

Abstract
An appropriate density of acetylcholine receptors (AChRs) and Na+ channels (NaChs) in the normal neuromuscular junction (NMJ) determines the magnitude of safety factor (SF) that guarantees fidelity of neuromuscular transmission. In myasthenia gravis (MG), an overall simplification of the postsynaptic folding secondary to NMJ destruction results in AChRs and NaChs depletion. Loss of AChRs and NaChs accounts respectively for 59% and 40% reduction of the SF at the endplate, which manifests as neuromuscular transmission failure. The extraocular muscles (EOM) have physiologically less developed postsynaptic folding, hence a lower baseline SF, which predisposes them to dysfunction in MG and development of fatigue during “high performance” eye movements, such as saccades. However, saccades in MG show stereotyped, conjugate initial components, similar to normal, which might reflect preserved neuromuscular transmission fidelity at the NMJ of the fast, pale global fibers, which have better developed postsynaptic folding than other extraocular fibers.
Keywords: safety factor, extraocular muscles, saccades, neuromuscular junction
Introduction
In myasthenia gravis (MG), the autoimmune attack is directed towards the acetylcholine receptors (AChRs) at the endplate with consequent complement-mediated destruction of the neuromuscular junction (NMJ). Approximately 50% of patients with MG present with ocular symptoms at onset while up to 80% develop them during the course of the disease. Several contributing factors might explain the common extraocular muscles (EOM) involvement in MG, including an intrinsic deficiency in complement-inhibitory proteins.1 Herea we summarize evidence from our previous studies on the role of safety factor reduction at the “myasthenic” NMJ and provide an explanation for some characteristic eye movement findings in MG.2–3
Role of the safety factor in the normal neuromuscular junction
In skeletal muscles, the normal postsynaptic architecture consisting of well developed primary and secondary folding is instrumental in guaranteeing fidelity of neuromuscular transmission. Thus, acetylcholine receptors (AChRs) are more concentrated at the crest of the primary synaptic folds while the secondary synaptic folds house most of the voltage-gated Na+ channels (NaChs). Following acetylcholine release from the synaptic vesicles in the synaptic space and its consequent binding to the AChRs, channel pores open up generating a localized potential, which results in a current conveyed towards the depths of the secondary synaptic folds where, in turn, the NaChs rapidly open up.4–8 The passage of the Na+ current through the NaChs ultimately allows the muscle fiber to depolarize. The efficiency of neuromuscular transmission at the skeletal NMJ largely depends on the presence of a safety factor (SF) which ensures that an action potential (AP) is always triggered following acethylcoline release. The SF can be defined by using the formula EPP/EAP, where EPP is the endplate potential amplitude and EAP is the voltage difference between the resting potential (RP) and the action potential threshold.2 Appropriate concentrations of AChRs and NaChs directly influence the SF magnitude. Thus, while density of AChRs located at the primary postsynaptic folds increases the size of the EPP, the density of NaChs located at the secondary postsynaptic folds reduces the AP threshold, both ultimately resulting in increased SF magnitude.2 Direct evidence for the critical role of endplate NaChs in determining an appropriate SF magnitude comes from electrophysiological studies. Thus, the values for EAP and the SF are respectively higher and lower at the extrajunctional membrane, where the NaChs are physiologically less concentrated than at the endplate. It is estimated that the NaChs concentration at the endplate directly accounts for 34% of the safety factor.2
Decrement of the safety factor at the myasthenic junction
