

Abstract
Herein we report the discovery and SAR of a novel series of non-MPEP site metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulators (PAMs) based on an aryl glycine sulfonamide scaffold. This series represents a rare non-MPEP site mGlu5 PAM chemotype.
Keywords: metabotropic glutamate receptor 5, mGlu5, positive allosteric modulator (PAM), non-MPEP
Allosteric modulation of metabotropic glutamate receptor subtype 5 (mGlu5), with positive allosteric modulators (PAMs) is an increasingly popular approach for selective receptor activation.1–5 Targeting NMDA hypofunction,6 as opposed to classical hyperdopaminergia,7 mGlu5 PAMs have provided robust preclinical validation in multiple schizophrenia and cognition models.8–15 Recently, mGlu5 PAMs have been reported representing diverse chemotypes (Fig. 1);16 however, the majority of these, such as 1–6, bind at the MPEP (an mGlu5 negative allosteric modulator, or NAM) binding site,1–5,8–14 and in vivo efficacy has yet to be demonstrated for a non-MPEP site PAM, such as 717,18 or 8.19 Moreover, a new phenomenon has emerged where very subtle structural changes, i.e., a `molecular switch', to multiple MPEP-site PAMs can modulate either the mode of pharmacology (PAM to NAM) or mGlu subtype selectivity, raising concerns over the pharmacology of metabolites in vivo.11,13,20–22 Importantly, this phenomenon of `molecular switches' has not been observed with the two known non-MPEP site PAM chemotypes, CPPHA (7)17,18 and VU0357121 (8),19 though the SAR has proven to be far steeper than the MPEP site congeners. Thus, our lab has been focused on the identification and optimization of additional non-MPEP site mGlu5 PAMs to ascertain the presence or absence of `molecular switches', and to determine if non–MPEP site ligands afford the same in vivo efficacy profile as MPEP-site PAMs.
Figure 1.

Representative MPEP site (1–6) and non-MPEP site (7 and 8) mGlu5 PAMs.
Herein, we describe the synthesis and SAR of a novel series of potent, non-MPEP site mGlu5 PAMs based on an aryl glycine sulfonamide scaffold identified in a functional HTS.12
A high-throughput functional screen, employing a triple-add calcium mobilization assay of 160,000 compounds, identified 1,400 confirmed mGlu5 PAMs, and over 60 with EC50s below 500 nM in our high-expressing rat mGlu5 HTS cell line.12 From this, we identified VU0034403 (9), an unprecedented mGlu5 PAM chemotype with potent (rat EC50 = 98 nM, 48% Glu Max, 7.9-fold shift) ago-PAM activity (Fig. 2 top panel), and amenable to chemical optimization via an iterative library approach. Significantly, 9 was selective versus the other mGluRs (mGlu1–4,6–8 > 10 μM), and afforded minimal displacement of [3H]-methoxyPEPy (Ki > 10 μM, Fig. 2 bottom panel), suggesting that 9 binds at a site distinct from the MPEP site.
Figure 2.

