

Abstract
Human mesenchymal stromal cells (hMSCs) represent an attractive cell source for clinic applications. Besides being multi-potent, recent clinical trials suggest that they secrete both trophic and immunomodulatory factors, allowing allogenic MSCs to be used in a wider variety of clinical situations. The yield of prospective isolation is however very low, making expansion a required step toward clinical applications. Unfortunately, this leads to a significant decrease in their stemness. To identify the mechanism behind loss of multi-potency, hMSCs were expanded until replicative senescence and the concomitant molecular changes were characterized at regular intervals. We observed that, with time of culture, loss of multi-potency was associated with both the accumulation of DNA damage and the respective activation of the DNA damage response pathway, suggesting a correlation between both phenomena. Indeed, exposing hMSCs to DNA damage agents led to a significant decrease in the differentiation potential. We also showed that hMSCs are susceptible to accumulate DNA damage upon in vitro expansion, and that although hMSCs maintained an effective nucleotide excision repair activity, there was a progressive accumulation of DNA damage. We propose a model in which DNA damage accumulation contributes to the loss of differentiation potential of hMSCs, which might not only compromise their potential for clinical applications but also contribute to the characteristics of tissue ageing.
Keywords: ageing, senescence, human mesenchymal stem cells, DNA damage, in vitro expansion, clinical application
Introduction
Human mesenchymal stromal cells (hMSCs) represent an attractive cell source for clinical applications. They can be isolated from different sources and are able to differentiate into several lineages such as the osteogenic, adipogenic, chondrogenic, myogenic and neurogenic lineages [1–5]. hMSCs are generally isolated from easily accessible tissues in a reproducible way and have a high expansion potential, which makes them good candidates for the repair and regeneration of a large variety of tissues. Currently, hMSCs from bone marrow are the most common source of cells for therapeutic applications. They are used in clinical trials mainly due to proven efficacy in pre-clinical studies [6–9]. Potential applications are repair of cardiovascular and central nervous system damage, for pancreatic, renal, hepatic and solid organ transplantation, for the use in the gastrointestinal tract, and for both orthopaedic and haematopoietic applications (reviewed by Brooke et al.) [10]. Recently, new insights based on clinical experimentation also suggest that MSCs secrete large quantities of bioactive factors with both trophic and immunomodulatory activity, thus preventing autoimmunity allowing allogenic MSCs to be used in a wide variety of clinical situations [11]. Because the yield of prospective hMSC isolation is very low, expansion is a necessary step for clinical application. Thus, successful clinical applications of hMSCs will rely on the ability to extensively expand them while maintaining their multi-potency. Unfortunately, in vitro expansion of hMSCs is associated with a gradual decrease and loss of differentiation capacity of hMSCs, which currently represent one of the major bottlenecks in cell-based clinical applications. Although normally stem cells cultured in low oxygen proliferate faster than in traditional 20% oxygen environments, there are several problems associated with growing cells in a hypoxic environment. Low oxygen levels tend to favour certain differentiation pathways in detriment of others [12] and there are even reports about attenuation of differentiation after cultivation of hMSCs in low oxygen [13, 14]. Furthermore, from a clinical/commercial point of view, the cultivation of hMSCs in low oxygen would require much more specialized equipment and would be more labour intensive. Although it has been shown that hMSCs can be expanded in vitro for several population doublings (PD), they ultimately stop dividing, a phenomenon known as replicative senescence or the Hayflick limit after the discovery by Leonard Hayflick that human diploid cell strains in culture divide about 50 times before entering a senescence phase [15]. The cellular senescence program is characterized by clear morphological alterations culminating in growth arrest. This phenomenon can be triggered by multiple mechanisms including telomere shortening and uncapping, DNA damage accumulation, proto-oncogene activation, epigenetic changes, stress and other inducers like cytokines [16, 17]. Senescence was already implicated in the loss of regenerative potential in ageing tissues [16, 18] and although establishing a direct causal relationship between cellular senescence and ageing might be complex, there is circumstantial evidence linking both processes. For clinical applications, another important concern is the fact that hMSCs lose their differentiation capacity (referred to as functional senescence in this article) very soon during in vitro expansion as previously described [19]. Despite several reports about the mechanisms that can drive cellular senescence with ageing, little is known about mechanisms that drive the loss of multi-potency in the early expansion phase of tissue cultured hMSCs.
Some mutations in DNA repair pathways are associated with a decrease in lifespan, an increase in the incidence of cancer and genomic instability and symptoms of premature ageing. Examples are the Ku80, TTDXpdR722W, Brca1 and Trp53 mutant mice [20, 21]. Three independent research teams recently published on the link between age-related accumulation of DNA damage and decline of stem-cell function [22–24]. The conclusion was that with age, adult haematopoietic stem cell function declines and that DNA damage and epigenetic modification may limit the regenerative potential of those cells. They agree that ageing may result in an accumulation of DNA mutations since stem cells are not protected from age-induced damage.
However, several issues remained unclear which lead us to investigate whether DNA damage plays a role in early loss of multi-potency observed during in vitro expansion of hMSC, the susceptibility of adult mesenchymal stromal cells to accumulate DNA damage, how effective DNA-repair activities are in these cells, and to what extent this balance might contribute to the characteristics of tissue ageing.
Materials and methods
Isolation and culture of hMSCs
Bone marrow aspirates were obtained from donors with written informed consent and hMSCs were isolated and proliferated as previously described [25]. Briefly, aspirates were resuspended using 20 G needles, plated at a density of 5×105 cells/cm2 and cultured in hMSC proliferation medium (PM) containing α-minimal essential medium (α-MEM, Life Technologies, Carlsbad, CA, USA), 10% foetal bovine serum (FBS, Cambrex), 0.2 mM ascorbic acid (Asap, Life Technologies), 2 mM L-glutamine (Life Technologies), 100 U/ml penicillin (Life Technologies), 10 mg/ml streptomycin (Life Technologies) and 1 ng/ml basic fibroblast growth factor (bFGF, Instruchemie). Cells were grown at 37°C in a humid atmosphere with 5% CO2. Medium was refreshed twice a week and cells were used for further subculturing or cryopreservation when reaching 80–90% confluency. hMSC basic medium/control medium (BM) was composed of hMSC PM without bFGF, hMSC osteogenic medium (OM) was composed of hMSC BM supplemented with 10−8 M dexamethasone (dex, Sigma-Aldrich, St. Louis, MO, USA) and hMSC mineralization medium (MM) was composed of BM supplemented with 10−8 M dex and 0.01 M β-glycerophosphate (Sigma). After plating of the bone marrow aspirate, hMSCs were expanded in PM and the cells obtained from the first trypsinization were considered as PD (population doubling) 0. Relative population doublings (RPD) refer to the number of population doublings that cells had undergone, relative to PD 0. Donors’ information is represented in Table S1.
