92
The Future of Vaccine Therapy
Although encouraging, results from single agent immunotherapy clinical trials in PDA patients have been underwhelming and disappointing. Most gastrointestinal tumors are immune tolerant and therefore require a combinatorial therapeutic approach which includes chemotherapy, radiation, surgery and immunotherapy. As more and more tumor antigens are identified, more specific and potent vaccines will be developed. The ideal vaccine will target multiple antigens that are crucial to the tumors growth and progression. Incorporating immunologic checkpoint blockade inhibitors will allow for increased T cell activation and tumoricidal activity. Tumor microenvironment modulators will allow for more efficient DC activation thereby improving antigen presentation. Optimization of PDA vaccine therapy requires additional studies to understand which agents are most important for PDA (Fig 2). This information will come from a continued focus on scientifically driven clinical trials with access to tissue and from genetically engineered mouse models that recapitulate human disease.
Figure 3.
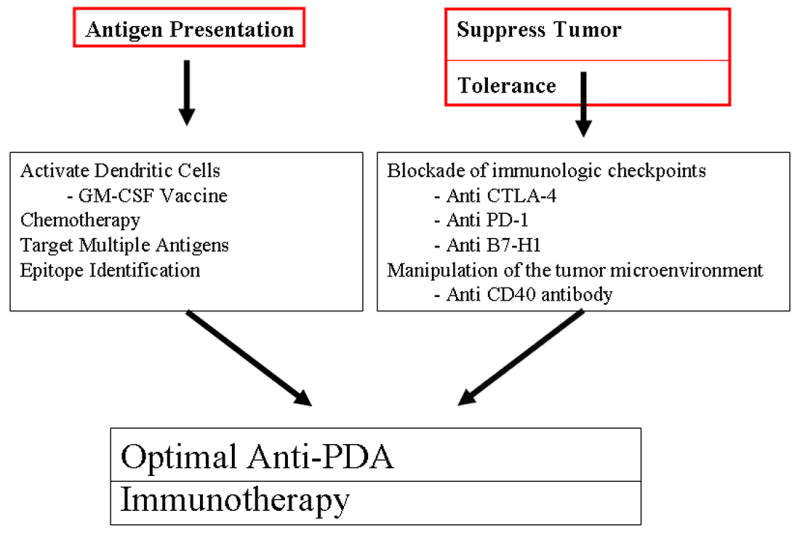
Combinatorial Immunotherapies Target Distinct Steps for an Optimal Anti-Tumor Immunity
Acknowledgments
Source of Funding: Under a licensing agreement between BioSante and the Johns Hopkins University (JHU), Dr. Jaffee and the University is entitled to milestone payments and royalty on sales of the GM-CSF secreting pancreatic tumor vaccine product described in this manuscript. Under licensing agreements between Aduro BioTech, Inc. and the JHU, Dr. Jaffee and JHU have the potential to receive royalties received on sales of the listeria-mesothelin vaccine described in this article.
Footnotes
Conflict of Interest For the remaining authors none were declared.
References
- 1.American Cancer Society. Society AC. Cancer Facts & Figures 2012. 2012 [Google Scholar]
- 2.Hruban RH, Goggins M, Parsons J, et al. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6(8):2969–2972. [PubMed] [Google Scholar]
- 3.Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett. 2009;279(1):1–7. doi: 10.1016/j.canlet.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 4.Kubuschok B, Xie X, Jesnowski R, et al. Expression of cancer testis antigens in pancreatic carcinoma cell lines, pancreatic adenocarcinoma and chronic pancreatitis. Int J Cancer. 2004;109(4):568–575. doi: 10.1002/ijc.20006. [DOI] [PubMed] [Google Scholar]
- 5.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19(6):728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 7.Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71(7):2411–2416. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- 9.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103(33):12493–12498. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esposito I, Menicagli M, Funel N, et al. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57(6):630–636. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albazaz R, Verbeke CS, Rahman SH, et al. Cyclooxygenase-2 expression associated with severity of PanIN lesions: a possible link between chronic pancreatitis and pancreatic cancer. Pancreatology. 2005;5(4-5):361–369. doi: 10.1159/000086536. [DOI] [PubMed] [Google Scholar]
- 12.Maitra A, Ashfaq R, Gunn CR, et al. Cyclooxygenase 2 expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia: an immunohistochemical analysis with automated cellular imaging. Am J Clin Pathol. 2002;118(2):194–201. doi: 10.1309/TPG4-CK1C-9V8V-8AWC. [DOI] [PubMed] [Google Scholar]
- 13.Schmitz-Winnenthal FH, Volk C, Z’Graggen K, et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005;65(21):10079–10087. doi: 10.1158/0008-5472.CAN-05-1098. [DOI] [PubMed] [Google Scholar]
- 14.Fukunaga A, Miyamoto M, Cho Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28(1):e26–31. doi: 10.1097/00006676-200401000-00023. [DOI] [PubMed] [Google Scholar]
- 15.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3(12):939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 16.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 17.Zheng L, Jaffee EM. Vaccine therapy and immunotherapy for pancreatic cancer. In: Neoptolemos J, Urrutia R, Abbruzzese L, et al., editors. Pancreatic Cancer. New York: Springer Science; 2010. pp. 1269–1318. [Google Scholar]