The AChRs antibodies involved in MG directly reduce the number of the endplate AChRs following a combination of complement-mediated membrane lysis and acceleration of AChR catabolism by receptor cross-linking.9–13 In addition, the voltage-gated NaChs are also depleted as a result of the overall simplification of the postsynaptic folding system that takes place in the disease.14–16 Experimental models of MG, induced by immunization with foreign or self AChRs (EAMG) or by passive transfer of myasthenogenic AChR-binding IgG (PTMG) in the rat, show that the combined loss of AChRs and NaChs results in a direct reduction of the endplate Na+ current at the muscle fibers.14,17–21
Loss of AChRs and NaChs account for decreased SF in myasthenia
Here we summarize evidence from our prior studies that diminished number of endplate AChRs and NaChs in myasthenia directly reduces the SF magnitude,2 which leaves an exiguous reserve for optimal neuromuscular transmission under demanding conditions (e.g., during exercise), leading to fatigue. The relative impact of AChRs versus NaChs loss in causing decrement of the endplate safety factor was investigated in human myasthenic fibers.2 Intercostal muscle fibers type IIb were donated by five male patients with myasthenia gravis (age 35–47 years) at the time of thymectomy. All patients had moderately severe generalized myasthenia gravis (MGFA class IIIa) and were seropositive for AChR binding antibodies. Seven control male subjects (age 35–52 years) with no evidence of neuromuscular disease, donated control intercostal muscle fibers at the time of a thoracotomy for treatment of cardiac or pulmonary disease. The protocol was approved by the Institutional Review Board of the Department of Veterans Affairs Medical Center in Cleveland. Resting potentials (RPs) in the human intercostal muscle fibers at the endplate and membrane potentials on extrajunctional membrane were measured using intracellular microelectrodes. Miniature endplate potentials (MEPPs) and endplate potentials (EPPs) were similarly measured at the endplate border. Action potential (AP) thresholds were measured using two microelectrodes, one to record the passing current and the other to record the membrane potential.
As expected, RPs were similar for control and MG patients. Both the MEPPs and the EPPs were reduced in fibers from patients with MG, being respectively 53% and 58% of the control value. The similar fractional reduction of MEPPs and EPPs is consistent with the notion that reduction in the EPP is due to decrease in the sensitivity of the postsynaptic membrane to acetylcholine. For control human muscle fibers, the depolarization threshold for initiating an AP was lower on the endplate border compared to the extrajunctional membrane, indicating that the density of NaChs was greater at the endplate. The values for AP threshold (EAP) measured on the extrajunctional membrane were similar for MG patients and controls. In contrast, the depolarization needed to trigger an action potential at the endplate was 13.5 mV for controls and 21.6 mV for MG patients.
Ultimately, the safety factor in the type IIb intercostal fibers from MG patients was only 37% of the safety factor in the control fibers, which was equal to 3. Comparison of EPP and EAP values between MG and control fibers made it possible to determine the relative impact on the SF magnitude of diminished number of AChRs and of NaChs. If the EPP of the MG fibers were the same as those of the control fibers, the safety factor for the MG fibers would have been 1.86 rather than the observed value of 1.09. If the EAP of MG fibers were the same as those of the control fibers, the safety factor for the MG fibers would have been 1.74. Thus, the reduction in EPP, which reflects the decreased concentration of AChRs, and the increase in EAP, which reflects the decreased concentration of NaChs, accounted respectively for 59% and 40% of the safety factor decrement in skeletal muscle fibers of patients with myasthenia.