Senescence associated-β-Galactosidase expression
hMSCs at different RPD were seeded at 15,000 cells/cm2 in 6-well plates and proliferated in PM until 70–80% confluence was reached. Then, cells were washed twice with PBS (Life Technologies), fixed with 4% paraformaldehyde for 10 min., washed again three times with PBS and stained overnight with a SA-β-gal Kit (Sigma) according to the manufacturer’s instructions. Cells were then counterstained with Mayer’s haematoxylin for 5 min. All chemicals were obtained from Sigma. Images were taken at a 100× magnification.
Immunophenotyping of hMSCs
To characterize the population of hMSCs during expansion, cell-surface marker expression was assessed. Low passage (RPD 6) and high passage (RPD 20) cells were harvested by trypsin and incubated for 30 min. in block buffer [PBS with 5% BSA (Sigma) and 0.05% NaN2], then incubated with primary antibodies (CD45, HLA-DR, CD19, CD90/Thy1, Endoglin/CD105, Integrin αM/CD11b from R&D Systems, CD34 and CD73 from Abcam) diluted in wash buffer (PBS with 1% BSA and 0.05% NaN2) for 1 hr or with isotype control antibodies. Cells were then washed three times with wash buffer and incubated with secondary antibody (anti-mouse IgM or anti-mouse IgG, conjugated with phycoerythrin from R&D systems) for 30 min. Samples were then washed three times and resuspended in 250 μl of wash buffer with 10 μl of Viaprobe (Pharmingen) for live/dead cell staining. Only live cells were used for further analysis. The percentage of cells expressing each marker and the respective overlays of the histograms (RPD6 versus RPD20) were assessed on FACSCalibur (Becton Dickinson Immunocytometry Systems).
Acidic phosphatase and Alamar blue
Acidic phosphatase (ACP) and Alamar blue assays were performed to measure the proliferative and metabolic status of the cells during the experiment, respectively. The Alamar blue solution (BioSource; Invitrogen, Carlsbad, CA, USA) was diluted 1:10 in culture medium, and cells were incubated for 4 hrs. The Alamar blue solution from each well was then transferred into 96-well plates, and fluorescence was measured using a Perkin Elmer Luminescence Spectrometer LS50B. Results were recorded by FL winlab 12. For the ACP determination, cells were lysed with a 0.2% Triton X-100 solution in 100 mM potassium phosphate buffer pH 7.8 and then incubated with 5 mM 4-nitrophenyl phosphate disodium salt dissolved in 0.1 M sodium acetate, 0.1% Triton-X, pH 5.5. After 1.5 hr at 37°C, the reaction was stopped by adding 0.5 N sodium hydroxide and allowed to equilibrate for 10 min. The absorbance was then measured at 405 nm.
Alkaline phosphatase (ALP) induction
For the biochemical ALP assay, hMSCs were seeded at 5000 cells/cm2 and allowed to adhere for 10–15 hrs in BM and then cells were grown in OM for 6 days. Each experiment was performed at least in triplicate with a negative control (cells grown in BM) and a positive control (cells grown in OM). To avoid donor variation, all the experiments were performed with cells from at least three independent donors. Results shown are usually from one donor, unless stated otherwise and are representative for all the donors tested. Briefly, cells were washed twice with PBS (Life technologies) and lysed using a 0.2% Triton X-100 solution in 100 mM PBS pH 7.8, supplemented with a protease inhibitor cocktail (Roche Applied Science). The lysate was incubated in the dark at 25°C with CDP-Star substrate (Roche) and allowed to react for 30 min. Luminescence was measured using a VICTOR3 luminometer (Perkin Elmer) at 25°C. The total ALP luminescence was normalized for cell number using ACP as readout. The data were then analysed using Student’s t-test at P < 0.05.
Mineralization and adipogenesis
Both assays were performed as described previously [19]. Experiments were performed at least in triplicate using BM as negative control. In the mineralization experiment, the calcium content was measured at 620 nm (Bio-tek instruments) after 28 days in MM and expressed as percentage reduction in calcium deposition compared to RPD 6 (passage 1), which was set to 100%.
Immunofluorescence
Cells were grown on glass coverslips for at least 48 hrs prior to immunostaining to minimize stress as a consequence of passaging. Cells were washed twice with PBS and fixed for 20 min. with freshly prepared 4% paraformaldehyde in PBS. After permeabilization for 20 min. with PBST (PBS with 0.2% Triton X-100), cells were blocked for 1 hr. The 53BP1 anti body (Novus) was diluted 1:500 in blocking buffer and incubated overnight at 4°C. Samples were then washed with TBST, incubated with Alexa-488 secondary antibody (Molecular Probes; Invitrogen, Carlsbad, CA, USA) and visualized by confocal laser scanning microscopy.