Top panel: Structure of non-MPEP mGlu5 PAM HTS hit VU0034403 (9) and the chemical optimization plan leading to analogs 10. Bottom panel: Radioligand displacement assay [[3H]-methoxyPEPy] using VU0034403 and MPEP.
Synthesis of first generation analogs 13 proceeded smoothly through a five step process (Scheme 1) surveying diversity at the Eastern amide moiety. Beginning with 2-Cl aniline 11, sulfonylation delivered 12 in quantitative yield. Alkylation of 12 with methyl bromoacetate provided ester 13, which was saponified to give acid 14. A two-step amide coupling procedure, employing a diverse collection of amines (aromatic, benzylic, 1°, 2°, aliphatic, with basic and acidic moieties) generated analogs 15 in isolated yields ranging from 26–68%.
Scheme 1.
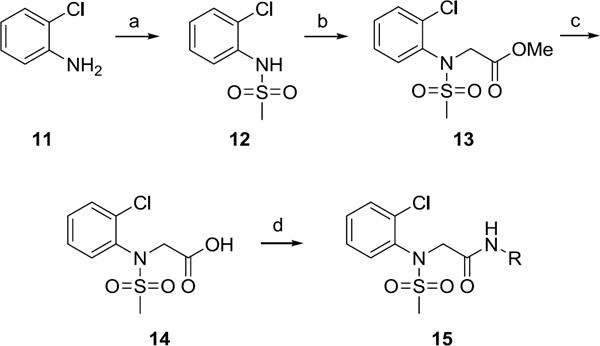
Library synthesis of analogs 15. Reagents and conditions: (a) MeSO2Cl, pyridine, CH2Cl2, 99%, (b) (i) NaH, DMF, (ii) methyl bromoacetate, DMF, 94–98%, (c) LiOH, MeOH, THF, 99%, (d) PS-HOBt, HATU, 2,6-lutidine, DMF,RR'NH, CH2Cl2, 26–68%. All library compounds were purified by mass-directed prep LC where required.
While it was important to employ the high expressing rat mGlu5 cell line in the HTS to ensure we identified even weak mGlu5 PAMs, the chemical optimization program transitioned to human mGlu5 cell lines with expression levels more closely aligned with native expression.12,23 Potency was maintained for freshly prepared 9 in the rat cell-line while maximum efficacy increased (rat EC50 = 78 nM, 74% Glu Max), and Table 1 shows data for selected analogs 15 in both rat and human mGlu5 cell lines. Against the human receptor, the potency of HTS lead 9 was ten-fold less potent relative to the rat receptor cell line with an EC50 of 780 nM.
Table 1.
Structures and activities of analogs 15.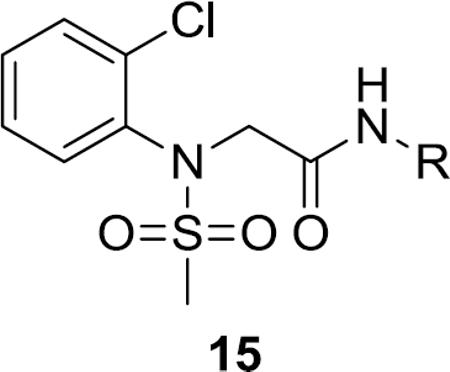
| Cmpd | R | rmGlu5 pEC50/EC50a | % Glu Max | hmGlU5 pEC50/EC50a | % Glu Max |
|---|---|---|---|---|---|
| 9 |

|
7.11 | 74 | 6.11 | 63 |
| 0.08 | 0.78 | ||||
| 15a |

|
7.14 | 61 | 5.86 | 77 |
| 0.07 | 1.38 | ||||
| 15b |

|
5.43 | 66 | NT | NT |
| 3.72 | |||||
| 15c |

|
5.76 | 64 | <5.0 | 42 |
| 1.74 | >10 | ||||
| 15d |

|
5.35 | 72 | <5.0 | 35 |
| 4.47 | >10 |
pEC50 are the average of three independent determinations and represent a coefficient of variation (CV) < 0.1, EC50 units are in μM;
NT = not tested.
Similar to the non-MPEP PAM CPPHA (7), SAR was extremely flat, with this first generation library affording very few active PAMs, and a ~3-fold difference between rat and human potency. Of the ~50 analogs tested, only four showed good PAM activity, and these are all closely related phenyl 15a (rat EC50 = 72 nM, 61% Glu Max), and fluoro-pyridine congeners 15b–d (rat EC50s 1.7–4.5 μM, 64–72% glu max). Basic and acidic analogs were inactive, as were 3° amide congeners including a constrained indoline. In general cyclic aliphatic amides were weakly active and acyclic amides were inactive. Like 9, 15a possessed strong allosteric agonist activity on rat mGlu5 (an ago-PAM) which precluded the determination of an accurate fold shift measurement; however, the fold-shift is estimated to be minimal (~1.2). Within this series no ago-PAM activity was detected in the lower expressing human mGlu5 cell line. Based upon previous studies wherein receptor expression level was found to impact allosteric agonist activity23 we believe this phenomenon to be expression level dependent rather than due to a species difference. While 15a possessed the potency, selectivity (mGlu1–4,6–8 > 10 μM) and free fraction (rat plasma protein binding, Fu = 0.13) required for in vivo studies in rats for a non-MPEP PAM ([3H]-methoxyPEPy Ki > 10 μM), 15a suffered poor metabolic stability (≤4% remaining after 15 minutes).24 The pyridyl congener 15b displayed an equivalent extent of plasma protein binding (rat Fu = 0.17, human Fu = 0.09) and improved metabolic stability in rat liver microsomes (39% remaining after 15 minutes); however, the rat mGlu5 potency was not optimal for in vivo studies.
The next library iteration surveyed branching at the α-position with alkyl substituents. Analogs were easily prepared (Scheme 2), beginning with alkylation of 12 using the appropriate ethylbromoacetate (Me, Et, and n-Pr branched) and saponification to afford acid intermediates 16a–c. Amide formation was performed as previously described to generate racemic analogs 17. The methyl derivative 17a was active (rat EC50 = 68 nM), but displayed diminished efficacy (43% Glu Max). Larger alkyl groups (Et 17b and n-Pr 17c) were weak (17b rat EC50 = 3.3 μM) or inactive (17c rat EC50 > 10 μM).
Scheme 2.