- 18.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6(7):506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 20.Goggins M, Kern SE, Offerhaus JA, et al. Progress in cancer genetics: lessons from pancreatic cancer. Ann Oncol. 1999;10(Suppl 4):4–8. [PubMed] [Google Scholar]
- 21.Harizi H, Gualde N. Pivotal role of PGE2 and IL-10 in the cross-regulation of dendritic cell-derived inflammatory mediators. Cell Mol Immunol. 2006;3(4):271–277. [PubMed] [Google Scholar]
- 22.Bellone G, Turletti A, Artusio E, et al. Tumor-associated transforming growth factor-beta and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155(2):537–547. doi: 10.1016/s0002-9440(10)65149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apostolopoulos V, Xing PX, McKenzie IF. Murine immune response to cells transfected with human MUC1: immunization with cellular and synthetic antigens. Cancer Res. 1994;54(19):5186–5193. [PubMed] [Google Scholar]
- 24.De Monte L, Reni M, Tassi E, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208(3):469–478. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4(5):336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 26.Laheru D, Jaffee EM. Immunotherapy for pancreatic cancer - science driving clinical progress. Nat Rev Cancer. 2005;5(6):459–467. doi: 10.1038/nrc1630. [DOI] [PubMed] [Google Scholar]
- 27.Argani P, Iacobuzio-Donahue C, Ryu B, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE) Clin Cancer Res. 2001;7(12):3862–3868. [PubMed] [Google Scholar]
- 28.Thomas AM, Santarsiero LM, Lutz ER, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004;200(3):297–306. doi: 10.1084/jem.20031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassan R, Ebel W, Routhier EL, et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007;7:20. [PMC free article] [PubMed] [Google Scholar]
- 30.Le DT, Brockstedt DG, Nir-Paz R, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 2012;18(3):858–868. doi: 10.1158/1078-0432.CCR-11-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng L, Foley K, Huang L, et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS One. 2011;6(4):e19390. doi: 10.1371/journal.pone.0019390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng L, Jaffee EM. Annexin A2 is a new antigenic target for pancreatic cancer immunotherapy. Oncoimmunology. 2012;1(1):112–114. doi: 10.4161/onci.1.1.18017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iacobuzio-Donahue CA. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut. 2012;61(7):1085–1094. doi: 10.1136/gut.2010.236026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Segal NH, Parsons DW, Peggs KS, et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68(3):889–892. doi: 10.1158/0008-5472.CAN-07-3095. [DOI] [PubMed] [Google Scholar]
- 36.Shapiro D, Tareen B. Current and emerging treatments in the management of castration-resistant prostate cancer. Expert Rev Anticancer Ther. 2012;12(7):951–964. doi: 10.1586/era.12.59. [DOI] [PubMed] [Google Scholar]
- 37.Gaudernack G. Prospects for vaccine therapy for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20(2):299–314. doi: 10.1016/j.bpg.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8(5):351–360. doi: 10.1038/nrc2373. [DOI] [PubMed] [Google Scholar]
- 39.Gjertsen MK, Bakka A, Breivik J, et al. Vaccination with mutant ras peptides and induction of T-cell responsiveness in pancreatic carcinoma patients carrying the corresponding RAS mutation. Lancet. 1995;346(8987):1399–1400. doi: 10.1016/s0140-6736(95)92408-6. [DOI] [PubMed] [Google Scholar]
- 40.Gjertsen MK, Bjorheim J, Saeterdal I, et al. Cytotoxic CD4+ and CD8+ T lymphocytes, generated by mutant p21-ras (12Val) peptide vaccination of a patient, recognize 12Val-dependent nested epitopes present within the vaccine peptide and kill autologous tumour cells carrying this mutation. Int J Cancer. 1997;72(5):784–790. doi: 10.1002/(sici)1097-0215(19970904)72:5<784::aid-ijc14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 41.Jaffee EM. Immunotherapy of cancer. Ann N Y Acad Sci. 1999;886:67–72. doi: 10.1111/j.1749-6632.1999.tb09401.x. [DOI] [PubMed] [Google Scholar]
- 42.Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90(8):3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gjertsen MK, Buanes T, Rosseland AR, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer. 2001;92(3):441–450. doi: 10.1002/ijc.1205. [DOI] [PubMed] [Google Scholar]