ImmunoFISH
The telomere dysfunction-induced (TIF) assay was performed as described previously [26]. Briefly, after immunofluorescent staining with 53BP1, cells were cross-linked with 4% paraformaldehyde in PBS for 20 min. Then, cells were dehydrated using a series of 70%, 90% and 100% ethanol and air-dried. Nuclear DNA was denatured for 5 min. at 80°C in hybridization buffer containing 0.5 μg/ml (C3TA2)3-Cy3-labelled peptide nucleic acid (PNA) telomeric probe (Applied Biosystems), 70% formamide, 12 mM Tris-HCl (pH 8), 5 mM KCl, 1 mM MgCl2, 0.001% Triton X-100 and 2.5 mg/ml acetylated BSA. After denaturation, incubation was continued for 2 hrs at room temperature in a humidified chamber. Cells were washed twice for 15 min. with 70% formamide/2 × SSC (0.3 M NaCl, 30 mM Na-citrate [pH 7]), followed by a 10 min. wash with 2 3 SSC and a 10 min. wash with TBST. To reinforce the 53BP1 signal, cells were incubated for 1 hr with an Alexa-488 conjugated anti-secondary antibody before mounting in a media containing DAPI.
Oxidative stress determination (8-OxoG and CM-H2DCFDA)
For the determination of oxidative stress load, two different methods were employed. The first method was based on the detection of 8-Oxoguanine (as part of the oxidized nucleotide 8-oxyguanosine) in fixed permeabilized cells using flow cytometry. Briefly, hMSCs from different RPD were fixed and permeabilized and then a FITC-labelled protein conjugate specific for the 8-oxyguanine moity was added for 1 hr, according to manufacturer’s instructions (Argutus Medical OxyDNA test – BD Biosciences). The presence of the oxidized DNA was then measured by FACS in at least 10,000 cells. The second method consisted in the direct detection of reactive oxygen metabolites as described previously [27]. Shortly, adherent cells were harvested with trypsin and combined with cells floating in the medium. Cells were then washed and treated with 10 μM (5-(and-6)-chloromethyl-2’7’-dichlorodihydrofluorescein diacetate acetyl ester) CM-H2DCFDA (Molecular Probes) for 30 min. at 37°C. Ten thousand cells were then analysed on a BD FACScan.
Western blot
For Western blot analysis, enzymatically dispersed cells were pelleted and lysed using a solution of 0.2% Triton X-100 in potassium phosphate buffer pH 7.8 containing a protease inhibitor cocktail (Roche Applied Science). Protein concentration was quantified spectrophotometrically using a BCA Protein Assay Kit (Pierce). Separated proteins were transferred electrophoretically to polyvinylidene difluoride membranes (Immobilon-P Millipore). Blots were incubated overnight at 4°C with primary antibodies: p16 (c-20, Santa Cruz Biotechnology); p21 (DCS60, Cell Signaling Technology); β-Actin, clone Ac-74 (Sigma) and the antibodies from the DNA Damage Antibody Sampler Kit (Cell Signaling Technology). Next, the membranes were incubated with peroxidase-conjugated secondary antibody, either Anti-Rabbit IgG or Anti-Mouse IgG (Dako, Glostrup, Denmark), for 1 hr at room temperature. Antigen-antibody complexes were visualized by SuperSignal West Dura Extended Duration Substrate (Thermo scientific).
Unscheduled DNA synthesis (UDS)
For testing the nucleotide excision repair (NER) capacity, hMSCs were seeded onto coverslips and grown in medium without thymidine and switched to medium without FBS 24 hrs prior to the start of the experiment. Cells were then washed with PBS, irradiated with 16 J/m2 UV-C (Philips TUV lamp) and subsequently incubated for 2.5 hrs in culture medium containing 10 μCi/ml 3H-thymidine (Amersham International PLC; GE Healthcare, Little Chalfont, UK). For analysis, cells were fixed in Bouin fixative and subjected to autoradiography as previously described [28]. Repair capacity was quantified by counting autoradiographic grains above the nuclei of ≥ 25 cells. UDS levels are expressed as the mean of grains per fixed area plus standard error for each PD.
Exposure to a DNA damage inducer
To assess the effects of oxidative stress on ALP expression, early passage cells (RPD 10) were plated at 2000 cells/cm2 and treated with hydrogen peroxide (H2O2, Sigma) or paraquat (Sigma) and grown either in BM or OM for 6 days. After this period, ALP expression was assessed using the biochemical method described above. In order to evaluate the DNA damage load, cells were expanded in PM and treated with either hydrogen peroxide or paraquat. The percentage of DNA damage positive cells and the average foci per cell were determined by immunofluorescence staining of 53BP1.
Results
Replicative senescence upon in vitro expansion of hMSCs
In order to investigate the differentiation capacity with respect to expansion, hMSCs from two donors were expanded until cessation of growth. As expected, hMSCs presented a continuous decrease in proliferation rate with increased time in culture (Fig. 1A). After an initial difference, both donors exhibited a similar level of PD/day until around 60 days in culture. At this point, the cells from donor 36 stopped proliferating while the cells from donor 24 displayed a temporary recovery followed by a permanent growth arrest. Although late passage cells did not proliferate, they were able to be maintained viable in culture for at least 3 more months before the experiment was ended. No spontaneous immortalization of hMSCs was ever observed and no clonal expansion was observed during this period of in vitro expansion.
Fig 1.
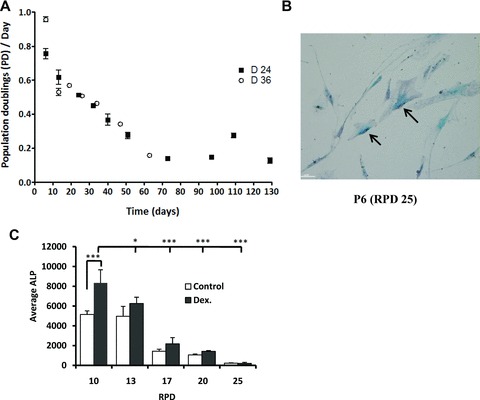
Long-term in vitro expansion of hMSCs leads to loss of multi-potency. (A) Growth profiles of hMSCs in vitro depicted by the number of population doublings per day plotted against culture period. (B) Phase-contrast photo of hMSCs at RPD 25. At RPD 25, most of the cells express SA-b-Galactosidase (black arrows). With increasing time of culture not only more cells were positive for SA- b-Galactosidase but also the staining was more intense (data not shown). (C) Effect of expansion in the expression of ALP, both in control and dexamethasone-treated cells. ALP expression was analysed by CDP-STAR base luminescence assay and corrected for the cell number. Error bars represent standard deviation. Statistical analysis was performed using one-way anova and Tukey post-test with a significance level of 0.05. * represents P < 0.01 to 0.05 and ***P < 0.001.