Library synthesis of analogs 17. Reagents and conditions: (a) NaH, RBr, DMF, 17-quant.% (b) LiOH, MeOH, THF, 99%, (c) PS-HOBt, HATU, 2,6-lutidine, DMF, (e) m-ClPhNH2, CH2Cl2, 73%. All library compounds were purified by mass-directed prep LC where required.
Based on these data, we then introduced a cyclic constraint between the α-position and the 2-Cl moiety to generate a racemic tetrahydroisoquinoline derivative 19 (Scheme 3). In the high expressing HTS rat mGlu5 cell line, this provided a moderately potent and efficacious PAM (EC50 = 1.0 μM, 60% Glu Max).
Scheme 3.

Synthesis of racemic tetrahydroisoquinoline derivative 19. Reagents and conditions: (a) MeSO2Cl, pyridine, CH2Cl2, 99%, (b) LiOH, MeOH, THF, 99%, (d) PS-HOBt, HATU, 2,6-lutidine, DMF, (e) m-ClPhNH2, CH2Cl2, 74%.
Based on the commercial availability of enantiopure (R)- and (S)-18, subsequent libraries (consisting of 80 analogs) explored stereochemistry as well as diversity at both the sulfonamide and the amide moieties following the chemistry outlined in Scheme 3. Enantioselective mGlu5 potentiation was observed, with the (S)-enantiomer generally >10-fold more potent than the (R)-enantiomer. Overall little improvement in potency was observed, SAR remained relatively flat, and metabolic stability remained an issue. Figure 3 highlights representative racemic and (S)-enantiomer analogs 20 – 23. Although efficacy would appear to be overall improved within the tetrahydroisoquinoline series as illustrated by these examples (68–78%), they displayed weak potency in the micromolar range.
Figure 3.

Structures of non-MPEP mGlu5 PAM (S)-tetrahydroisoquinoline analogs 20–22.
In parallel, additional libraries were focused on identifying alternative aryl R1 moieties (Fig. 2) and non-aromatic amides R2 in an attempt to improve metabolic stability while maintaining acceptable potency and efficacy following the routes outlined in Schemes 1 and 2. Similar to CPPHA 7, SAR was shallow, with only ~10% of compounds assayed displaying mGlu5 PAM activity. Of these, VU0400100 (24), an analog with an additional 5-CF3 on the western 2-ClPh ring and an eastern cyclopropyl methyl amide, was studied extensively (Fig. 4). PAM 24 was moderately potent on both human and rat mGlu5, did not displace [3H]-methoxyPEPy, was highly selective versus the other mGlus and displayed significantly reduced plasma protein binding (rat, human) and nonspecific binding (Fu, 0.10) in rat brain homogenate binding. Moreover, 24 lacked significant activity in a Ricerca radioligand binding panel of 68 GPCRs, ion channels and transporters (<50% inhibition at 10 μM) and 24 was devoid of functional activity in a panel of ion channels relevant to cardiovascular safety (hERG, Ca, and Na, IC50 >10 μM).
Figure 4.