In this study, the issue of whether anti-AChRs antibodies in MG reduce Na+ current at the endplate by a direct action towards the endplate NaChs was also addressed.2–3 With this respect, it has been previously established that the gating properties of NaChs away from the endplate are not altered in myasthenia gravis or PTMG and that, therefore, pathogenic antibodies in myasthenia gravis or PTMG do not target extrajunctional NaChs.14 Light microscopy and single channel Na+ current recording techniques were combined to study omohyoid nerve-muscle preparations from rats injected with monoclonal myasthenogenic IgG (McAb3) and inactive antibodies (McAb1), and from control rats. Injecting rats with the inactive McAb1 did not alter Na+ current at the endplate or on extrajunctional membrane compared with recordings from animals injected with no antibody. Currents recorded from the endplates of rat fibers injected with McAb3 and from rat fibers injected with McAb1 were similar to those recorded from the endplates of rats not treated with an antibody. Thus, rather than by direct impairment, anti-AchRs antibodies cause NaChs dysfunction through complement-mediated destruction of the endplate membrane.2
Suceptibility of extraocular muscles in myasthenia
Different structural and functional aspects of the extraocular muscles (EOM) need to be considered in order to explain their common involvement in myasthenia: (1) the EOM and orbital tissue in general, are intrinsically deficient in complement-inhibitory proteins, being therefore at risk for complement-mediated attack, as it happens in the typical immunological scenario of myasthenia gravis;1 (2) the EOM are physiologically subject to unusual demands imposed by the visual system, which require sustained and precise ocular alignment for single, binocular vision, in all conditions. Thus, even mild fatigue of the EOM can cause dramatic visual symptoms such as diplopia, whereas similar fatigue of skeletal muscle may be asymptomatic; (3) there are six types of neuromuscular junction fibers identified in the orbital and the global layers of the EOM (Fig. 1). Five of these fibers, whether singly or multiply innervated, lack the well-developed postsynaptic folding system which guarantees an appropriate safety factor and fidelity of neuromuscular transmission in regular skeletal muscles.22 In other words, EOM fibers are physiologically disadvantaged as the lack of a prominent postsynaptic folding system may result into an inherently lower safety factor, hence increased susceptibility to exercise-induced transmission failure in MG. However, the pale global singly innervated fibers (PGSF) represent an exception, as they indeed possess well developed postsynaptic folding (Fig. 1G). These fibers, which have fast-twitch properties and low fatigue resistance, are believed to briefly discharge at the beginning of a fast (saccadic) eye movement and provide the initial necessary drive to move the eyes toward the desired target.23
Figure 1.
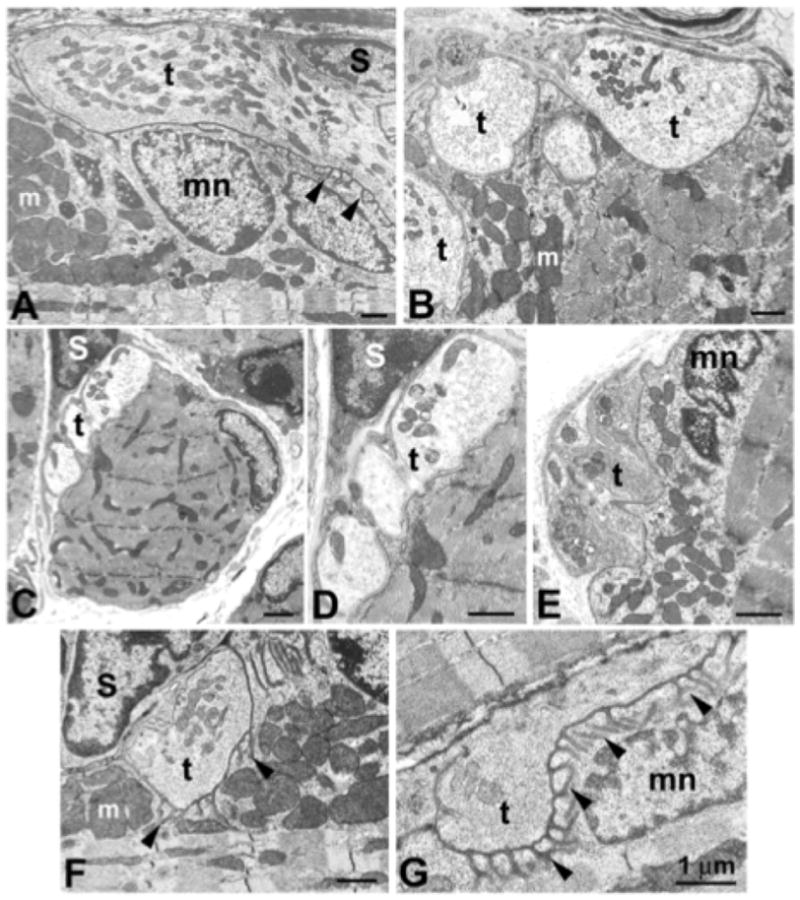
Electron photomicrographs of mouse extraocular muscle neuromuscular junction (NMJ) types. (A, B) Orbital singly innervated fibers (SIF) NMJs encircle individual myofibers, with long terminals (t) embedded in deep depressions of the myofiber surface. Terminals are capped by Schwann cells (S). Postjunctional folding is either absent or sparse (arrowheads), and postjunctional accumulations of myonuclei (mn) and mitochondria (m) is evident. Multiply innervated fibers (MIF) NMJs are small and do not lie in sarcolemmal depressions for both orbital (C, D) and global (E) fiber types. The range of variability in global SIF NMJs includes most junctions with few or no folds (F), but junctions with a high density of postjunctional folds (G) are found, primarily on the global pale SIF type. Reproduced, with permission from Khanna and Porter, IOVS, May 2003, Vol. 44, No. 5.