An evident change in cell morphology was observed with increasing time of culture (Fig. S1A). In RPD 0, cells presented a fibroblastic-like appearance that progressed toward a more spread morphology characterized by enlarged and flattened cytoplasm, increased granularity and heterogeneous cell shape. hMSCs from low RPD grew homogeneously while in higher RPD they grew in cluster with neighbouring cells, leaving large interspaces between the clusters (Fig. S1A, RPD25). Late passage cells also displayed an increased expression of senescence-associated b-galactosidase (SA-β-gal), a common marker for senescent cells (Fig. 1B).
hMSCs after minimally or extensively expansion were phenotypically similar, presenting the same percentage of marker expression (Fig. S2). hMSCs were negative for CD11b, CD19, CD34 and CD45 and positive for CD73, CD90 and CD105. HLA-DR was present in 6% of the cells independent of the passage.
Gradual loss of the differentiation capacity of hMSCs after in vitro expansion
To determine the differentiation capacity of hMSC during in vitro culture, we performed both in vitro osteogenic and adipogenic assays at different RPD. We observed a decrease in the levels of ALP of cells grown in BM mainly from RPD 13 to the following PDs. We also observed a decrease in the dexamethasone-induced ALP levels (an early marker for bone formation) with increasing RPD (Fig. 1C). At RPD 13 (passage 3) both the basal as well as the induced levels of ALP were already very low and the cells were no longer responsive to dexamethasone. A decline in calcium deposition (a late marker for in vitro osteogenesis) was also observed with culturing (Fig. S1B). This decrease was most pronounced (approximately 70% reduction in calcium deposition) between RPD 17 and RPD 20. After RPD 20, the cells lost their ability to mineralize in vitro. In addition, we also tested the ability of the expanded hMSC to differentiate into the adipogenic lineage. We observed loss of adipogenic potential during expansion (Fig. S1C). At RPD 20, a small subset of hMSC was still able to differentiate into the adipogenic lineage, but it was proportionally considerably less than in hMSC from early PDs.
DNA damage accumulation and DNA damage pathway activation during in vitro expansion
To evaluate the relation between DNA damage and in vitro expansion, immunofluorescence staining of p53 binding protein 1 (53BP1) and TIF assays were performed at several RPD [29–32]. We observed that with increasing PD there was a significant increase in the number of nuclei containing 53BP1 foci, a marker of DNA damage (Fig. 2A). Interestingly, we also observed an increase in telomere-associated DNA damage, demonstrated by the co-localization of 53BP1 staining with a telomeric probe (Fig. 2B). In cells after 27 RPD or more (passage 8), the telomeric signal could no longer be detected. To determine the changes in oxidative levels that hMSCs face during expansion, we quantified the presence of 8-oxoguanine (Fig. 2C) and the presence of reactive oxygen metabolites (Fig. 2D). Both assays demonstrated that during expansion hMSCs are exposed to increased oxidative stress levels. These data demonstrate that in vitro cultured hMSCs accumulate DNA damage upon expansion.
Fig 2.
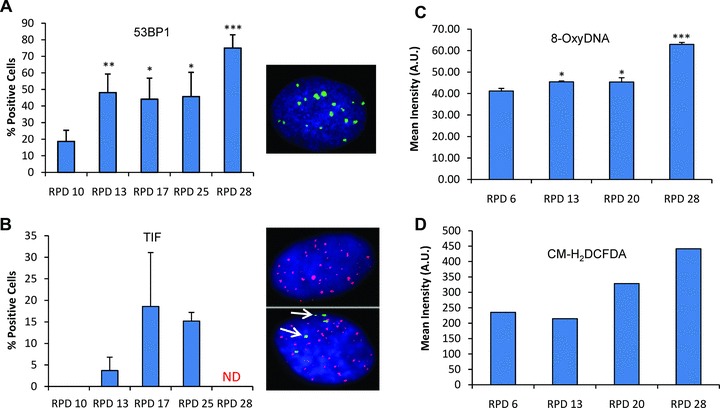
Accumulation of 53BP1, telomeric 53BP1 foci and oxidative stress after expansion of hMSCs. Cells were immunostained with 53BP1 antibody. At the indicated RPD, 53BP1 positive cells were quantified (A). Cells were processed by immunoFISH to visualize 53BP1 foci at the telomeres. At the indicated RPD, TIF-positive cells were quantified (B). A cell was considered TIF-positive if $50% of its 53BP1 foci co-localized with telomeres (PNA). Green, blue and red colour channels represent, respectively: 53BP1, DAPI and telomeres (PNA). Arrows represent co-localization of 53BP1 foci and telomeric probe. The level of 8-oxoguanine (C) and oxidative stress (D) was measured at the respective RPD by FACS in at least 10,000 cells. Error bars represent standard deviation. Statistical analysis was performed using one-way anova and Tukey post-test with a significance level of 0.05. *P < 0.05, **P < 0.01 and ***P < 0.001.
Next, we performed Western blot analysis on hMSCs at different RPDs and analysed the abundance of proteins involved in the DNA damage response pathway, such as phospho-p53 (Ser15), p16 (c-20) and p21Waf1/Cip1 (Fig. 3A). In accordance with the previous results, we observed an activation of the DNA damage response pathway with time of culture. There was a particularly strong increase from RPD 10 to RPD 17. We suggested that DNA damage accumulation is due to impaired DNA repair mechanisms in late passage cells. We analysed the capacity of NER because it is involved in the repair of many types of DNA damage including the ones induced by reactive oxygen species associated with premature ageing phenotypes. To this end, hMSC from different RPD were seeded onto coverslips and irradiated with UV after which we measured the incorporation of 3H-thymidine. With increased PD, there was no significant change in the NER capacity (Fig. 3B).
Fig 3.