- 44.Gilliam AD, Broome P, Topuzov EG, et al. An international multicenter randomized controlled trial of G17DT in patients with pancreatic cancer. Pancreas. 2012;41(3):374–379. doi: 10.1097/MPA.0b013e31822ade7e. [DOI] [PubMed] [Google Scholar]
- 45.Vonderheide RH, Hahn WC, Schultze JL, et al. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity. 1999;10(6):673–679. doi: 10.1016/s1074-7613(00)80066-7. [DOI] [PubMed] [Google Scholar]
- 46.Bernhardt SL, Gjertsen MK, Trachsel S, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br J Cancer. 2006;95(11):1474–1482. doi: 10.1038/sj.bjc.6603437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cole DJ, Wilson MC, Baron PL, et al. Phase I study of recombinant CEA vaccinia virus vaccine with post vaccination CEA peptide challenge. Hum Gene Ther. 1996;7(11):1381–1394. doi: 10.1089/hum.1996.7.11-1381. [DOI] [PubMed] [Google Scholar]
- 48.Goydos JS, Elder E, Whiteside TL, et al. A phase I trial of a synthetic mucin peptide vaccine. Induction of specific immune reactivity in patients with adenocarcinoma. J Surg Res. 1996;63(1):298–304. doi: 10.1006/jsre.1996.0264. [DOI] [PubMed] [Google Scholar]
- 49.Kaufman HL, Kim-Schulze S, Manson K, et al. Poxvirus-based vaccine therapy for patients with advanced pancreatic cancer. J Transl Med. 2007;5:60. doi: 10.1186/1479-5876-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arlen PM, Gulley JL, Madan RA, et al. Preclinical and clinical studies of recombinant poxvirus vaccines for carcinoma therapy. Crit Rev Immunol. 2007;27(5):451–462. doi: 10.1615/critrevimmunol.v27.i5.40. [DOI] [PubMed] [Google Scholar]
- 51.Singh R, Paterson Y. Listeria monocytogenes as a vector for tumor-associated antigens for cancer immunotherapy. Expert Rev Vaccines. 2006;5(4):541–552. doi: 10.1586/14760584.5.4.541. [DOI] [PubMed] [Google Scholar]
- 52.Singh R, Dominiecki ME, Jaffee EM, et al. Fusion to Listeriolysin O and delivery by Listeria monocytogenes enhances the immunogenicity of HER-2/neu and reveals subdominant epitopes in the FVB/N mouse. J Immunol. 2005;175(6):3663–3673. doi: 10.4049/jimmunol.175.6.3663. [DOI] [PubMed] [Google Scholar]
- 53.Brockstedt DG, Giedlin MA, Leong ML, et al. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci U S A. 2004;101(38):13832–13837. doi: 10.1073/pnas.0406035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshimura K, Laird LS, Chia CY, et al. Live attenuated Listeria monocytogenes effectively treats hepatic colorectal cancer metastases and is strongly enhanced by depletion of regulatory T cells. Cancer Res. 2007;67(20):10058–10066. doi: 10.1158/0008-5472.CAN-07-0573. [DOI] [PubMed] [Google Scholar]
- 55.Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117(5):1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lepisto AJ, Moser AJ, Zeh H, et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008;6(B):955–964. [PMC free article] [PubMed] [Google Scholar]
- 57.Kimura Y, Tsukada J, Tomoda T, et al. Clinical and immunologic evaluation of dendritic cell-based immunotherapy in combination with gemcitabine and/or S-1 in patients with advanced pancreatic carcinoma. Pancreas. 2012;41(2):195–205. doi: 10.1097/MPA.0b013e31822398c6. [DOI] [PubMed] [Google Scholar]
- 58.Simons JW, Jaffee EM, Weber CE, et al. Bioactivity of autologous irradiated renal cell carcinoma vaccines generated by ex vivo granulocyte-macrophage colony-stimulating factor gene transfer. Cancer Res. 1997;57(8):1537–1546. [PMC free article] [PubMed] [Google Scholar]
- 59.Nemunaitis J. Vaccines in cancer: GVAX, a GM-CSF gene vaccine. Expert Rev Vaccines. 2005;4(3):259–274. doi: 10.1586/14760584.4.3.259. [DOI] [PubMed] [Google Scholar]
- 60.Hege KM, Jooss K, Pardoll D. GM-CSF gene-modifed cancer cell immunotherapies: of mice and men. Int Rev Immunol. 2006;25(5-6):321–352. doi: 10.1080/08830180600992498. [DOI] [PubMed] [Google Scholar]
- 61.Jaffee EM, Hruban RH, Biedrzycki B, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19(1):145–156. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 62.Lutz E, Yeo CJ, Lillemoe KD, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann Surg. 2011;253(2):328–335. doi: 10.1097/SLA.0b013e3181fd271c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laheru D, Lutz E, Burke J, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res. 2008;14(5):1455–1463. doi: 10.1158/1078-0432.CCR-07-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ghiringhelli F, Menard C, Puig PE, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56(5):641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mandell RB, Flick R, Staplin WR, et al. The alphaGal HyperAcute((R)) Technology: enhancing immunogenicity of antiviral vaccines by exploiting the natural alphaGal-mediated zoonotic blockade. Zoonoses Public Health. 2009;56(6-7):391–406. doi: 10.1111/j.1863-2378.2008.01191.x. [DOI] [PubMed] [Google Scholar]