Dynamics of saccades in ocular myasthenia
Here we summarize the evidence from our prior studies to support the hypothesis that the PGSF are relatively spared in ocular myasthenia, because of their structural characteristics, which in turn accounts for some typical eye movement findings.3 We studied interocular conjugacy (i.e., how well the eyes move together) of fast eye movements (saccades) from a group of patients affected by ocular myasthenia. As previously shown, interocular conjugacy of horizontal saccades in patients with myasthenia gravis affected by disturbance of horizontal gaze was consistently preserved in the initial portion of the eye movement, despite the later component being markedly and variably dysconjugate.24–25 This was in contrast to consistent normal conjugacy of saccades made by control subjects and abnormal conjugacy of saccades made by patients with lesions at a site other than the NMJ (i.e., abducense nerve palsy or internuclear ophthalmoparesis),25 who showed instead early and sustained interocular dysconjugacy. However, a minimal dysconjugacy of horizontal saccades exists even in normal subjects, due the physiological delay of signal transmission imposed by the presence of the medial longitudinal fasciculus (MLF) between one set of burst neurons and one group of motoneurons for the medial rectus muscle.26–27 Thus, in a later study we focused on vertical saccades in patients with myasthenia gravis who showed disturbances of vertical gaze,3 as in healthy people vertical saccades are tightly yoked (burst neurons in the midbrain rostral interstitial nucleus of the medial longitudinal fasciculus directly project to motoneurons innervating yoke muscle pairs).
In particular, we recorded horizontal and vertical saccades from six patients with MG (age range 40–73 years; median 63), three age-matched patients with cranial nerve palsies (1 oculomotor, 1 trochlear, 1 abducens) and 10 age-matched control subjects. All patients and control subjects gave informed written consent in accordance with the Declaration of Helsinki and the Institutional Review Board of the Cleveland Veterans Affairs Medical Center. On clinical examination of patients with MG, in three cases dysconjugate saccades were evident horizontally; in two cases dysconjugate saccades were evident both horizontally and vertically; and in one case saccades were dysconjugate vertically. During the experimental protocol, subjects viewed a laser spot projected onto a tangent screen at 1.2 m. Saccades were tested as the visual target jumped at 0.25 Hz with amplitude ranging 5–40 degrees horizontally and 5–20 degrees vertically; the direction of target jumps was non-predictable. Binocular eye movements were recorded using the magnetic search coil technique; coil signals were filtered (0–150 Hz) before digitization at 500 Hz; eye velocity and acceleration were computed as previously described.28 The interocular conjugacy of horizontal and vertical saccades was assessed using the binocular phase-plane technique.26 For each saccade, the displacement (change in position) and velocity of each eye were normalized by assigning a value of 1.0 to the maximum displacement, and to the peak velocity, of the eye making the larger movement. Thus, the phase plane plotted the normalized velocity of each eye against the normalized displacement in 1% (0.01) position increments; this way, we were able to compare the velocity of each eye for the same normalized eye displacement during the entire saccadic movement as a velocity dysconjugacy plot. We applied this technique to over a thousand saccades from the 10 age-matched normal subjects and used linear regression to define 5% to 95% prediction intervals (PIs). Finally, for each patient, the average velocity dysconjugacy was calculated from at least 10 saccades and was then compared with the PI of the control subjects.