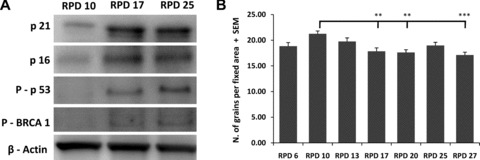
Activation of the DNA damage signalling pathway and efficiency of NER. Samples from different RPD were analysed by Western blotting for p16 and p21 protein expression and p53 and BRCA1 protein phosphorilation (A). β-actin served as loading control. NER efficiency was determined at several RPD after in vitro expansion using the unscheduled DNA synthesis assays. Error bars represent standard deviation. Statistical analysis was performed using one-way anova and Tukey post-test with a significance level of 0.05. **P < 0.01 and ***P < 0.001.
DNA damage induction is associated with loss of multi-potency
The data described above show a correlation between DNA damage accumulation and loss of hMSC multi-potency but whether this is a causal relation is not known. To mimic the increased DNA damage load in culture expanded hMSCs, we induced DNA damage using hydrogen peroxide (H2O2) and paraquat (PQ). Hydrogen peroxide has been the most commonly used source of oxidative stress and known to induce features similar to replicative senescence like: morphology changes, senescence-associated galactosidade activity, cell cycle regulation [33–36]. Being paraquat another extensively used compound to induce superoxide production causing extensive mitochondrial damage [37–39].
First, we established a dose-response curve of hydrogen peroxide on proliferation of hMSCs. Up to 50 μM H2O2, the differences observed were not statistically significant (Fig. 4A). For paraquat, a concentration of 5 μM and 10 μM was used based on our previous experiments (data not shown). Based on this, we choose two concentrations of H2O2 (30 and 50 μM) and two of paraquat (5 and 10 μM) to treat hMSCs, where after 6 days, both DNA damage accumulation and ALP induction was assessed and compared to the control situation. Under these conditions, DNA damage accumulation occurred evidenced by the increase in 53BP1 positive cells [33.33 ± 4.9% in the control situation to 52.83 ± 6% in 30 μM H2O2 treated hMSCs and 51.8 ± 4.7 in 5 μM paraquat treated cells, (Fig. 4B)] and the number of 53BP1 foci per cell which increased from 0.50 ± 0.05 to 1.68 ± 0.19 (H2O2, 30 μM) or 0.77 ± 0.06 in (paraquat, 5 μM) in treated cells (Fig. 4C). When a stronger concentration of DNA damage inducer was used, both the percentage of damaged cells (53BP1 positive, Fig. 4B) and average foci per cell (Fig. 4C) increased even further. When a DNA-damage inducer was used, an increase in the β-galactosidase activity was also observed (Fig. S3). Exposure of hMSCs for 6 days to DNA damage inducers did not have an effect on ALP activity in BM (except for cells treated with 50 μM H2O2), but it did significantly inhibit dexamethasone-induced ALP expression in all the conditions tested (Fig. 4D). Mineralization was also significantly repressed by the usage of any of the inducers (Fig. 4E).
Fig 4.
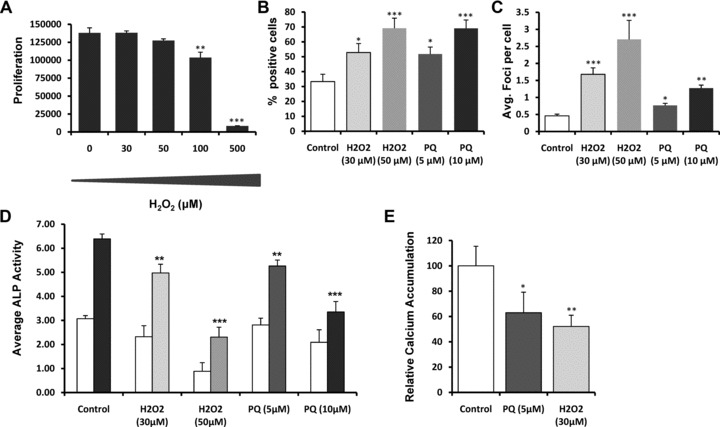
Effects of the exposure of DNA damage inducers on the differentiation potential of hMSCs. Effect of variant concentrations of hydrogen peroxide (H2O2) on the proliferation of hMSCs after 6 days of exposure (A). Effect of continuous exposure (6 days) of H2O2 and paraquat on the differentiation potential (ALP levels and mineralization) of hMSCs (D, E) and on the DNA damage load (B, C). DNA damage load was assessed by quantifying the percentage of 53BP1 positive cells (B) and 53BP1 foci per cell (C). Data are expressed as total ALP normalized for cell number repeated in at least three different donors. Error bars represent standard deviation. Statistical analysis was performed using one-way anova and Tukey post-test with a significance level of 0.05. *P < 0.05, **P < 0.01 and ***P < 0.001.
Discussion
Human mesenchymal stromal cells are multi-potent cells, which are able to differentiate into several lineages [1–5]. They can be isolated from bone marrow and other tissues and appear not to require tissue matching between MSC donor and recipient [40]. Therefore, they are promising candidates for cell-based therapies for a number of regenerative conditions [10]. However, the attractiveness of this cell source for regenerative medicine depends largely on the possibility to expand, differentiate and induce the desired effect in vivo after the expansion period. It is therefore crucial to analyse the effect of long-term in vitro expansion.
There is one report claiming spontaneous immortalization of adipose-derived MSCs [41], although we never observed it in cells from at least six donors that we followed up for 3 months after cell cycle arrest. Our observation is supported by a number of other studies [19, 42, 43]. For example, the consequences of long-term in vitro expansion have also been evaluated in MSCs derived from bone marrow and adipose tissue of both human beings and rhesus macaques [44]. The authors observed that, unlike rhesus MSCs, human MSCs did not develop polyploidy throughout the prolonged culture and no chromosomal rearrangements were detected in any of the human MSC samples analysed at any passage. The only observation we made that may resemble spontaneous transformation was a transient and partial recovery in cell morphology and proliferation in cells from one donor. However, after a few cell divisions, the cells permanently arrested.