- 66.Hardacre JM, Mulcahy MF, Talamonti M, et al. Effect of hyperacute immunotherapy in addition to standard adjuvant therapy for resected pancreatic cancer on disease-free and overall survival: Preliminary analysis of phase II data. ASCO Annual Meeting; 2010. [Google Scholar]
- 67.Ascierto PA, Marincola FM, Ribas A. Anti-CTLA4 monoclonal antibodies: the past and the future in clinical application. J Transl Med. 2011;9:196. doi: 10.1186/1479-5876-9-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ribas A, Hanson DC, Noe DA, et al. Tremelimumab (CP-675,206), a cytotoxic T lymphocyte associated antigen 4 blocking monoclonal antibody in clinical development for patients with cancer. Oncologist. 2007;12(7):873–883. doi: 10.1634/theoncologist.12-7-873. [DOI] [PubMed] [Google Scholar]
- 69.Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12(7):864–872. doi: 10.1634/theoncologist.12-7-864. [DOI] [PubMed] [Google Scholar]
- 70.Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–833. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le DT, Lutz E, Huang L, et al. Phase Ib study of ipilimumab alone or in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene (vaccine) in pancreatic cancer. Gastrointestinal Cancers Symposium; 2012. [Google Scholar]
- 72.Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 73.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shevach EM. Special regulatory T cell review: How I became a T suppressor/regulatory cell maven. Immunology. 2008;123(1):3–5. doi: 10.1111/j.1365-2567.2007.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169(5):2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 79.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 80.Woo EY, Chu CS, Goletz TJ, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61(12):4766–4772. [PubMed] [Google Scholar]
- 81.Hiraoka N, Onozato K, Kosuge T, et al. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12(18):5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 82.Huang CT, Workman CJ, Flies D, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21(4):503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 83.Le DT, Jaffee EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res. 2012;72(14):3439–3444. doi: 10.1158/0008-5472.CAN-11-3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202(12):1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chu Y, Wang LX, Yang G, et al. Efficacy of GM-CSF-producing tumor vaccine after docetaxel chemotherapy in mice bearing established Lewis lung carcinoma. J Immunother. 2006;29(4):367–380. doi: 10.1097/01.cji.0000199198.43587.ba. [DOI] [PubMed] [Google Scholar]
- 86.Machiels JP, Reilly RT, Emens LA, et al. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 2001;61(9):3689–3697. [PubMed] [Google Scholar]
- 87.Plate JM, Plate AE, Shott S, et al. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol Immunother. 2005;54(9):915–925. doi: 10.1007/s00262-004-0638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Motoyoshi Y, Kaminoda K, Saitoh O, et al. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol Rep. 2006;16(1):141–146. [PubMed] [Google Scholar]
- 89.Lutsiak ME, Semnani RT, De Pascalis R, et al. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105(7):2862–2868. doi: 10.1182/blood-2004-06-2410. [DOI] [PubMed] [Google Scholar]
- 90.Ercolini AM, Ladle BH, Manning EA, et al. Recruitment of latent pools of high-avidity CD8(+) T cells to the antitumor immune response. J Exp Med. 2005;201(10):1591–1602. doi: 10.1084/jem.20042167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sistigu A, Viaud S, Chaput N, et al. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011;33(4):369–383. doi: 10.1007/s00281-011-0245-0. [DOI] [PubMed] [Google Scholar]
- 92.Emens LA, Asquith JM, Leatherman JM, et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J Clin Oncol. 2009;27(35):5911–5918. doi: 10.1200/JCO.2009.23.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]