Representative records of an upward saccade from a control subject, a patient with ocular myasthenia, and a patient with oculomotor nerve palsy are shown in Figure 2. To the left in each row of the panels is a time plot; to the right are corresponding phase-plane plots of the upward saccades shown in the time plots. The vertical saccade made by the control subject (Fig. 2A) is tightly conjugate, as expected. The vertical saccade made by the patient with ocular myasthenia (Fig. 2B) is quite disjunctive, with greatly reduced movement of his left eye. However, as shown in the corresponding phase plane plot, the upward saccade was initially conjugate (indicated by black arrow). In contrast, the saccade from the patient with oculomotor nerve palsy (Fig. 2C) was disjunctive throughout its course, including the onset (indicated by gray arrow on the phase-plane plot).
Figure 2.

Representative records of a vertical upward saccade made by one healthy control subject (row A), a patient with ocular myasthenia manifest as left pseudo-oculomotor nerve palsy (row B), and a patient with right oculomotor nerve palsy (row C). Each row comprises a time plot of the upward saccade (left panel) and the correspondent phase-plane (right panel). The normal subject shows tight interocular saccadic conjugacy. The saccade from the patient with ocular myasthenia is initially conjugate (black arrow) but subsequently very disjunctive. The saccade from the patient with oculomotor nerve palsy is disjunctive from the onset (gray arrow). Eye movements were recorded using the magnetic search coil technique. Positive values on time plots indicate upward movements.
The three distinct patterns of vertical saccades were consistent from saccade to saccade when we systematically compared normalized phase planes of patients and control subjects: saccades of patients with ocular myasthenia invariably fell within the normal 95% prediction intervals for at least the first 10% of the movement (although later components did not), but saccades of patients with cranial nerve palsies always fell outside the normal 95% prediction intervals from their start. Similarly to what previously shown with horizontal saccades,26 patients with ocular myasthenia exhibited a consistent initial interocular conjugacy of vertical saccades, from saccade to saccade, even though later components of saccades varied, reflecting fatigue.
Conclusions
Basic knowledge of physiological and pathological neuromuscular transmission can be applied to account for typical disorders of saccades encountered in ocular myasthenia. In health, the well-developed system of postsynaptic folding, which houses AChRs and NaChs, allows fidelity of neuromuscular transmission by determining an optimal magnitude of safety factor. Thus, the normal folding architecture of the postsynaptic membrane is instrumental in directing the current resulting from AChRs opening (at the crest of the primary folds) to an area of the membrane rich in NaChs (at the bottom of the secondary folds).8 In myasthenia gravis, the complement-mediated destruction of the postsynaptic membrane results in simplification of the folding system and depletion of AChRs and NaChs. This is ultimately responsible for the decrement of the endplate safety factor which translates in exercise-induced failure of neuromuscular transmission.2–3 As opposed to regular skeletal muscle, the NMJ of five out of six categories of extraocular muscle fibers show physiologically poorly developed postsynaptic folding,22 which would result in a lower baseline safety factor even in health. This could be an important factor to account for the common occurrence of abnormal eye movements in myasthenia.24 However, one type of EOM fibers, the pale global singly-innervated fibers, have better developed synaptic folding (Fig. 1) and their baseline safety factor may be higher than other EOM fiber types. The pale global fibers are believed to exert their function at the beginning of the eye movement, when their firing rate briefly increases, and are probably responsible for the initial high-acceleration component of saccades. The observation that the initial component of saccades remains binocularly conjugate in MG, supports the hypothesis that the activity of the PGSF is relatively preserved in the disease due to their structural characteristics. Finally, the relative preserved functioning of the PGSF in myasthenia gravis might also account for the pathognomic clinical finding of “quiver movements”, where eye movements are typically still fast despite having a markedly restricted range of motion.29
Acknowledgments
The Office of Research and Development, Medical Research Service, Department of Veterans Affairs (Drs. Serra, Ruff, and Leigh); NIH grant EY06717 and the Evenor Armington Fund (Dr. Leigh); Center of Excellence support from the Office of Rehabilitation Research of the Research and Development Service of the Department of Veterans Affairs (Dr. Ruff).