Although immortalization was not observed, in vitro expansion did negatively affect the hMSCs from an early time-point of culture, seen by the decrease in both the proliferation rate and differentiation potential. The decrease in proliferation rate seems constant until senescence hits in after about 20 population doublings but starts right from the beginning of the expansion period. There are various stresses that can induce replicative senescence, i.e. oxidative stress, oncogene overexpression and telomere shortening. The response to oncogene overexpression is thought to be a tumour-suppressive mechanism, by which cells prevent the uncontrolled proliferation due to oncogene activation. For example, normal cells senesce in response to the activation of certain oncogenes, such as RAS or of its downstream effectors [45, 46]. Oncogene-mediated senescence is most likely to occur late during the expansion phase, because mutagenic events have to occur. However, we observed an instant decline in proliferative capacity in cultured hMSCs, which makes the oncogene theory not likely, although we cannot exclude it.
The decline in proliferation and differentiation can also be caused by telomere shortening. During cell expansion, a gradual shortening of the average length of telomeres is observed eventually leading to telomere uncapping. Once unprotected, dysfunctional telomeres activate a DNA damage response resulting in permanent cell cycle arrest. We formerly reported that there is a direct association between the age of primates and the levels of telomere-dysfunction found in their skin fibroblasts, providing a clear association between telomere dysfunction and senescence in vivo [47]. In this respect, it is important to note that telomere length varies among the MSC population and therefore, initiation of the senescence program is expected to start at different numbers of population doublings for different subclones in an MSC culture. However, we never observed clonal expansion in culture. Although telomere length was not measured, we could not detect the telomeres at RPD27, which suggested to us that telomere shortening contributes to the last stages of senescence and to terminal cell cycle arrest.
In this article, we also describe that with increasing time of culture, there is an increase in the DNA damage load, even though the capacity of the NER path ways remains unchanged. Recently, in a complementary study, the initial step of base excision repair in uncultured and cultured adipose-tissue derived mesenchymal stem cells (AT-MSCs) was compared [48]. By analysing the base removal activities (8-oxoG, uracil, 5-ohU, and ϵA lesions) and expression levels of relevant DNA glycosylases, the authors concluded that the DNA glycosylase activities tested were stable throughout culturing, suggesting that high DNA repair activity (base excision repair) is maintained, although a slight decrease in 8-oxoG removal activity was observed at senescence. The gene expression analyses on the DNA glycosylases showed no substantial changes in mRNA expression. There are, however, reports where genes related to DNA repair were repressed after in vitro culturing, although, most refer to experiments performed in non-Human cells, so possible species variation must be taken into consideration [49–51]. These results suggest that the DNA damage load which the hMSCs are facing in culture is higher than what is possible to be repaired, leading to a constant DNA damage accumulation, activation of the DNA damage signalling pathway and ultimately to loss of differentiation capacity and cellular senescence. Previous reports demonstrated that DNA damage accumulation due to deficiencies in DNA damage repair not only induces premature ageing phenotypes [52] but also limit the function of haematopoietic stem cells with age [23, 24]. A recent publication also suggests that the senescence growth arrest in hMSCs after in vitro expansion may result from activation of stress signalling pathways and consequent onset of stress responses, due in part to (reactive oxygen species) ROS production during prolonged in vitro culture [53]. The authors by introducing Wip1 (wild-type p53 inducible phosphatase-1), a stress signalling modulator, manage to extend the life span of hMSCs and to overcome oncogenic stress mediated growth arrest.
In addition to the decline in proliferation, we observed a decrease in the differentiation potential. In contrast to two previous reports which claim maintenance or increased propensity for osteogenic differentiation of hMSCs after in vitro expansion [2, 54], our data show that osteogenesis is more affected by the in vitro culture period than adipogenesis. These results are confirmed by several other reports [19, 42–43, 55]. For instance, it was shown that after 20 population doublings, hMSCs were unable to mineralize and presented a negligible ALP induction by dexamethasone. Moreover, the amount of bone formed by hMSCs after implantation in nude mice displayed a dependence on the in vitro age of the cells [42]. Interestingly, the loss of differentiation capacity observed during in vitro expansion of hMSCs precedes terminal senescence and coincides with the activation of the DNA damage signalling pathways. We suggest that with expansion, there is an increase in DNA damage content, and therefore activation of the DNA damage signalling pathways, which results in a decrease in differentiation potential. In accordance, we used hydrogen peroxide and paraquat to increase the level of DNA damage in hMSCs at a concentration that did not affect significantly the proliferation. These concentrations proved still to be enough to increase DNA damage levels and affect the differentiation capacity. These results, in combination with the knowledge that, with age, adult haematopoietic stem cell function declines and that DNA damage and epigenetic modification limit the regenerative potential of those cells, seem to further substantiate the possible link between DNA damage accumulation and loss of stemness [22–24].
The significant drop in quality of culture expanded hMSCs poses some concerns considering the possibility to use these cells for clinical applications. We conclude that hMSCs are susceptible to accumulate DNA damage upon in vitro expansion and that this DNA damage accumulation correlates directly with the observed loss of multi-potency after hMSC in vitro expansion. Although they maintained an effective NER activity, there was still a progressive DNA damage accumulation, which might be one of the mechanisms underlying age-related loss of tissue function.
These results suggest that hMSCs should be minimally expanded and leave a window of possibility for the use and screen of molecules that might circumvent or reduce the in vitro DNA damage accumulation, leading to a possible extension of the differentiation capacity and making them even more suitable for clinical applications.
Acknowledgments
We thank Dr. L. Creemers, Dr. W. Dhert (University Medical Center Utrecht) for kindly providing us with bone marrow aspirates. We also thank Ruud Licht and Anouk Leusink for technical support and The Netherlands Technology Foundation (STW grant TGT.6745) for financial support.