Structure, pharmacological, and DMPK profile of VU0400100, 24, a non-MPEP site mGlu5 PAM.
Consistent with the in vitro predicted clearance (Fig. 4), rat pharmacokinetics indicated 24 suffered extensive first-pass hepatic metabolism (EH, 0.98) following an oral administration, as significantly lower exposure was observed in systemic supply relative to hepatic portal vein blood supply (AUCHPV: AUCplasma, 52). Exposures were improved with intraperitoneal or subcutaneous dosing routes, achieving CNS exposure relative to the systemic circulation (AUCbrain:AUCplasma, 0.4) sufficient to suggest brain penetration is possible for this class of non-MPEP based mGlu5 PAMs.
Currently, in vitro studies are underway to establish if the pan-P450 inactivator, 1-aminobenzotriazole (ABT), will have an impact on the clearance of 24 in rat microsomes.25 If sufficient reduction of P450-mediated clearance of 24 is observed in vitro, it may be possible to enhance exposure in vivo and establish proof-of-concept in rat behavioral models with a non-MPEP site mGlu5 PAM by administering 24 to rats that receive an oral dose of ABT.
It is interesting to note that to our knowledge this is a rare example of a chemical series of mGlu5 PAMs containing a sulfonamide moiety. Analogs of 9 wherein the sulfonamide was replaced with N-alkyl, N-aryl or a N-acetyl moiety were devoid of PAM activity. These observations prompted us to replace the amide moiety in the known mGlu5 MPEP-site PAM series 4 and 5 (Fig. 1) with sulfonamides (Fig. 5). While potency was diminished at least an order of magnitude relative to the direct amide counterpart scaffolds (4 and 5, Fig. 1),12,15 the sulfonamide congeners 25 and 26 were weak to moderately active PAMs in the high expressing rat cell line, suggesting additional avenues for optimization. Within the human cell line the acetylene PAM 25 was inactive and the Addex analog 26 a weak PAM. Competition binding studies using membranes isolated from rmGlu5 demonstrated no displacement of [3H]-methoxyPEPy with up to 30 μM 25 or 26. Although more potent analogs are needed to establish an allosteric binding site for these hybrid PAMs, the activity observed suggests that it may be possible to identify additional sulfonamide containing mGlu5 PAMs.
Figure 5.

Structures and activities of representative MPEP-site mGlu5 PAMs with the amide moiety replaced with the analogous sulfonamide linker.
In addition to the above exercise preparing sulfonamide hybrids using known MPEP-site mGlu5 PAM scaffolds we have also undertaken a computational evaluation of low energy, ligand-based conformational ensembles of MPEP and non-MPEP ligands. Mutual flexible low energy shape-based alignment of sulfonamide 15a with CPPHA (7) using the Surflex-Sim algorithm suggests a preferred fit for CPPHA (7) versus other MPEP modulators (Fig. 6).26 Studies are ongoing to develop a comprehensive model based upon this approach as an entry point to identify additional novel non-MPEP modulators.
Figure 6.