Footnotes
Portions of this paper have been previously published in Annals of the New York Academy of Sciences 1233(1):26-33(2011).
References
- 1.Soltys J, Gong B, Kaminski HJ, Zhou Y, Kusner LL. Extraocular muscle suceptibility to myasthenia gravis: unique immunological environment? Ann NY Acad Sci. 2008;1132:220–224. doi: 10.1196/annals.1405.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruff RL, Lennon VA. How myasthenia gravis alters the safety factor for neuromuscular transmission. J Neuroimmunol. 2008;15:201–202. doi: 10.1016/j.jneuroim.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serra A, Ruff R, Kaminski H, Leigh RJ. Factors contributing to failure of neuromuscular transmission in myasthenia gravis and the special case of the extraocular muscles. Ann N Y Acad Sci. 2011;1233(1):26–33. doi: 10.1111/j.1749-6632.2011.06123.x. [DOI] [PubMed] [Google Scholar]
- 4.Angelides KJ. Fluorescently labeled Na+ channels are localized and immobilized to synapses of innervated muscle fibers. Nature. 1986;321:63–66. doi: 10.1038/321063a0. [DOI] [PubMed] [Google Scholar]
- 5.Flucher BE, Daniels MP. Distribution of Na+ channels and ankyrin in neuromuscular junctions is complementary to that of acetylcholine receptors and the 43 kD protein. Neuron. 1989;3:163–175. doi: 10.1016/0896-6273(89)90029-9. [DOI] [PubMed] [Google Scholar]
- 6.Haimovich B, Schotland DL, Barchi RL. Localization of sodium channel subtypes in rat skeletal muscle using channel-specific monoclonal antibodies. J Neurosci. 1987;7:2957–2966. doi: 10.1523/JNEUROSCI.07-09-02957.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leteut T, Boudier JL, Jover E, Cau P. Localization of voltage-sensitive sodium channels on the extrasynaptic membrane surface of mouse skeletal muscle by autoradiography of scorpion toxin binding sites. J Neurocytol. 2011;19:408–420. doi: 10.1007/BF01188407. [DOI] [PubMed] [Google Scholar]
- 8.Slater CR. Structural factors influencing the efficacy of neuromuscular transmission. Ann NY Acad Sci. 2007;1132:1–12. doi: 10.1196/annals.1405.003. [DOI] [PubMed] [Google Scholar]
- 9.Engel AG, Fumagalli G. Mechanisms of acetylcholine receptor loss from the neuromuscular junction. Ciba Found Symp. 1982;90:197–224. doi: 10.1002/9780470720721.ch12. [DOI] [PubMed] [Google Scholar]
- 10.Engel AG, Lindstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of the acetylcholine receptors in myasthenia gravis and in its experimental autoimmune model. Neurology. 1977;27:307–315. doi: 10.1212/wnl.27.4.307. [DOI] [PubMed] [Google Scholar]
- 11.Fambrough DM, Drachman DB, Satyamurti S. Neuromuscular junction in Myasthenia Gravis: Decreased acetylcholine receptors. Science. 1973;182:293–295. doi: 10.1126/science.182.4109.293. [DOI] [PubMed] [Google Scholar]
- 12.Kaminski HJ, Ruff RL. Structure and kinetic properties of the acetylcholine Receptor. In: Engel AG, editor. Myasthenia Gravis and Myasthenic Syndromes. Oxford University Press; New York: 1999. pp. 40–64. [Google Scholar]
- 13.Kao I, Drachman D. Myasthenic immunoglobulin accelerates acetylcholine receptor degradation. Science. 1977;196:526–528. doi: 10.1126/science.850793. [DOI] [PubMed] [Google Scholar]
- 14.Ruff RL, Lennon VA. Endplate voltage-gated sodium channels are lost in clinical and experimental myasthenia gravis. Ann Neurol. 1998;43:370–279. doi: 10.1002/ana.410430315. [DOI] [PubMed] [Google Scholar]