Supporting Information
Fig. S1 hMSCS morphology and differentiation is affected by in vitro expansion (A) Phase-contrast photos of hMSCs at different RPD, 340 magnification. An evident change in cell size and morphology can be observed with increased time of culture. (B) Change in the mineralization potential after in vitro expansion. Relative calcium accumulation by hMSCs in MM after 28 days, expressed as percentage reduction in calcium deposition ability over passages compared to RPD 6. No detectable calcium was measured in hMSCs cultured in BM (data not shown). (C) Phase-contrast images of hMSC in adipogenic medium at different RPD. Adipocytes are stained with Oil red O and visualized at 340 magnification. Error bars represent standard deviation. Statistical analysis was performed using one-way ANOVA and Tukey post-test with a significance level of 0.05. * represents P < 0.01 to 0.05and *** P < 0.001.
Fig. S2 Immunophenotype of hMSCs (RPD 6 versus RPD 20). Representative overlay of flow cytometry histograms of hMSCs from human bone marrow are presented. Black histogram, RPD ± and green line, histogram of RPD 20.
Fig. S3 Senescence-associated b-galactosidase expression after DNA damage induction. RPD ± cells were exposed to 0, 30 or50 mM of H2O2 for ± days after which cells were stained for SA-b-galactosidase.
Table S1 Information relative to the hMSCs donors used in this article.
References
- 1.Haynesworth SE, Goshima J, Goldberg VM, et al. Characterization of cells with osteogenic potential from human marrow. Bone. 1992;13:81–8. doi: 10.1016/8756-3282(92)90364-3. [DOI] [PubMed] [Google Scholar]
- 2.Bruder SP, Jaiswal N, Haynesworth SE. Growth kinetics, self-renewal, and the osteogenic potential of purified human mesenchymal stem cells during extensive subcultivation and following cryopreservation. J Cell Biochem. 1997;64:278–94. doi: 10.1002/(sici)1097-4644(199702)64:2<278::aid-jcb11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 3.Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res. 2004;95:9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- 4.Xu W, Zhang X, Qian H, et al. Mesenchymal stem cells from adult human bone marrow differentiate into a cardiomyocyte phenotype in vitro. Exp Biol Med. 2004;229:623–31. doi: 10.1177/153537020422900706. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez-Ramos J, Song S, Cardozo-Pelaez F, et al. Adult bone marrow stromal cells differentiate into neural cells in vitro. Exp Neurol. 2000;164:247–56. doi: 10.1006/exnr.2000.7389. [DOI] [PubMed] [Google Scholar]
- 6.Meijer GJ, De Bruijn JD, Koole R, et al. Cell-based bone tissue engineering. PLoS Med. 2007;4:e9. doi: 10.1371/journal.pmed.0040009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quarto R, Mastrogiacomo M, Cancedda R, et al. Repair of large bone defects with the use of autologous bone marrow stromal cells. N Engl J Med. 2001;344:385–6. doi: 10.1056/NEJM200102013440516. [DOI] [PubMed] [Google Scholar]
- 8.Cancedda R, Mastrogiacomo M, Bianchi G, et al. Bone marrow stromal cells and their use in regenerating bone. Novartis Found Symp. 2003;249:133–43. ; discussion 43–7, 70–4, 239–41. [PubMed] [Google Scholar]
- 9.Schimming R, Schmelzeisen R. Tissue-engineered bone for maxillary sinus augmentation. J Oral Maxillofac Surg. 2004;62:724–9. doi: 10.1016/j.joms.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Brooke G, Cook M, Blair C, et al. Therapeutic applications of mesenchymal stromal cells. Semin Cell Dev Biol. 2007;18:846–58. doi: 10.1016/j.semcdb.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 11.Caplan AI. Why are MSCs therapeutic? New data: new insight. J Pathol. 2009;217:318–24. doi: 10.1002/path.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Csete M, Walikonis J, Slawny N, et al. Oxygen-mediated regulation of skeletal muscle satellite cell proliferation and adipogenesis in culture. J Cell Physiol. 2001;189:189–96. doi: 10.1002/jcp.10016. [DOI] [PubMed] [Google Scholar]
- 13.Fehrer C, Brunauer R, Laschober G, et al. Reduced oxygen tension attenuates differentiation capacity of human mesenchymal stem cells and prolongs their lifespan. Aging Cell. 2007;6:745–57. doi: 10.1111/j.1474-9726.2007.00336.x. [DOI] [PubMed] [Google Scholar]
- 14.Potier E, Ferreira E, Andriamanalijaona R, et al. Hypoxia affects mesenchymal stromal cell osteogenic differentiation and angiogenic factor expression. Bone. 2007;40:1078–87. doi: 10.1016/j.bone.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 15.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 16.Campisi J, D’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 17.Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–13. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 18.Cichowski K, Hahn WC. Unexpected pieces to the senescence puzzle. Cell. 2008;133:958–61. doi: 10.1016/j.cell.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siddappa R, Licht R, Van Blitterswijk C, et al. Donor variation and loss of multipotency during in vitro expansion of human mesenchymal stem cells for bone tissue engineering. J Orthop Res. 2007;25:1029–41. doi: 10.1002/jor.20402. [DOI] [PubMed] [Google Scholar]
- 20.Hasty P, Vijg J. Accelerating aging by mouse reverse genetics: a rational approach to understanding longevity. Aging Cell. 2004;3:55–65. doi: 10.1111/j.1474-9728.2004.00082.x. [DOI] [PubMed] [Google Scholar]
- 21.De Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21:453–60. doi: 10.1093/carcin/21.3.453. [DOI] [PubMed] [Google Scholar]
- 22.Chambers SM, Shaw CA, Gatza C, et al. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nijnik A, Woodbine L, Marchetti C, et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447:686–90. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 24.Rossi DJ, Bryder D, Seita J, et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–9. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 25.Both SK, Van Der Muijsenberg AJ, Van Blitterswijk CA, et al. A rapid and efficient method for expansion of human mesenchymal stem cells. Tissue Eng. 2007;13:3–9. doi: 10.1089/ten.2005.0513. [DOI] [PubMed] [Google Scholar]
- 26.Herbig U, Jobling WA, Chen BP, et al. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–13. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 27.Ebert R, Ulmer M, Zeck S, et al. Selenium supplementation restores the antioxidative capacity and prevents cell damage in bone marrow stromal cells in vitro. Stem Cells. 2006;24:1226–35. doi: 10.1634/stemcells.2005-0117. [DOI] [PubMed] [Google Scholar]