Suflex-Sim overlay of CPPHA (7, grey surface, ball and stick) and 15a (gold surface, capped sticks).
Significantly, this is only the third known non-MPEP site mGlu5 PAM chemotype, and like CPPHA (7) and VU0357121 (8), SAR was steeper than that of MPEP site PAMs. Importantly, no `molecular switches' were observed within this non-MPEP site series.16 While DMPK properties preclude in vivo studies, key PAMs within this series are valuable tools for detailed in vitro pharmacological and electrophysiology studies. Additional studies and refinements are in progress and will be reported in due course.
Acknowledgments
This work was supported in part by grants from the NIH (NS031373, MH062646, and MH89870) and from an industry sponsored contract from Johnson & Johnson. The authors thank Daryl F. Venable and Kiran K. Gogi for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Williams DL, Jr., Lindsley CW. Curr. Topics in Med. Chem. 2005;5:825–838. doi: 10.2174/1568026054750290. [DOI] [PubMed] [Google Scholar]
- 2.Lindsley CW, Wolkenberg SE, Shipe W, Williams DL., Jr. Curr. Opin. in Drug Disc. Dev. 2005;8(4):449–457. [PubMed] [Google Scholar]
- 3.Conn PJ, Christopolous A, Lindsley CW. Nat. Rev. Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conn PJ, Lindsley CW, Jones C. Trends in Pharm. Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wroblewski JT, Pin JP. Neuropharmacology. 2011;60:1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindsley CW, Shipe WD, Wolkenberg SE, Theberge CR, Williams DL, Jr., Sur C, Kinney GG. Curr. Topics in Med. Chem. 2006;8:771–784. doi: 10.2174/156802606777057599. [DOI] [PubMed] [Google Scholar]
- 7.Seeman P, Lee T, Chau-Wong M, Wong K. Nature. 1976;261:717–719. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Lindsley CW, Sur C, Pettibone DJ, Conn J, Williams DL. Mol. Pharmacol. 2003;64:731–741. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 9.Lindsley CW, Wisnoski DD, Leister WH, O'Brien JA, Lemiare W, Williams DL, Jr., Burno M, Sur C, Kinney GG, Pettibone DJ, Miller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. J. Med. Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 10.Kinney GG, O'Brien, Lemaire W, Burno M, Bickel DJ, Clements MK, Wisnoski DD, Lindsley CW, Tiller PR, Smith S, Jacobson MA, Sur C, Duggan ME, Pettibone DJ, Williams DW., Jr. J. Pharmacol. Exp. Therapeut. 2005;313(1):199–208. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- 11.Sharma S, Kedrowski J, Rood JM, Smith RL, Jones CK, Rodriguez AL, Conn PJ, Lindsley CW. J. Med. Chem. 2009;52:4103–4108. doi: 10.1021/jm900654c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon U, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Mol. Pharm. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y, Manka J, Rodriguez AL, Weaver CD, Jones CK, Conn PJ, Lindsley CW, Stauffer SR. ACS Med. Chem. Lett. 2010;1:433–438. doi: 10.1021/ml100181a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong H, Brugel TA, Balestra M, Brown DG, Brush KA, Hightower C, Hinkley L, Hoesch V, Kang J, Koether GM, McCauley JP, Jr., McLaren FM, Panko LM, Simpson TR, Smith RW, Woods JM, Brockel B, Chhajlani V, Gadient RA, Spear N, Sygowski LA, Zhang M, Arora J, Breysse N, Wilson JM, Isaac M, Slassi A, King MM. Bioorg. Med. Chem. Lett. 2010;20:7381–7385. doi: 10.1016/j.bmcl.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 15.Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, Smith D, Olsen M, Kouranova E, Lai M, Pruthi F, Pulcicchio C, Day M, Gilbert A, Pausch MH, Brandon NJ, Beyer CE, Comery TA, Logue S, Rosenzweig-Lipson S, Marquis KL. J. Pharmacol. Exp. Therapeut. 2008;327:827–839. doi: 10.1124/jpet.108.136580. [DOI] [PubMed] [Google Scholar]
- 16.Stauffer SR. ACS Chem. Neurosci. 2011;2:450–470. doi: 10.1021/cn2000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Sur C, Duggan ME, Pettibone DJ, Conn J, Williams DL. J. Pharmacol. Exp. Therapeut. 2004;309(2):568–579. doi: 10.1124/jpet.103.061747. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Z, Wisnoski DD, O'Brien JA, Lemiare W, Williams DL, Jr., Jacobson MA, Wittman M, Ha S, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg. Med. Chem.Lett. 2007;17:1386–1389. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 19.Hammond AS, Rodriguez AL, Niswender CM, Lindsley CW, Conn PJ. ACS Chem. Neurosci. 2010;1:702–716. doi: 10.1021/cn100051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma S, Rodriguez A, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2008;18:4098–4101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. Biochemistry. 2011;50:2403–2140. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamb JA, Engers DW, Niswender CM, Venable D, Conn PJ, Lindsley CW. Bioorg. Med.Chem. Lett. 2011;21:2711–2714. doi: 10.1016/j.bmcl.2010.11.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, Xiang Z, Daniels, Daniels JS, Lindsley CW, Weaver CD, Conn PJ. Mol. Pharm. 2012;81:120–133. doi: 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.See reference 12 for a description of in vitro DMPK assay methods (plasma protein binding, metabolic stability)
- 25.Balani SK, Zhu T, Yang TJ, Liu Z, He B, Lee FW. Drug. Metab. Dispos. 2002;30:1059–1062. doi: 10.1124/dmd.30.10.1059. [DOI] [PubMed] [Google Scholar]
- 26.Jain AN. J. Med. Chem. 2004;47:947–961. doi: 10.1021/jm030520f. Analysis and surface figure generated using SybylX v1.2 on a Linux workstation. For Suflex Sim background. [DOI] [PubMed] [Google Scholar]