- 15.Engel AG, Santa T. Histometric analysis of the ultrastructure of the neuromuscular junction in myasthenia gravis and the myasthenic syndrome. Ann NY Acad Sci. 1971;183:46–63. doi: 10.1111/j.1749-6632.1971.tb30741.x. [DOI] [PubMed] [Google Scholar]
- 16.Santa T, Engel AG, Lambert EH. Histometric study of neuromuscular junction ultrastructure. I. Myasthenia gravis. Neurology. 1972;22:71–82. doi: 10.1212/wnl.22.1.71. [DOI] [PubMed] [Google Scholar]
- 17.Kaminski HJ, Ruff RL. The myasthenic syndromes. In: Schultz SG, Andreoli TE, Brown AM, Fambrough D, Hoffman JF, Welsh M, editors. Physiology of Membrane Disorders. Plenum Press; New York: 1996. pp. 565–593. [Google Scholar]
- 18.Lennon VA, Lambert EH. Myasthenia gravis induced by monoclonal antibodies to acetylcholine receptors. Nature. 1980;285:238–240. doi: 10.1038/285238a0. [DOI] [PubMed] [Google Scholar]
- 19.Lindstrom JM, Einarson BL, Lennon VA, Seybold ME. Pathological mechanisms in experimental autoimmune myasthenia gravis. I. Immunogenicity of syngeneic muscle acetylcholine receptor and quantitative extraction of receptor and anti-receptor complexes from muscle of rats with experimental autoimmune myasthenia gravis. J Exp Med. 1976;144:726–738. doi: 10.1084/jem.144.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindstrom JM, Engel AG, Seybold ME, Lennon VA, Lambert EH. Pathological mechanisms in experimental autoimmune myasthenia gravis. II. Passive transfer of experimental autoimmune myasthenia gravis in rats with anti-acetylcholine receptor antibodies. J Exp Med. 1976;144:739–753. doi: 10.1084/jem.144.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maselli RA, Richman DP, Wollmann RL. Inflammation at the neuromuscular junction in myasthenia gravis. Neurology. 1991;41:1497–1504. doi: 10.1212/wnl.41.9.1497. [DOI] [PubMed] [Google Scholar]
- 22.Spencer RF, Porter JD. Biological organization of the extraocular muscles. Prog Brain Res. 2006;151:33–79. doi: 10.1016/S0079-6123(05)51002-1. [DOI] [PubMed] [Google Scholar]
- 23.Scott AB, Collins CC. Division of labor in human extraocular muscle. Arch Ophthalmol. 1973;90:319–322. doi: 10.1001/archopht.1973.01000050321017. [DOI] [PubMed] [Google Scholar]
- 24.Khanna S, Liao K, Kaminski HJ, Tomsak RL, Joshi A, Leigh RJ. Myasthenia revisited: New insights from pseudo-internuclear ophthalmoplegia. J Neurology. 2007;254:1569–1574. doi: 10.1007/s00415-007-0591-y. [DOI] [PubMed] [Google Scholar]
- 25.Serra A, Liao K, Matta M, Leigh RJ. Diagnosing disconjugate eye movements: phase-plane analysis of horizontal saccades. Neurology. 2008;71:1167–1175. doi: 10.1212/01.wnl.0000327525.72168.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leigh RJ, Zee DS. The Neurology of Eye Movements. 4. Oxford University Press; New York: 2006. Book/DVD. [Google Scholar]
- 27.Moschovakis AK, Scudder CA, Highstein SM. A structural basis for Hering’s law: Projections to extrocular motoneurons. Science. 1990;248:1118–1119. doi: 10.1126/science.2343316. [DOI] [PubMed] [Google Scholar]
- 28.Ramat S, Somers JT, Das VE, Leigh RJ. Conjugate ocular oscillations during shifts of the direction and depth of visual fixation. Invest Ophthalmol Vis Sci. 1999;40:1681–1686. [PubMed] [Google Scholar]
- 29.Cogan DG, Yee RD, Gittinger J. Rapid eye movements in myasthenia gravis. I. Clinical observations. Arch Ophthalmol. 1976;94:1083–1085. doi: 10.1001/archopht.1976.03910040003001. [DOI] [PubMed] [Google Scholar]