- 28.Vermeulen W, Scott RJ, Rodgers S, et al. Clinical heterogeneity within xeroderma pigmentosum associated with mutations in the DNA repair and transcription gene ERCC3. Am J Hum Genet. 1994;54:191–200. [PMC free article] [PubMed] [Google Scholar]
- 29.Schultz LB, Chehab NH, Malikzay A, et al. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151:1381–90. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rappold I, Iwabuchi K, Date T, et al. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153:613–20. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol. 2001;21:1719–29. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia Z, Morales JC, Dunphy WG, et al. Negative cell cycle regulation and DNA damage-inducible phosphorylation of the BRCT protein 53BP1. J Biol Chem. 2001;276:2708–18. doi: 10.1074/jbc.M007665200. [DOI] [PubMed] [Google Scholar]
- 33.Chen QM, Bartholomew JC, Campisi J, et al. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem J. 1998;332:43–50. doi: 10.1042/bj3320043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci USA. 1994;91:4130–4. doi: 10.1073/pnas.91.10.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frippiat C, Dewelle J, Remacle J, et al. Signal transduction in H2O2-induced senescence-like phenotype in human diploid fibroblasts. Free Radic Biol Med. 2002;33:1334–46. doi: 10.1016/s0891-5849(02)01044-4. [DOI] [PubMed] [Google Scholar]
- 37.Krall J, Bagley AC, Mullenbach GT, et al. Superoxide mediates the toxicity of paraquat for cultured mammalian cells. J Biol Chem. 1988;263:1910–4. [PubMed] [Google Scholar]
- 38.Jung T, Hohn A, Catalgol B, et al. Age-related differences in oxidative protein-damage in young and senescent fibroblasts. Arch Biochem Biophys. 2009;483:127–35. doi: 10.1016/j.abb.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 39.McCarthy S, Somayajulu M, Sikorska M, et al. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble Coenzyme Q10. Toxicol Appl Pharmacol. 2004;201:21–31. doi: 10.1016/j.taap.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 40.Ringden O, Uzunel M, Rasmusson I, et al. Mesenchymal stem cells for treatment of therapy-resistant graft-versus-host disease. Transplantation. 2006;81:1390–7. doi: 10.1097/01.tp.0000214462.63943.14. [DOI] [PubMed] [Google Scholar]
- 41.Rubio D, Garcia-Castro J, Martin MC, et al. Spontaneous human adult stem cell transformation. Cancer Res. 2005;65:3035–9. doi: 10.1158/0008-5472.CAN-04-4194. [DOI] [PubMed] [Google Scholar]
- 42.Stenderup K, Justesen J, Clausen C, et al. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone. 2003;33:919–26. doi: 10.1016/j.bone.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 43.Bonab MM, Alimoghaddam K, Talebian F, et al. Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 2006;7:14. doi: 10.1186/1471-2121-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Izadpanah R, Kaushal D, Kriedt C, et al. Long-term in vitro expansion alters the biology of adult mesenchymal stem cells. Cancer Res. 2008;68:4229–38. doi: 10.1158/0008-5472.CAN-07-5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 46.Zhu J, Woods D, McMahon M, et al. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herbig U, Ferreira M, Condel L, et al. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 48.Hildrestrand GA, Duggal S, Bjoras M, et al. Modulation of DNA glycosylase activities in mesenchymal stem cells. Exp Cell Res. 2009;315:2558–67. doi: 10.1016/j.yexcr.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 49.Kenyon J, Gerson SL. The role of DNA damage repair in aging of adult stem cells. Nucleic Acids Res. 2007;35:7557–65. doi: 10.1093/nar/gkm1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagner W, Bork S, Horn P, et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS ONE. 2009;4:e5846. doi: 10.1371/journal.pone.0005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galderisi U, Helmbold H, Squillaro T, et al. In vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem Cells Dev. 2009;18:1033–42. doi: 10.1089/scd.2008.0324. [DOI] [PubMed] [Google Scholar]
- 52.De Boer J, Andressoo JO, De Wit J, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–9. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- 53.Lee JS, Lee MO, Moon BH, et al. Senescent growth arrest in mesenchymal stem cells is bypassed by Wip1-mediated downregulation of intrinsic stress signaling pathways. Stem Cells. 2009;27:1963–75. doi: 10.1002/stem.121. [DOI] [PubMed] [Google Scholar]
- 54.Wagner W, Horn P, Castoldi M, et al. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PLoS ONE. 2008;3:e2213. doi: 10.1371/journal.pone.0002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Izadpanah R, Trygg C, Patel B, et al. Biologic properties of mesenchymal stem cells derived from bone marrow and adipose tissue. J Cell Biochem. 2006;99:1285–97. doi: 10.1002/jcb.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 hMSCS morphology and differentiation is affected by in vitro expansion (A) Phase-contrast photos of hMSCs at different RPD, 340 magnification. An evident change in cell size and morphology can be observed with increased time of culture. (B) Change in the mineralization potential after in vitro expansion. Relative calcium accumulation by hMSCs in MM after 28 days, expressed as percentage reduction in calcium deposition ability over passages compared to RPD 6. No detectable calcium was measured in hMSCs cultured in BM (data not shown). (C) Phase-contrast images of hMSC in adipogenic medium at different RPD. Adipocytes are stained with Oil red O and visualized at 340 magnification. Error bars represent standard deviation. Statistical analysis was performed using one-way ANOVA and Tukey post-test with a significance level of 0.05. * represents P < 0.01 to 0.05and *** P < 0.001.
Fig. S2 Immunophenotype of hMSCs (RPD 6 versus RPD 20). Representative overlay of flow cytometry histograms of hMSCs from human bone marrow are presented. Black histogram, RPD ± and green line, histogram of RPD 20.
Fig. S3 Senescence-associated b-galactosidase expression after DNA damage induction. RPD ± cells were exposed to 0, 30 or50 mM of H2O2 for ± days after which cells were stained for SA-b-galactosidase.
Table S1 Information relative to the hMSCs donors used in this article.